Advances in Biological Chemistry
Vol.3 No.3A(2013), Article ID:33483,6 pages DOI:10.4236/abc.2013.33A002
Different effects of the inhibition of Src activity on Akt/PKB in melanoma cells with wild BRAF and mutated BRAF V600E
![]()
Institute of Medical Biochemistry and Laboratory Diagnostics, First Faculty of Medicine, Charles University in Prague, Prague, Czech Republic
Email: tuhack@img.cas.cz
Copyright © 2013 Zdena Tuháčková et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 26 April 2013; revised 28 May 2013; accepted 6 June 2013
Keywords: HBL Melanoma Cells; SK-MEL-5 Melanoma Cells; BRAF V600E mutation; Src Kinase Activity; Akt/PKB Signaling
ABSTRACT
Src regulates cell adhesion, invasiveness, motility and growth in cancer cells. In melanoma, accumulating data show that Src inhibition can be effective and may enhance the effects of other agents. Increased Src expression and activity thus has recently become a target for drug therapy. Several melanoma cell lines were exposed to inhibitors of Src activity despite their broad specificity. To examine the particular activity of Src in human melanoma cells, we used SU6656, the selective inhibitor of Src family protein kinases. The activity of Src and cell proliferation were suppressed in HBL human cells, wild type melanoma cells and in SK-MEL-5 human melanoma cells harboring mutant BRAF V600E, upon their treatment with SU6656. The suppression of Src kinase activity had not inhibitory effects on Akt/PKB activity in SK-MEL-5 cells, which we have previously found in HBL cells. This may indicate that changes of Src involvement in the control of Akt/PKB activity and its downstream signaling could be induced by BRAF V600E mutation in SK-MEL-5 cells.
1. INTRODUCTION
Acquired resistance that is often developed to targeted therapies, has become a great clinical problem and is currently in the center of intensive clinical and basic research. Inhibitors of several protein kinase cascades in intracellular signaling serve as the therapeutic agents and some of them participate in various feedback loops that may induce drug resistance.
The drug rapamycin is a specific and efficient inhibitor of the mTOR-dependent signaling. Hyperactivation of the mTOR-dependent signaling pathway occurs in many human diseases, and rapamycin and its synthetic analogs, are currently used in cancer therapy. However, the inhibition of mTORC1 by rapamycin was shown also to induce phosphorylation and activation of Akt/PKB and its downstream signaling in some cancer cells [1], indicating the induction of a negative feedback mechanism that may reduce the anti-tumor effects of mTOR inhibition and develop drug resistance [2].
Another efficient inhibitor, vemurafenib is a smallmolecule drug that inhibits BRAF [3] and achieves clinical responses in patients with Braf—mutant melanoma. Over 50% of melanomas harbor activating V600E mutations in Braf gene (BRAF V600E), an oncogene known to be critical for the proliferation and survival of melanoma cells through activation of the RAF/MEK/MAPK pathway [4]. Despite the impressive initial responses, however, most patients treated with vemurafenib develop resistance after a relatively short period of treatment. Also other BRAF inhibitors may induce invasion and cell proliferation through paradoxical activation of MAPK pathway in melanoma cells [5,6].
The mitogen activated protein kinase (MAPK) are serinethreonine kinases activated by phosphorylation catalyzed by MEK, the MAPK kinase. MEKs are phosphorylated and activated by the RAF protein kinases. The RAF family is composed of three members (ARAF, BRAF, and CRAF) that exhibit a high degree of homology within three conserved regions. RAF family members are intermediate molecules in the MAPK signaling pathway that has been implicated in the development of 60% to 90% of melanomas, activating BRAF mutations playing the most prominent role in this process.
Activation of the BRAF/MEK/MAPK pathway has been shown to increase tumor cell proliferation, survival, invasion, and tumor angiogenesis [7]. It is a signal transduction pathway that conveys extracellular signals from the cell membrane to nucleus through a series of phosphorylation events and leads to the expression of genes associated with cell growth and survival. Suppression of BRAF expression in cultured melanoma cells inhibits the MAPK cascade and leads to growth arrest and promotes apoptosis in vitro and inhibits tumor development in animals [8].
