Advances in Biological Chemistry
Vol. 2 No. 3 (2012) , Article ID: 21799 , 9 pages DOI:10.4236/abc.2012.23026
Activation of peroxisome proliferator activated receptor-gamma results in an atheroprotective apolipoprotein profile in HepG2 cells
![]()
Department of Chemistry and Biochemistry, California State University Northridge, Northridge, USA
Email: *jheem.medh@csun.edu, diala.dahabreh.54@my.csun.edu
Received 14 June 2012; revised 19 July 2012; accepted 27 July 2012
Keywords: PPAR-γ; Ciglitazone; Thiazolidinediones; Apolipoproteins; Hepatocytes; HepG2
ABSTRACT
Background: Insulin resistance is linked to dyslipidemia, characterized by a decrease in high density lipoproteins and an increase in low density lipoproteins. Thiazolidinediones (TZDs) are insulin-sensitizing agents used to improve glycemic control in patients with type 2 diabetes. Recently, the safety of certain TZD regimens has been questioned because of associated adverse effects on the plasma lipid profile. We examined the effect of a TZD, Ciglitazone, on apolipoprotein synthesis and secretion in human liver HepG2 cells. Methods and Results: The effect of Ciglitazone treatment on apolipoprotein synthesis was addressed at the level of transcription, translation and secretion. RT-PCR showed that Ciglitazone increased the transcription of apoE and apoAI but reduced the levels of apoCI and apoB mRNA. Western blot analysis showed an increase in apoAI and apoE secreted in the cell culture media, whereas the amounts of apoB100 and apoCI were reduced. To confirm that Ciglitazone regulates apolipoprotein translation, its effect on de novo protein synthesis was evaluated by metabolic labeling with [35S]-methionine/cysteine, and a similar pattern of regulation was observed. The change in apolipoprotein levels was not secondary to cholesterol biosynthesis or clearance, since Ciglitazone did not regulate the transcription of HMGCoA reductase, or the LDL receptor. However, mRNA levels for both PPAR-γ and LXRα were induced, suggesting a role for either or both receptors in modulating the hepatic apolipoprotein profile. The involvement of these nuclear receptor transcription factors was confirmed since direct activation of these receptors by endogenous PPAR-γ ligand, 15d-prostaglandin J2, or LXRα ligand, 22(R)hydroxy-cholesterol, similarly upregulated apoAI and apoE, but down-regulated apoB100 protein synthesis. Conclusion: Our results suggest that Ciglitazone treatment results in an atheroprotective lipopro- tein profile in liver cells. Thus, while the adipose and muscle tissues may be primary targets in TZD-mediated glucose homeostasis, liver PPAR-γ contributes significantly to the regulation of plasma lipoprotein profile.
1. INTRODUCTION
Thiazolidinediones (TZDs) are synthetic insulin-sensitizing agents used to treat type 2 diabetes [1]. TZDs activate peroxisome proliferator-activated receptor gamma (PPAR- γ), a member of the nuclear receptor family of transcription factors, to exert a wide range of plasma glucose-lowering strategies, including increased expression of glucose transporters, increased glucose uptake, and reduced hepatic glucose production [2].
Besides lowering plasma glucose levels, TZDs also modulate plasma lipid and lipoprotein profiles [2-4]. There are significant differences in the lipid effects of different TZDs [5,6]. Of the currently prescribed TZDs, Pioglitazone (Actos) significantly improves the plasma lipid profile by reducing fasting plasma triglycerides (TG), and increasing high density lipoprotein (HDL) cholesterol, whereas Rosiglitazone (Avandia) increases fasting plasma TG and induces a lower increase in plasma HDL levels [5,6]. These data are consistent with the conclusion that Rosiglitazone significantly increases the risk of myocardial infarction, while Pioglitazone is athero-neutral. The lipid effects of PPAR-γ agonists are thus important determinants of their suitability as therapeutic agents.
The molecular mechanisms responsible for the lipid effects of TZDs are not clear. The reduction in plasma TG levels may be secondary to the insulin-sensitizing effect of TZDs, due to reduced adipose lipolysis and lower hepatic very low density lipoprotein (VLDL)-TG production [4]. TZDs may divert plasma triglycerides for storage by stimulating adipose lipoprotein lipase (LPL) expression [7]. The TZD-dependent increase in HDL cholesterol may be mediated by up-regulation of macrophage ABCA1 expression, and a reduction in CETP-mediated exchange of HDL cholesterol with VLDL triglycerides [4,7]. Here, we have investigated if regulation of biosynthesis in the liver may contribute to the lipid effects of TZDs.
Most studies so far have focused on systemic effects rather than changes at the cellular level. The adipose tissue is a primary target of TZDs, but TZDs also regulate gene expression in the liver, muscle and macrophages [8,9]. The systemic effects are a sum total of the sometimes contradictory changes in different tissues. The liver contributes a majority of plasma lipids and apolipoproteins. Thus, it is important to investigate the effects of PPAR-γ activation on lipid and lipoprotein synthesis in the liver. In this study, we have investigated the effects of Ciglitazone, a selective PPAR-γ ligand, on apolipoprotein synthesis and secretion by human hepatoma HepG2 cells. Our results show that Ciglitazone increases the expression of apolipoprotein (apo)AI and apoE, but reduces the synthesis of apoB100 and apoCI, suggesting an atheroprotective contribution of the PPAR-γ-activated liver.