We have recently found that pharmacological disruption of the Src tyrosine kinase activity in HBL melanoma cells, the wild type cells for BRAF mutation, resulted in the inhibition of mTORC1 signaling, similarly as by rapamycin treatment [9]. Moreover, unlike rapamycin, the inhibition by SU6656, a selective inhibitor of Src kinase activity [10], was not accompanied by the activation of Akt/PKB, but, on the contrary, by its inhibition in HBL melanoma cells [9].
SU6656, which is 2-oxo-3-(4,5,6,7-tetrahydro-1H-indol- 2-ylmethylene)-2,3-dihydro-1H-indole-5-sulfonic acid dimethylamide, was developed as a selective inhibitor of the ubiquitously expressed Src family kinases (Src, Fyn, and Yes). It has proved to be a useful tool to study Src protein function in cells without having to resort to transfection or microinjection systems. SU6656 had the same in vivo effects as other inhibitors of Src family kinases, such as dominant-negative constructs and neutralizing antibodies, at least in NIH 3T3 cells [10].
In this study, we used the small-molecule inhibitor SU6656, to analyze a role of Src tyrosine kinase activity in the deregulation of intracellular signaling in SK-MEL-5 human melanoma cells, harboring mutant BRAF V600E.
2. MATERIALS AND METHODS
All general reagents were from Sigma (St. Louis, MO) unless otherwise stated. ECL Prime WB detection reagent was obtained from GE Healthcare Bio-Sciences (Uppsala, Sweden). All primary and secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA).
2.1. Cell Culture and Preparation of Cell Extracts
Human melanoma cell lines used in this study were described previously [11]. HBL cells were cultivated in Dulbecco’s Modified Eagle’s Medium containing 10% fetal bovine serum (FBS), antibiotics, and L-glutamine. SK-MEL-5 cells were grown in Eagle’s minimal essential medium containing 10% FCS, antibiotics, L-glutamine, sodium pyruvate and non-essential amino acids. Normal human melanocytes were purchased from Cascade Biologics (Invitrogen, Carlsbad, CA) and maintained in the supplemented 254 medium as recommended by the manufacturer. Cells were brought to quiescence by an overnight incubation with 2% FCS and then treated with the inhibitor rapamycin or SU6656 added to a final concentration of 20 nM and 5 μM (DMSO solution diluted in a medium, 1:106), respectively, or DMSO carrier alone for time periods as indicated. The cell lysates were prepared in ice-cold extraction buffer [50 mM TRIS pH 7.4, 100 mM NaCl, 2 mM EDTA, 1% Triton X-100, 40 mM β-glycerophosphate, proteinase inhibitors aprotinin, leupetin and pepstatin (0.4 μg/ml each), 1 mM PMSF and PhosSTOP (Roche, Mannheim, Germany)]. Cell debris was pelleted by centrifugation at 20,000 g for 10 minutes, and supernatants were assayed for protein concentration.
2.2. SDS-PAGE and Western Blot Analysis
SDS-PAGE followed by immunoblot analysis were performed as described [12]. Briefly, 5× Laemmli sample buffer was added to clear cell lysates (15 μg). Samples were resolved on 10% or 12% SDS gels and proteins were transferred to Immobilon P (Millipore, Bedford, MA, USA). The membranes were blocked in TBS-T (TBS containing 0.1% Tween-20) with 5% milk, incubated with the indicated antibody, washed in TBS-T, incubated with relevant horseradish peroxidase-conjugated secondary antibody, washed again in TBS-T and revealed by enhanced chemiluminiscence followed by film exposure.
2.3. Proliferation Rates
HBL and SK-MEL-5 melanoma cells were seeded in triplicate wells, grown for periods of time as indicated, fixed in 3% paraformaldehyde in PBS for 10 min. Then they were stained with 0.1% crystal violet for 30 min, washed and unstained. The dye was quantified by photometry, absorbance was measured at 600 nm.