2. MATERIALS AND METHODS
2.1. Cell Culture and Treatments
HepG2 cells were obtained from ATCC and maintained in minimum essential medium Eagle (MEM) containing 1 mM sodium pyruvate, 1500 mg/L sodium bicarbonate, and 10% fetal bovine serum (FBS). Confluent monolayers in 6-well plates were treated with or without 5 μM Ciglitazone in MEM media supplemented with 1 mg/mL bovine serum albumin (BSA) instead of FBS. After 4 days, the cell culture media was collected and saved for detection of secreted apolipoproteins. The cells were washed with phosphate buffered saline (PBS) and harvested in TRI reagent (Sigma Chemical Co.) for RNA isolation.
2.2. RNA Isolation and RT-PCR
Total RNA was isolated using TRI reagent (Sigma) according to the manufacturer’s protocol and it was treated with RQ1 DNase to remove any contaminating genomic DNA. Two μg of RNA was used to synthesize cDNA using Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT), dNTPs, and oligodT primers (Promega).
The cDNA was subjected to conventional end-point PCR using primer pairs shown (Table 1) and GoTaq PCR mastermix (Promega). The PCR products were resolved on a 2% agarose gel and the DNA bands were quantified by ImageJ (NIH) analysis. Hydroxymethylbilane synthase (HMBS) or β-actin was used as the reference amplicon [10]. The cDNA was also subjected to real time Quantitative PCR using a Smart cycler (Cepheid Inc), SyBr green mix (Fisher Scientific), and primer pairs shown (Table 1). The threshold cycle (CT) values were used to calculate fold change in transcript levels using the 2–ΔΔCT method [11] allowing correction for the reference gene as follows: Fold change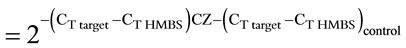 . All experiments were performed at least in triplicates with similar results.
. All experiments were performed at least in triplicates with similar results.
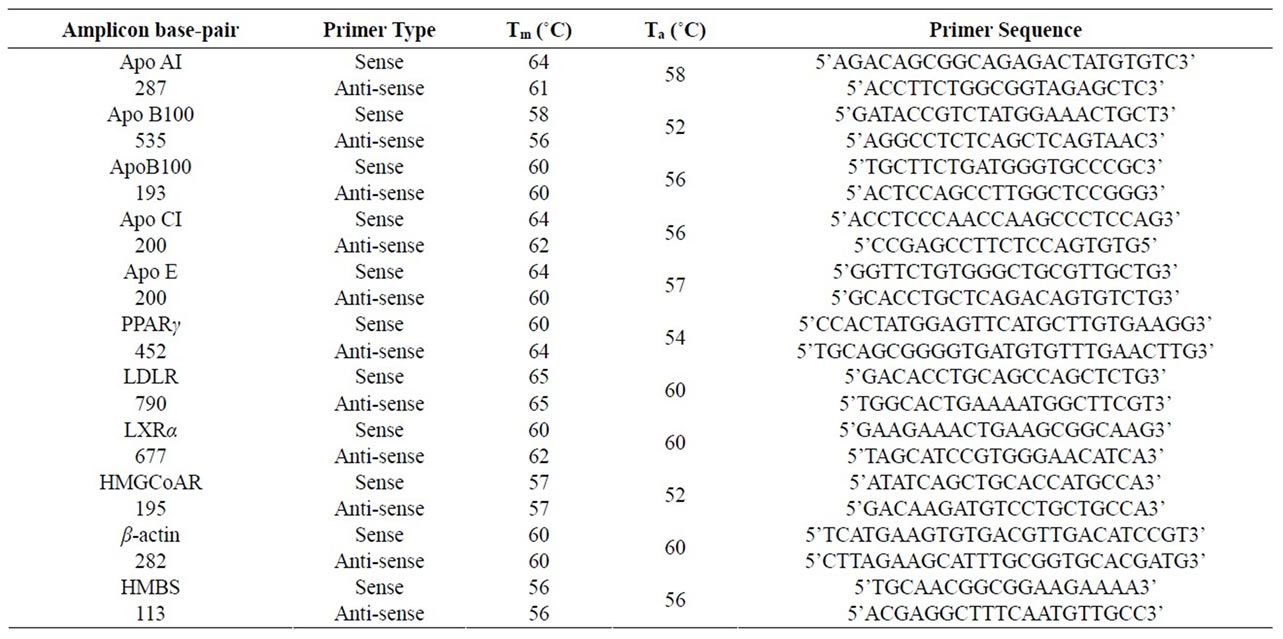
Table 1. PCR primers used in this study. Tm is the melt-temperature of each primer and Ta is the annealing temperature used for PCR. For apoB100, the primer pair giving the shorter amplicon was used for quantitative PCR, and the primer pair giving the longer amplicon was used for end-point PCR.
2.3. Western Blot Analysis of Apolipoproteins
Since apolipoproteins are secreted proteins, their levels were quantified in the cell culture media by specific immuno-precipitation and Western blot analysis. Rabbit polyclonal antibodies against human apoAI, apoE, and apoCI were prepared and characterized earlier. Each antibody was immobilized by covalently coupling to cyanogen bromide activated Sepharose 4B.
ApoB100 has a high affinity for heparin, hence it was precipitated using heparin-sepharose.
Briefly, cell culture media was collected after treatments described above and adjusted to a final concentration of 20% glycerol and 0.05% Triton X-100. One mL of this was mixed with a 50% suspension (100 μL) of the antibody-sepharose beads in immunoprecipitation (IP) buffer (20% glycerol, 0.15 M NaCl and 10 mM phosphate, pH 7.0). After incubation at 4˚C for 2 hr, the beads were washed several times with cold IP buffer and the adsorbed proteins were eluted with 50 μl of 1× Lammeli’s SDS-PAGE sample buffer. Equal volumes of immunoprecipitated samples were resolved by 10% SDS-PAGE (5% for apoB100) and transferred onto polyvinylidine fluoride membrane (ImmobilonTM-P) in transfer buffer (25 mM Tris, 192 mM glycine, 20% v/v methanol, pH 8.5). The blot was first incubated in blocking buffer (20 mM Tris, 150 mM NaCl, 2 mM CaCl2, 1% gelatin, 0.05% TWEEN-20, pH 7.4) overnight at 37˚C and then probed with corresponding anti-apolipoprotein-specific antibodies from SantaCruz BioTechnology. The antibodies used were apoAI:sc-23605, apoB100:sc-11795, apoCI:sc-101263, apoE:sc-31822. The SuperSignal West Pico Chemiluminescent Substrate kit (Thermo Scientific) was used to detect the protein bands. Results of Western Blot analysis were quantitated using the ImageJ software available from NIH.
2.4. Metabolic Labeling of Apolipoproteins
HepG2 cells were grown and treated with 5 μM Ciglitazone for 4 days as described above. In some experiments, cells were treated with the LXR ligand (25 μM 22Rhydroxycholesterol (Sigma) + 10 μM 9-cis-retinoic acid, (Sigma)) or the PPAR-γ ligand (3 μM 15-deoxy-Δ-12,14- prostaglandin J2 (15d-PGJ2, Cayman) + 10 μM 9-cis-retinoic acid). Next, cells were equilibrated for 10 min in methionine-free medium (ATCC) containing 1 mg/mL BSA.
The medium was then supplemented with 100 μCi/ml of TRANS-[35S] (MP Biomedicals) and the cells were incubated for 90 min at 37˚C. Synthesis of nascent radiolabeled apoproteins was chased to completion by addition of 10 mM unlabelled methionine, and further incubation for 30 min. Radiolabeled apoB100, apoE, apoAI and apoCI were precipitated from the culture media as described above, and resolved by SDS-PAGE. The gels were fixed for 30 min in 50% methanol/10% acetic acid, soaked in 1M salicylic acid/15% glycerol for 30 min, dried, and exposed to autoradiography film at –70˚C for 2 days.
2.5. Statistical Analysis
All experiments were repeated at least 3 times. The intensity of DNA and/or protein bands were quantitated using Image J software (NIH). The data were corrected for the housekeeping gene and the percentage change was calculated in treated samples compared to controls. The gel images from one representative experiment are shown alongside bar graphs showing averages and standard deviation of 3 or more experiments.
3. RESULTS
Recently, there was significant debate about the cardiovascular risk of TZDs [5,6]. Pioglitazone is favored due to its similar glucose-lowering ability, but lower cardiovascular risk than Rosiglitazone. It is hypothesized that Pioglitazone’s desirable characteristics may result from its ability to activate PPAR-α at higher concentrations [2]. In this study, we have investigated the specific effects of PPAR-γ activation on apolipoprotein synthesis at the cellular level in hepatocytes. We chose to use Ciglitazone which is a selective PPAR-γ ligand [12], and a precursor compound of all three TZDs listed above [2].
3.1. Ciglitazone Regulates Apolipoprotein mRNA Expression in HepG2 Cells
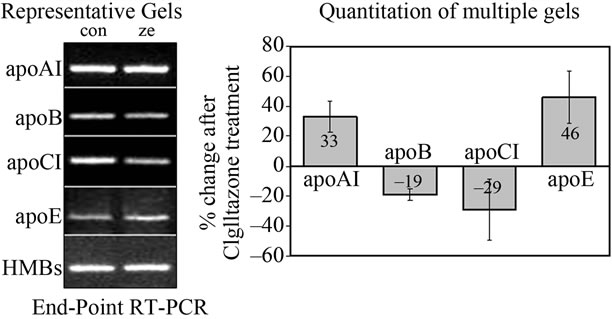
We examined the ability of Ciglitazone to regulate apolipoprotein transcription and translation in HepG2 cells. Treatment with Ciglitazone for 96 hr did not alter the cellular morphology relative to control cells. For end-point PCR, the level of hydroxymethylbilane synthase (HMBS) transcript was used as a control for RNA loading on the gel, and remained unchanged after treatment (Figure 1). Activation of PPAR-γ with 5 μM Ciglitazone increased apoE transcript levels by 46% over untreated cells, while there was a 33% increase in the level of apoAI mRNA compared to untreated cells (Figure 1(b)). The levels of apoB and apoCI transcripts were reduced to 81% and 71% of control levels, respectively (i.e. a reduction of 19% and 29% respectively). Results are averages of multiple experiments with n = 3 for apoAI and apoE, n = 4 for apoCI and n = 5 for apoB.
Since end-point PCR is not strictly quantitative, apolipoprotein levels in control and Ciglitazone-treated cells were compared by quantitative real-time (Q-RT) PCR using a Cepheid SmartCycler. HMBS was used as the reference gene. The CT values were obtained using primer pairs shown in Table 1 and Sybr Green as the intercalating dye. The melt curves showed a melting temperature (Tm) of 85˚C or above indicating specific amplification. The Tm for apoB was 82˚C. The fold change over control was calculated using the 2–ΔΔCT method [13] as follows:
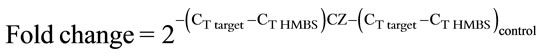
Accordingly, Ciglitazone treatment induced a 5-fold increase in the level of apoAI transcript and almost 10-fold increase in apoE transcript (Table 2). There was no significant change in apoCI mRNA levels. The level of apoB mRNA was dramatically reduced after ciglitazone treatment to just 2% of untreated cells in the experiment shown; in repeat experiments, the apoB CT value remained undetermined in Ciglitazone-treated samples, indicating negligible template levels. For all other genes, a similar pattern of regulation was observed in three independent experiments. The increase in apoAI and apoE transcripts ranged from 3- to 8-fold and 5- to 15-fold, respectively. The fold-change for apoCI varied between 0.62 and 1.28.