3. RESULTS
Elevated tyrosine kinase activity of the protein product of c-src protooncogene was found in a number of human tumors, also in melanoma cell lines and in melanoma tumors in vivo. Increased Src expression and activity thus have become a target for drug therapy. Several melanoma cell lines were exposed to inhibitors of Src activity alone or in combination with standard chemotherapy agents, despite of broad specificity of these inhibitors. To examine activity of Src in paticular, we used SU6656, the selective inhibitor of the Src family protein kinases [10] in our studies.
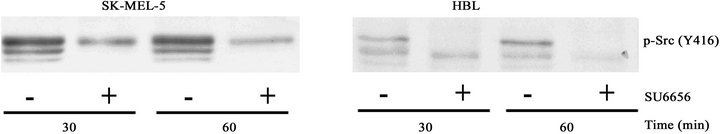
Inhibitory effects of SU6656 in HBL human cells, the wild type melanoma cells, and in SK-MEL-5 human melanoma cells harboring mutant BRAF V600E, are demonstrated in Figure 1. Activity of Src was suppressed in both cell types upon their treatment with SU6656 as indicated by the decreased binding of phospho-specific antibodies to the Src catalytic Tyr416 amino acid residues in these cells (Figure 1(a)).
Also growth of the analyzed cells was sensitive to SU6656. Exposure of HBL and SK-MEL-5 cells to SU6656, caused inhibition of the cells proliferation at 2.5 µM, 5 µM and 10 µM SU6656. The suppression of HBL cells proliferation by SU6656 shown to be more effective than the growth inhibition of SK-MEL-5 cells (Figure 1(b)).
In number of melanoma cell lines we have found a highly elevated activity of the mTOR-dependent signaling pathway, in contrast to normal human melanocytes (not shown). Treatment of HBL melanoma cells with rapamycin, the specific inhibitor of mTORC1 signaling pathway, inhibited the hyperactivity of mTORC1 signaling. However, rapamycin also increased phosphorylation of the regulatory amino acid residues, Ser473 and Thr308, in the Akt molecule and thus stimulation of Akt/PKB activity in HBL cells [9]. Similarly, increased phosphorylation of Akt/PKB at Ser473 and Thr308, and thus activation of Akt upon prolonged rapamycin treatment for 1 and 2 hrs, is shown in SK-MEL-5 cells (Figures 2(a) and (b)), indicating that a negative feedback mechanism has been induced by rapamycin treatment in melanoma cells independently on BRAF mutation.
We have previously demonstrated an important implication of Src tyrosine kinase activity in the regulation of the mTOR signaling pathway in v-src-transformed hamster fibroblasts [12] and in HBL human melanoma cells [9]. Treatment of these cells with SU6656 resulted in the decreased phosphorylation of signaling proteins in the mTORC1-dependent pathway and also strongly decreased phosphorylation of Akt at Ser473 and Thr308 in these cells. In SK-MEL-5 cells that carry BRAF V600E mutations, however, the suppression of Src kinase activity had not such inhibitory effects on Akt/PKB activity. Phosphorylation of Akt at Ser473 and Thr308 was not significantly inhibited in SK-MEL-5 cells upon SU6656 treatment, as shown in Figure 2.