3.2. Ciglitazone Treatment Regulates Secretion of Apolipoproteins from HepG2 Cells
Activation of HepG2 cells with Ciglitazone also regulated the amount of apolipoprotein protein secreted in the culture medium, as determined by immunoprecipitation and Western blot analysis (Figure 2). Consistent with the mRNA data, Ciglitazone induced apoAI and apoE protein levels by 23% and 53% respectively, but reduced the levels of apoCI and apoB100 by 14% and 8% respec-
 (a) (b)
(a) (b)
Figure 1. Ciglitazone treatment regulates apolipoprotein transcription in HepG2 human hepatoma cells. HepG2 cells were cultured as described in “Methods”. Cells were incubated in media alone (control) or treated with 5 mM Ciglitazone (CZ) for 4 days. Total RNA was isolated using TRI reagent, and subjected to end-point RT-PCR using specific primer pairs as shown in Table 1. After resolution of the PCR products on a 1% agarose gel (a), the relative intensity of each band was determined by Image J analysis. Data from multiple experiments were used to calculate the change in intensity of apolipoprotein band in CZtreated cells, relative to control, and corrected for HMBS (b). n = 3 for apoAI and apoE, n = 4 for apoCI, and n = 5 for apoB.
tively, in the culture medium. The amount of apolipoprotein in the culture medium is a sum total of protein synthesis, secretion and degradation. Thus, it appears that apoB100 mRNA levels were drastically reduced, but the amount of apoB100 protein is reduced only marginally.
3.3. Ciglitazone Regulates De Novo Synthesis of Apolipoproteins
To directly investigate the effect of Ciglitazone on apolipoprotein translation, newly synthesized proteins were labeled with TRANS-[35S], a mix of [35S]-L-Methionine
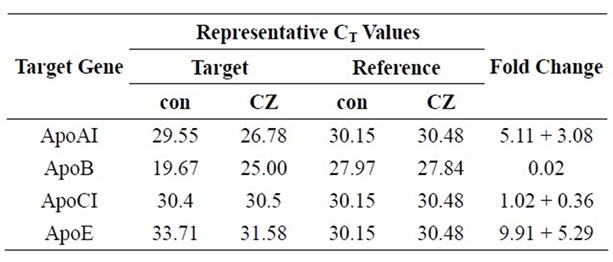
Table 2. Quantitative-RT-PCR results. Quantitative real-time RTPCR was performed on a Cepheid SmartCycler using primer pairs for each apolipoprotein (Table 1) or HMBS. The threshold cycle (CT) values from a representative experiment are shown. The CT values were used to calculate the amount of apolipoprotein cDNA in CZ-treated cells relative to control using the 2–ΔΔCT method as follows: Fold change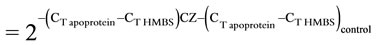 . The last column shows the average of 3 independent experiments, except for apoB, which showed a measurable CT value in only one experiment.
. The last column shows the average of 3 independent experiments, except for apoB, which showed a measurable CT value in only one experiment.
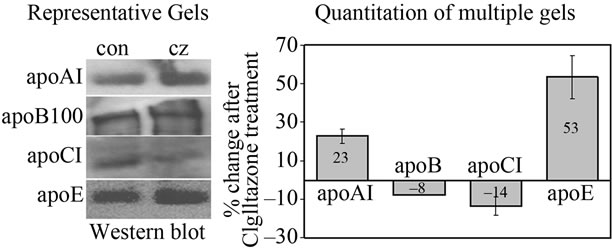 (a) (b)
(a) (b)
Figure 2. Ciglitazone modulates the level of apolipoproteins accumulated in cell culture media of HepG2 cells. HepG2 cells were incubated in the presence or absence of 5 μM CZ for 4 days. ApoAI, apoCI and apoE were immunoprecipitated from the cell culture media using specific anti-apoprotein antibodies immobilized by conjugation to sepharose beads. ApoB100 was precipitated using heparin-sepharose. The precipitated proteins were resolved on 10% SDS-PAGE (apoAI, apoCI and apoE) or 5% SDS-PAGE (apoB100), transferred to PVDF and subjected to Western blot analysis as described in “Methods”. A representative gel is shown in (a) The band intensity in CZ samples is calculated as a percent of the intensity in control samples, and data from multiple experiments is averaged. Averages of multiple experiments are shown in (b) n = 3 for apoAI, apoCI and apoE; n = 1 for apoB100.
and [35S]-L-Cysteine. Secreted apolipoproteins were immunoprecipitated and detected by SDS-PAGE and autoradiography. The direction of regulation was consistent with the RT-PCR and Western blot data. Ciglitazone significantly up-regulated de novo synthesis of apoAI and apoE by 50% and 44%, respectively, but down-regulated the synthesis of apoB100 by 30% (Figure 3). ApoCI could not be detected using this method, probably because it contains only one methionine and no cysteine [14]. Its small size may also cause it to diffuse out of the gel during the fixing and equilibration steps.
3.4. Ciglitazone Treatment Increases Expression of PPAR-γ and LXR-α
The plasma lipoprotein profile can be altered by influencing cholesterol synthesis or cholesterol clearance. The liver is responsible for both processes, which are regulated, respectively, by HMGCoA Reductase, the rate-limiting enzyme in cholesterol biosynthesis, and by the low density lipoprotein (LDL) receptor, which mediates hepatic uptake of circulating LDL. As shown in Figure 4, Ciglitazone treatement did not alter the transcription of either of these two genes. However, there was a 49% and 42% increase, respectively (n = 4), in the level of PPAR-γ and liver X receptor (LXR)-α transcripts suggesting that either or both nuclear receptors/transcription factors may directly contribute to Ciglitazone-mediated regulation of apolipoprotein expression.
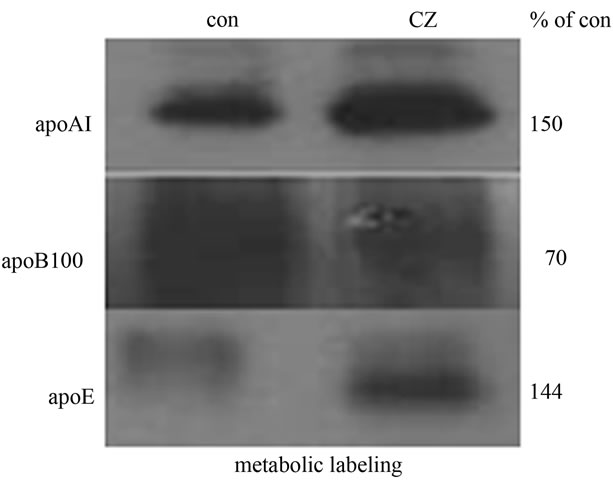
Figure 3. Ciglitazone treatment regulates de novo apolipoprotein synthesis. Confluent HepG2 cells were treated without any agonist (con), or with 5 μM Ciglitazone (CZ). Cells were metabolically labeled with 100 Ci/ml of TRANS-[35S] (MP Biomedicals) as described. Radiolabeled apoAI, apoB100, and apoE were isolated from the culture media by immunoprecipitation, resolved by SDS-PAGE, and detected by autoradiography. Image J analysis was used to calculate The intensity of bands in CZtreated cells relative to control were calculated using Image J and are shown to the right.
3.5. PPAR-γ and LXRα Contribute to Regulation of Apolipoprotein Synthesis in HepG2 Cells
Activation of PPAR-α is known to regulate lipoprotein metabolism in the liver [4]. Ciglitazone is a PPAR-γ-specific ligand and does not activate PPAR-α [12]. However, to confirm that PPAR-γ contributes to apolipoprotein synthesis, HepG2 cells were treated with a non-TZD, endogenous ligand of PPAR-γ, 15-deoxy-Δ-12,14-prostaglandin J2 (15dPGJ2). Metabolic labeling experiments showed an up-regulation of apoAI and apoE, and a down-regulation of apoB100 de novo synthesis (Figure 5), confirming a direct role for PPAR-γ in regulation of apolipoprotein biosynthesis.
Since Ciglitazone treatment induced the expression of LXRα in HepG2 cells (Figure 4), it is likely that LXRα may contribute to regulation of apolipoproteins by Ciglitazone. To investigate the effect of LXRα activation on apolipoprotein synthesis, HepG2 cells were treated with the specific LXRα ligand, 22(R)hydroxyl cholesterol (HC) in the presence of 9-cis retinoic acid. Metabolic labeling with TRANS-[35S] revealed a similar pattern of regulation as with Ciglitazone (Figure 5). Incorporation of radiolabel in apoE was significantly enhanced, and apoB synthesis was down-regulated. There was no remarkable effect on apoAI synthesis. Thus, since LXRα has a PPAR response element (PPRE) [15], it is likely that the effect of Ciglitazone on apolipoprotein synthesis is mediated by upregulation of PPAR-γ followed by binding of PPAR-γ- Ciglitazone complex to LXRα, resulting in increased expression of LXRα which then is activated by endogenous ligands (oxysterols) resulting in regulation of apolipoprotein synthesis.
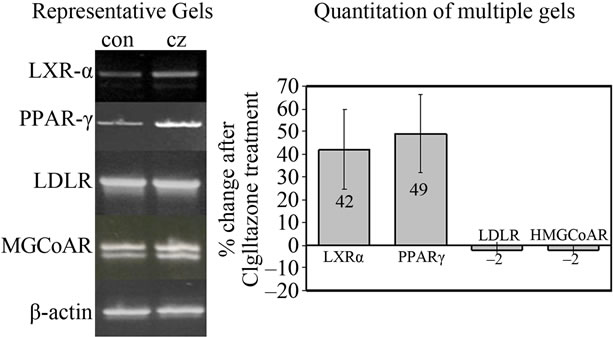 (a) (b)
(a) (b)
Figure 4. Ciglitazone up-regulates PPAR-γ and LXR-α mRNA levels in HepG2 cells. HepG2 cells were treated with or without 5 μM CZ for 4 days and total RNA was isolated as described in Figure 1. End-point RT-PCR was performed using primer pairs shown in Table 1. Representative gel images are shown in (a) Image J analysis was used to calculate the intensity of bands in CZ-treated cells relative to control. Average results from multiple experiments (n = 4) after correction for β-actin used as the housekeeping gene are shown in (b).