 (a)
(a)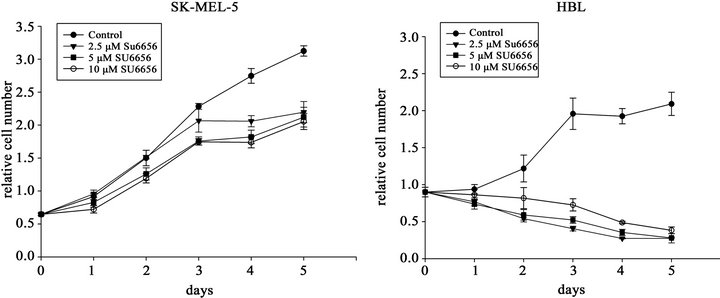 (b)
(b)
Figure 1. The inhibitory effects of the Src inhibitor SU6656 on Src phosphorylation (a) and proliferation rate (b) of SK-MEL-5 and HBL melanoma cells. (a) Phosphorylation of Src at Tyr416 in SK-MEL-5 and HBL cells. Western blots of the crude cell extracts (15 mg) of the melanoma cells SK-MEL-5 and HBL that were incubated with SU6656 (5 mM) (+) or with vehicle, DMSO (−), for periods of time as indicated. Phosphorylation of Src was examined using the specific anti-phosphoTyr416-Src antibody; (b) Cell proliferation rate of SK-MEL-5 cells and HBL cells. SK-MEL-5 and HBL cells were grown in the presence of DMSO (●), 2.5 mM SU6656, (▼) 5 mM SU6656 (■) or 10 mM SU6656 (○) for the time periods as indicated. The relative cell number was estimated by staining as described in Materials and Methods. The values are means of two experiments performed in triplicates ± SE.
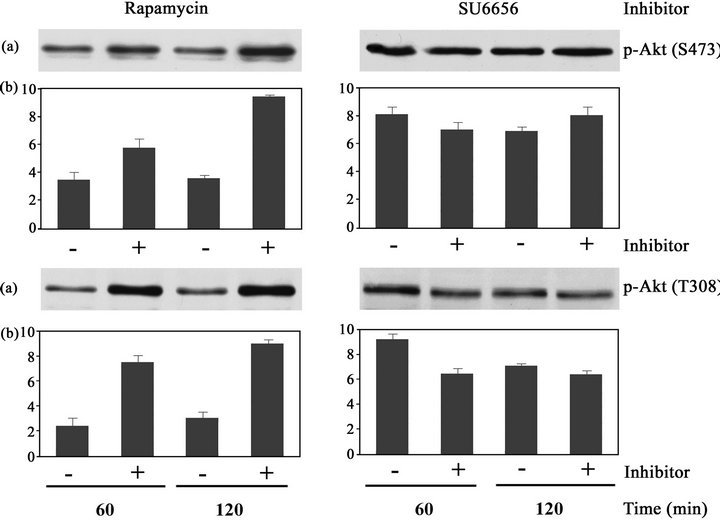
Figure 2. Phosphorylation of Akt/PKB at Ser473 and Thr308 in SK-MEL-5 cells that were treated with rapamycin or SU6656. Crude extracts (15 mg) of SK-MEL-5 melanoma cells that were incubated with SU6656 (5 mM) (+) or with rapamycin (20 nM) (+) or with vehicle, DMSO (−), for periods of time as indicated, were analyzed by Western blotting. (a) Phosphorylation of Akt/PKB at Ser473 and Thr308 was examined using the specific anti-phospho Ser473-Akt antibody and anti-phospho Thr308- Akt antibody. (b) Densitometric evaluation of the relevant protein bands. Data are the means of at least three independent experiments ± SE, from which one experiment is depicted.
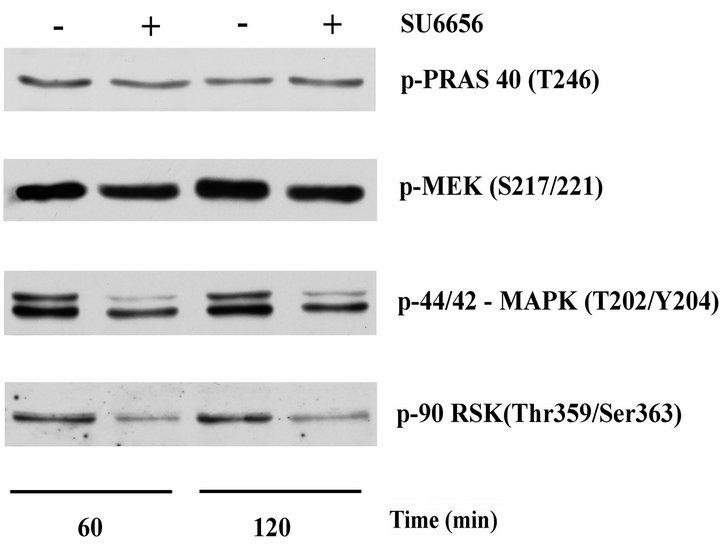
Figure 3. Changes in the phosphorylation of PRAS 40, MEK, MAPK, p90rsk, in SK-MEL-5 cells upon treatment with SU6656. Crude extracts (15 μg) of SK-MEL-5 melanoma cells that were incubated with SU6656 (5 μM) (+) or with vehicle, DMSO (-) for periods of time as indicated were analyzed by Western blotting. Phosphorylation of PRAS 40, MEK, MAPK (p44/42) and p90rsk was examined using the specific anti-phospho Thr246- PRAS40 antibody, anti-phospho Ser217/221-MEK anti-body, anti-phospho Thr202/Tyr204-MAPK (p44/p42) anti-body and anti-phospho Thr359/Ser363-p90rsk antibody. One experiment is depicted from at least three similar independent experiments.