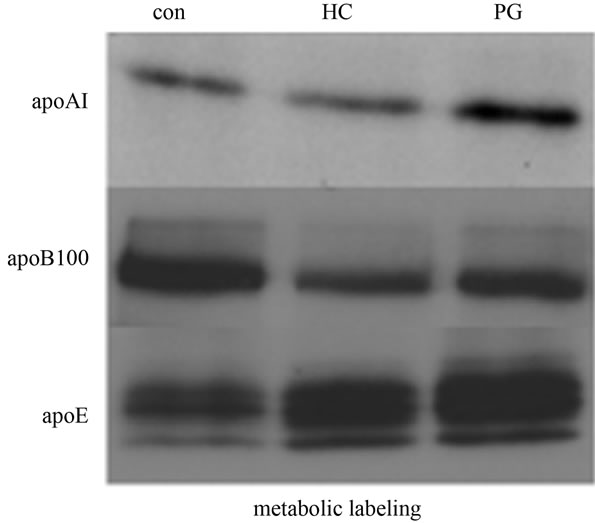
Figure 5. Activation of PPAR-γ or LXRα regulates de novo apolipoprotein synthesis. Confluent HepG2 cells were treated without any agonist (con), or with 25 μM 22R-hydroxycholesterol + 10 μM 9-cis-retinoic acid (HC), or 3 μM 15-deoxy- Δ-12,14-prostaglandin J2 + 10 μM 9-cis-retinoic acid (PG). Cells were metabolically labeled with 100 μCi/ml of TRANS-[35S] (MP Biomedicals) as described. Radiolabeled apoAI, apoB100, and apoE were isolated from the culture media, resolved by SDS-PAGE, and detected by autoradiography.
4. DISCUSSION
Insulin resistance and dyslipidemia are related metabolic disorders. Insulin resistance is correlated to lipid abnormalities including increased triglycerides, and decreased HDL cholesterol levels [16]. Thus, it is not surprising that insulin-sensitizing agents such as TZDs also correct dyslipidaemia in human subjects or animal models [5,6]. By improving insulin sensitivity, TZDs promote fat storage in adipose tissue and reduce fat utilization and accumulation in the muscle and liver. The deficiency of lipids may, in turn, regulate lipoprotein synthesis and secretion to present an atheroprotective lipid profile. Such a mechanism of correcting dyslipidemia requires cross-talk and regulation of metabolism between different tissues.
Adipose PPAR-γ is the primary target of TZD treatment, but liver, muscle and macrophages also contribute to the systemic TZD response [8,9]. The liver plays a dominant role in lipoprotein metabolism, being the primary site for the synthesis of cholesterol, triglycerides and various apolipoproteins including apoB100, apoAI, apoCI and apoE. As protein components of plasma lipoproteins, apolipoproteins dictate how much lipoprotein is assembled and secreted outside the cells. TZD regulation of hepatic lipid metabolism is significant because cardiovascular risk is correlated to lipid metabolism; and hepatotoxicity is a probable side effect of TZD therapy [5,6]. In order to determine how the liver adjusts its apolipoprotein expression profile in response to TZD treatment, we investigated the effect of ciglitazone on apolipoprotein synthesis and secretion in HepG2 cells.
Our data show that Ciglitazone treatment results in upregulation of atheroprotective apolipoproteins apoE and apoAI, and a down-regulation of atherogenic apolipoproteins apoCI and apoB100. The regulation is at the level of transcription, translation, and secretion, as evident from RT-PCR, Western blot analysis and metabolically labeled proteins in the HepG2 culture media. The difference seen in the end-point PCR and real-time PCR data for apoCI could be due to variability in assay sensitivity.
ApoE is a component of triglyceride-rich VLDL particles and HDL particles. It plays a prominent role in reverse cholesterol transport and is thought to inhibit platelet aggregation; thus it is athero-protective in nature. The liver is a primary site for apoE synthesis, though it is also expressed in the brain and macrophages. The gene for apoE is located on chromosome 19 and is part of a apolipoprotein gene cluster that includes apoE, apoCI, apoCII and apoCIV. A functional PPRE consensus sequence was identified in the apoE/apoCI intergenic region [10]; this region contains regulatory elements for both apoE and apoCI genes. Accordingly, it was demonstrated that apoE expression was significantly induced upon PPAR-γ activation in macrophages.
The apoE gene also contains a LXR response element (LXRE) in the apoE/apoCI intergenic region [11]. All four apolipoproteins in the apoE/CI gene cluster were induced upon activation of macrophages with LXR ligands, and the regulation of apoE and apoCI was abolished in LXRα/β null mice [11]. A recent report demonstrates that induction of apoE in adipocytes by PPAR-γ activation requires the presence of a LXRE within the apoE/apoCI intergenic regulatory region [17].
The upregulation of hepatic apoE in our study is consistent with TZD-mediated upregulation of macrophage and adipocyte apoE. Since Ciglitazone treatment upregulated hepatic LXRα expression, regulation of apoE and apoCI by Ciglitazone may be mediated via PPRE and/or LXRE. It is interesting that apoE and apoCI are regulated in opposite directions by Ciglitazone whereas Mak et al. have shown LXR-dependent upregulation of all 4 apolipoproteins from the apoE gene cluster in macrophages [11]. The down-regulation of apoCI may be a hepato-specific event, or it could a dose-related response. The downregulation of apoCI is consistent with the anti-diabetic role of TZDs since, apoCI is an inhibitor of lipoprotein lipase and over-expression of apoCI is associated with hypertriglyceridemia [18].
ApoAI is the primary apolipoprotein of HDL and its gene is regulated by a number of nuclear receptor transcription factors including hepatocyte nuclear factor 4a (HNF-4a), apoAI regulatory protein-1 (ARP-1), and retinoid X receptor (RXRα) [19]. The apoAI gene promoter contains a PPRE, and activators of PPARα, such as fibrates, increase plasma HDL and apoAI levels in humans [20]. Similarly, TZD therapy is correlated with a significant increase in plasma HDL levels [4]. Previous studies have shown that Pioglitazone increases apoAI transcription in HepG2 cells [21], and it was shown to be a weak activetor of PPARα [2,21]. Thus, it has been suggested that the apoAI inducing effect of TZDs may be mediated via PPARα.
In the present study, Ciglitazone induced the expression of apoAI from HepG2 cells. So far, there is no report of Ciglitazone’s ability to bind and activate PPARα [13]. Also, metabolic labeling experiments showed that apoAI synthesis was up-regulated by the endogenous PPAR-γ- specific ligand, 15d-PGJ2, thus confirming a direct role for PPARγ in apoAI regulation.
Plasma apoB100 levels are modulated by its synthesis, degradation, and assembly into VLDL particles by the liver. Synthesis and stability of apoB100 is directly correlated with the availability of triglycerides. Insulin resistance is accompanied by accumulation of triglycerides and increased production of apoB100 [16], and insulin sensitization reduces TG levels [5,6]. Although the apoB gene lacks a PPRE, there is a definite correlation between PPAR-γ and apoB100 metabolism. Activation of PPAR-γ by Pioglitazone reduces plasma VLDL/TG levels, possibly because of it’s ability to increase lipoprotein lipase-mediated conversion of VLDL to LDL. Such a mechanism is suggested by the finding that TZD treatment results in a steady increase in plasma LDL levels [5]. Few studies have investigated the correlation between TZD treatment and plasma apoB100 levels (most have studied plasma LDL levels), and a direct effect of TZD on the synthesis of hepatic apoB100 has not yet been established.
As demonstrated here, activation of PPAR-γ in liver cells leads to a down-regulation of apoB100 synthesis. This was shown with Ciglitazone and 15d-PGJ2, two different classes of PPAR-γ ligands. In results not shown here, Ciglitazone was also found to down-regulate hepatic triglyceride synthesis by 40%. Thus, the combined effect on triglycerides and apoB100 would ensure a lowering of VLDL assembly and secretion.
TZDs are known to affect a number of genes in various tissues, often with conflicting functions. Thus, in human or animal studies, the systemic effect is the sum total of multiple tissues and target genes. In the cell culture assay, there is no interference of other organs/tissues, hence, it is possible to study the primary regulation of individual genes by specific transcription factors such as PPARγ, and LXRα.
Based on the data presented here, it is clear that hepatic PPAR-γ is a direct target of TZD and that activation of PPAR-γ in HepG2 cells results in an atheroprotective lipoprotein profile. A role for liver PPAR-γ in TZD action was reported earlier by Gavrilova et al. in normal and lipoatrophic mice [22]. They reported that Rosiglitazone’s ability to reduce serum triglyceride and serum glucose levels was abolished when the liver PPAR-γ was knocked-out in lipoatrophic mice, but not in wild-type mice. Thus, in the absence of adipose tissue, the liver is a major target of Rosiglitazone. But, in the presence of adipose tissue, liver PPAR-γ is not required for Rosiglitazone’s hypoglycemic effect. However, liver PPAR-γ was found to be a contributor of fat tolerance and triglyceride homeostasis [22]. Our studies suggest that liver PPAR-γ may help reduce hypertriglyceridemia by reducing its synthesis/secretion (downregulation of apoB100) and increasing its clearance (upregulation of apoE and LPL). Thus, while the adipose and muscle tissues may be primary targets in TZD-mediated glucose homeostasis, liver PPAR-γ contributes significantly to the regulation of lipid and lipoprotein profile.
5. CONCLUSION
Our data clearly demonstrate that Ciglitazone upregulates apoAI and apoE mRNA and protein expression in HepG2 cells, but it down-regulates apoB100 and apoCI transcription and translation. Since both PPAR-γ and LXR-α are upregulated by Ciglitazone, either or both receptors may contribute to Ciglitazone-mediated regulation of apolipoprotein expression. This study suggests that the liver may be a direct and important target of therapeuticcally administered TZDs.
6. ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health Awards S06GM8680, R15HL083946, and 1SC3GM095413.
![]()
![]()
REFERENCES
- Olefsky, J.M. (2000) Treatment of insulin resistance with peroxisome proliferators-activated receptor γ agonists. Journal of Clinical Investigation, 106, 467-472. doi:10.1172/JCI10843
- Smith, U. (2002) Thiazolidinedione-induced effects beyond glycemic control. British Journal of Diabetes & Vascular Disease, 2, S24-S27.
- Walczak, R. and Tontonoz, P. (2002) PPRadigms and PPARadoxes: Expanding roles for PPAR-gamma in the control of lipid metabolism. Journal of Lipid Research, 43, 177-186.