Consequently, phosphorylation of Thr246 at PRAS40, one of the direct downstream targets of Akt [2], was unchanged by treatment of SK-MEL-5 cells with SU6656 (Figure 3). On the other hand, phosphorylation of MEK and their substrates, MAPK and protein kinase p90rsk, significantly decreased upon SU6656 treatment, indicating regulatory effects of Src tyrosine kinase activity on MEK/MAPK/p90rsk signaling in SK-MEL-5 melanoma cells carrying mutant BRAF V600E.
4. DISCUSSION
Mutant V600E is the most common activating BRAF mutation in melanoma. It is critical for tumor initiation and growth because BRAF gene mutations occur early during tumor progression and were shown to increase melanoma cell survival under hypoxic conditions [13]. Selective BRAF inhibitors potently suppress RAF-MEKMAPK signaling in BRAF-mutant melanoma cells. However, despite clinical efficacy of BRAF inhibitors, the same inhibitors unexpectedly activate RAF-MEK-MAPK signaling pathway in some different types of melanoma cells [14].
It has been recently reported that binding of BRAF inhibitors to wild-type BRAF in cells that carry an oncogenic mutation in the KRAS gene as well as in the cells in which the RAS-RAF-MEK-MAPK pathway is activated by other oncogenes such as HER2 [4] promotes formation of dimeric RAF complexes enhanced by the presence of an oncogenic (RAS or HER2) mutation [15]. It was suggested that the inhibitor might induce conformational changes in BRAF, which specifically impairs BRAF binding to other RAF kinases. Thus, inhibitors thought to be selective for mutant BRAF can activate wild CRAF, through the formation of dimeric RAF comexes, and consequently the MEK-MAPK pathway.
To improve effectivity of the BRAF selective inhibitors, combining therapy with other inhibitors of cellular signaling was used. Combined treatment with IGF-1R/PI3K and MEK inhibitors induced death of BRAF inhibitor-resistant cells [16], and Src and MEK inhibitors cooperated to block invasion in drug-sensitive melanoma cells [17]. The growth of resistant cells was blocked by a combination of EGFR and BRAF inhibitors in vitro and in vivo [18]. The EGFR-STAT3 signaling pathway was shown to mediate resistance to vemurafenib, the BRAF selective inhibitor, in BRAF-mutant melanomas, and BRAF and EGFR or SFK (Src family tyrosine kinases) inhibittion blocked proliferation and invasion of the resistant tumors [18,19].
The SFKs probably play a key role in mediating resistance to BRAF inhibitors in melanoma cells, as suggested by previous studies linking Src to BRAF inhibitor resistance in cell lines [20-22]. Activation of SFKs was found in a resistant tumor and dasatinib, the broadspecificity tyrosine kinase inhibitor, blocked the growth and metastasis of this tumor in immunocompromised mice, and also in the resistant cells in vitro and in vivo [23]. Preclinical evaluation of dasantinib shows that it has anti-proliferative, pro-apoptotic and anti-invasive effects in some melanoma cell lines, and combining dasatinib with chemotherapeutic agents improved response in melanoma cell lines [24].
It has been suggested that broad-specificity tyrosine kinase inhibitors—such as dasatinib—that target both non-receptor and receptor tyrosine kinases, could overcome some forms of drug resistance [18]. Src regulates cell adhesion, invasiveness and motility in cancer cells and specific inhibition of Src by small-molecule inhibitors or by genetic manipulation may induce cell death and/or reduce growth.
In this study we used the specific small-molecular inhibitor of Src kinase activity, SU6656 [9,10,12,25], to examine the role of Src kinase activity in the signaling in SK-MEL-5 melanoma cells carrying BRAF V600E associated mutation. We have found that unlike in HBL cells (wild-type melanoma cells), the disruption of Src activity did not result in the inhibition of Akt/PKB phosphorylation in BRAF mutated SK-MEL-5 cells. On the other hand, activity of the signaling pathway MEK/ MAPK was reduced upon treatment with SU6656 in both HBL (wild) and SK-MEL-5 (mutated) melanoma cells. This may indicate that changes of Src involvement in the control of Akt/PKB activity and its downstream signaling were induced by BRAF V600E mutation in SKMEL-5 cells and probably might contribute to drug resistance. Therefore, it would be especially important to establish the exact role of Src inhibition at the growthpromoting signaling pathways in different types of melanoma cells.
5. ACKNOWLEDGEMENTS
The study was supported by grant NT11231-3/2010 from the Ministry of Health of the Czech Republic
REFERENCES
- Chen, X.G., Liu, F., Song, X.F., Wang, Z.H., Dong, Z.Q., Hu, Z.Q., et al. (2010) Rapamycin regulates Akt and ERK phosphorylation through mTORC1 and mTORC2 signaling pathways. Molecular Carcinogenesis, 49, 603- 610. doi:10.1002/mc.20628
- Laplante, M. and Sabatini, D.M. (2012) mTOR signaling in growth control and disease. Cell, 149, 274-293. doi:10.1016/j.cell.2012.03.017
- Yang, H., Higgins, B., Kolinsky, K., Packman, K., Bradley, W.D., Lee, R.J., et al. (2012) Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Research, 72, 779-789. doi:10.1158/0008-5472.CAN-11-2941
- Hatzivassiliou, G., Song, K., Yen, I., Brandhuber, B.J., Anderson, D.J., Alvarado, R., et al. (2010) RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature, 464, 431-435. doi:10.1038/nature08833
- Bollag, G., Hirth, P., Tsai, J., Zhang, J., Ibrahim, P.N., Cho, H., et al. (2010) Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature, 467, 596-599. doi:10.1038/nature09454
- Shao, Y. and Aplin, A.E. (2010) Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Research, 70, 6670-6681. doi:10.1158/0008-5472.CAN-09-4471
- Ceol, C.J., Houvras, Y., Jane-Valbuena, J., Bilodeau, S., Orlando, D.A., Battisti, V., et al. (2011) The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature, 471, 513-517. doi:10.1038/nature09806
- Su, F., Bradley, W.D., Wang, Q., Yang, H., Xu, L., Higgins, B., et al. (2012) Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Research, 72, 969-978. doi:10.1158/0008-5472.CAN-11-1875
- Ondrušová, L., Réda, J., Žáková, P. and Tuháčková, Z. (2013) Inhibition of mTORC1 by SU6656, the selective Src kinase inhibitor is not accompanied by activation of Akt/ PKB signaling in melanoma cells. Folia Biologica, 59.