- Kersten, S. (2008) Peroxisome proliferator activated receptors and lipoprotein metabolism. PPAR Research, 2008, 132960. doi:10.1155/2008/132960
- Goldberg, R.B., Kendall, D.M., Deeg, M.A., Buse, J.B., Zagar, A.J., Pinaire, J.A. Tan, M.H., Khan, M.A., Perez, A.T. and Jacober, S.J. (2005) A comparison of lipid and glycemic effects of pioglitazone and rosiglitazone in patients with type 2 diabetes and dyslipidemia. Diabetes Care, 28, 1547-1554. doi:10.2337/diacare.28.7.1547
- Van Wijk, J.P.H., De Koning, E.D.P., Martens, E.P. and Rabelink, T.J. (2003) Thiazolidinediones and blood lipids in type 2 diabetes. Arteriosclerosis, Thrombosis, and Vascular Biology, 23, 1744-1749. doi:10.1161/01.ATV.0000090521.25968.4D
- Lee, C., Olson, P. and Evans, R.M. (2003) Minireview: Lipid metabolism, metabolic diseases, and peroxisome proliferators-activated receptors. Endocrinology, 144, 2201- 2207. doi:10.1210/en.2003-0288
- Wolf, G. (2004) Tissue-specific knockout defines peroxisome proliferators-activated receptor gamma function in muscle and liver. Nutrition Reviews, 62, 253-255. doi:10.1301/nr2004.jun253-255
- Glass, C.K. (2001) Potential roles of the peroxisome proliferator-activated receptor-γ in macrophage biology and atherosclerosis. Journal of Endocrinology, 169, 461-464. doi:10.1677/joe.0.1690461
- Galetto, R., Albajar, M., Polanco, J.I., Zakin, M.M., Rodriguez-Rey, J.C. (2001) Identification of a peroxisome proliferator-activated receptor response element in the apolipoprotein E gene control region. Biochemical Journal, 357, 521-527. doi:10.1042/0264-6021:3570521
- Mak, P.A., Laffitte, B.A., Desrumaux, C., Joseph, S.B., Curtiss, L.K., Mangelsdorf, D.J., Tontonoz, P. and Edwards, P.A. (2002) Regulated expression of the apolipoprotein E/C-I/C-IV/C-II gene cluster in murine and human macrophages. Journal of Biological Chemistry, 277, 31900-31908. doi:10.1074/jbc.M202993200
- Willson, T.M., Cobb, J.E., Cowan, D.J., Wiethe, R.W., Correa, I.D., Prakesh, S.R., Beck, K.D., Moore, L.B., Kliewer, S.A. and Lehmann, J.M. (1996) The structureactivity relationship between peroxisome proliferator-activated receptor-γ agonism and the antihyperglycemic activity of thiazolidinediones. Journal of Medicinal Chemistry, 39, 665-668. doi:10.1021/jm950395a
- Livak, K.J. and Schmittgen, T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔC T method. Methods, 25, 402-408. doi:10.1006/meth.2001.1262
- Jackson, R.L., Sparrow, J.T., Baker, H.N., Morrisett, J.D., Taunton, O.D. and Gotto, A.M. Jr. (1974) The primary structure of apolipoprotein-serine. Journal of Biological Chemistry, 249, 5308-5313.
- Laffitte, B.A., Joseph, S.B., Walczak, R., Pei, L., Wilpitz, D.C., Collins, J.L. and Tontonoz, P. (2001) Autoregulation of the human liver X receptor α promoter. Molecular and Cellular Biology, 21, 7558-7568. doi:10.1128/MCB.21.22.7558-7568.2001
- Goldberg, I.J. (2001) Diabetic dyslipidemia: Causes and consequences. Journal of Clinical Endocrinology & Metabolism, 86, 965-971. doi:10.1210/jc.86.3.965
- Yue, L. and Mazzone, T. (2009) Peroxisome proliferatoractivated receptor-γ stimulation of adipocyte apoE gene transcription mediated by the liver receptor X pathway. Journal of Biological Chemistry, 284, 10453-10461. doi:10.1074/jbc.M808482200
- Berbee, J.F.P., Van der Hoogt, C.C., Sundararaman, D., Havekes, L.M. and Rensen, P.C.N. (2005) Severe hypertriglyceridemia in human apoC1 transgenic mice is caused by apoC-I-induced inhibition of LPL. Journal of Lipid Research, 46, 297-306. doi:10.1194/jlr.M400301-JLR200
- Malik, S. (2003) Transcriptional regulation of the apolipoprotein AI gene. Frontiers in Bioscience, 8, 360-368. doi:10.2741/1005
- Staels, B. and Auwerx, J. (1998) Regulation of apoA-I gene expression by fibrates. Atherosclerosis, 137, S19- S23. doi:10.1016/S0021-9150(97)00313-4
- Qin, S., Liu, T., Kamanna, A.S. and Kashyap, M.L. (2007) Pioglitazone stimulates apolipoprotein A-I production without affecting HDL removal in HepG2 cells. Arteriosclerosis, Thrombosis, and Vascular Biology, 27, 2428- 2434. doi:10.1161/ATVBAHA.107.150193
- Gavrilova, O., Haluzik, M., Matsusue, K., Cutson, J.J., Johnson, L., Dietz, K.R., Nicol, C.J., Vinson, C., Gonzales, F.J. and Reitman, M.L. (2003) Liver peroxisome proliferator-activated receptor-γ contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. Journal of Biological Chemistry, 278, 34268-34276. doi:10.1074/jbc.M300043200
LIST OF ABBREVIATIONS
15d-PGJ2: 15-deoxy-Δ-12,14-prostaglandin J2; ABCA1: ATP-binding cassette protein A1; apo: apolipoprotein, BSA: bovine serum albumin; cDNA: complementary DNA; CETP: cholesterol ester transfer protein, CT: threshold cycle; CZ: Ciglitazone; dNTP: deoxy nucleotide triphosphates; FBS: fetal bovine serum; HC: 22R-hydroxycholsterol; HDL: high density lipoprotein; HMBS: hydroxylmethylbilane synthase; IP: immunoprecipitation, LDL, low density lipoprotein; LPL: lipoprotein lipase, LXR: liver X receptor; LXRE: LXR regulatory element; MEM: minimum essential medium Eagle; PBS: phosphate buffered saline; PCR: polymerase chain reaction; PG: 15-deoxy-Δ-12,14-prostaglandin J2; PPAR: peroxisome proliferator activated receptors; PPRE: PPAR regulatory element; RT: reverse transcription; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; TG: triglycerides; TZD: thiazolidinediones; VLDL: very low density lipoproteins.
NOTES
*Corresponding author.