- Blake, R.A., Broome, M.A., Liu X., Wu, J., Gishizky, M., Sun, L., et al. (2000) SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Molecular and Cellular Biology, 20, 9018-9027. doi:10.1128/MCB.20.23.9018-9027.2000
- Vachtenheim, J., Novotná, H. and Ghanem, G. (2001) Transcriptional repression of the microphthalmia gene in melanoma cells correlates with the unresponsiveness of target genes to ectopic microphthalmia-associated transcription factor. The Journal of Investigative Dermatology, 117, 1505-1511. doi:10.1046/j.0022-202x.2001.01563.x
- Vojtěchová, M., Turečková, J., Kučerová, D., Šloncová, E., Vachtenheim, J. and Tuháčková, Z. (2008) Regulation of mTORC1 signaling by Src kinase activity is Akt1- independent in RSV-transformed cells. Neoplasia, 10, 99- 107. doi:10.1593/neo.07905
- Kumar, S.M., Yu, H., Edwards, R., Chen, L., Kazianis, S., Brafford, P., et al. (2007) Mutant V600E BRAF increases hypoxia inducible factor-1α expression in melanoma. Cancer Research, 67, 3177-3184. doi:10.1158/0008-5472.CAN-06-3312
- Zheng, B., Jeong, J.H., Asara, J.M., Yuan,Y.Y., Granter, S.R., Chin, L., et al. (2009) Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Molecular Cell, 33, 237-247. doi:10.1016/j.molcel.2008.12.026
- Poulikakos, P.I., Zhang, C., Bollag, G., Shokat, K.M., Rosen, N., et al. (2010) RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature, 464, 427-430. doi:10.1038/nature08902
- Villanueva, J., Vultur, A., Lee, J.T., Somasundaram, R., Fukunaga-Kalabis, M., Cipolla, A.K., et al. (2010) Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell, 18, 683- 695. doi:10.1016/j.ccr.2010.11.023
- Ferguson, J., Arozarena, I., Ehrhardt, M., Wellbrock, C. et al. (2013) Combination of MEK and SRC inhibition suppresses melanoma cell growth and invasion. Oncogene, 32, 86-96. doi:10.1038/onc.2012.25
- Girotti, M.R., Pedersen, M., Sanchez-Laorden, B., Viros, A., Turajlic, S., Niculescu-Duvaz, D., et al. (2013) Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discovery, 3, 158-167. doi:10.1158/2159-8290.CD-12-0386
- Niu, G., Bowman, T., Huang, M., Shivers, S., Reintgen, D., Daud, A., et al. (2002) Roles of activated Src and Stat3 signaling in melanoma tumor cell growth. Oncogene, 21, 7001-7010. doi:10.1038/sj.onc.1205859
- Homsi, J., Cubitt, C. and Daud, A. (2007) The Src signaling pathway: A potential target in melanoma and other malignancies. Expert Opinion Therapeutic Targets, 11, 91-100. doi:10.1517/14728222.11.1.91
- Homsi, J., Cubitt, C.L., Zhang, S., Munster, P.N., Yu, H., Sullivan, D.M., et al. (2009) Src activation in melanoma and Src inhibitors as therapeutic agents in melanoma. Melanoma Research, 19, 167-175. doi:10.1097/CMR.0b013e328304974c
- Ribas, A., Hersey, P., Middleton, M.R., Gogas, H., Flaherty, K.T., Sondak, V.K., et al. (2012) New challenges in endpoints for drug development in advanced melanoma. Clinical Cancer Research, 18, 336-341. doi:10.1158/1078-0432.CCR-11-2323
- Wu, W., Sun, Z., Wu, J., Peng, X., Gan, H., Zhang, C., et al. (2012) Trihydrophobin 1 phosphorylation by c-Src regulates MAPK/ERK signaling and cell migration. PLoS One, 7, e29920. doi:10.1371/journal.pone.0029920
- Eustace, A.J., Crown, J., Clynes, M., ODonovan, N., et al. (2008) Preclinical evaluation of dasatinib, a potent Src kinase inhibitor, in melanoma cell lines. Journal of Translational Medicine, 6, 53-56. doi:10.1186/1479-5876-6-53
- Jin, W., Yun, C., Hobbie, A., Martin, M.J., Sorensen, P.H., Kim, S.J., et al. (2007) Cellular transformation and activation of the phosphoinositide-3-kinase-Akt cascade by the ETV6-NTRK3 chimeric tyrosine kinase requires c-Src. Cancer Research, 67, 3192-3200. doi:10.1158/0008-5472.CAN-06-3526