Open Journal of Physical Chemistry
Vol. 3 No. 1 (2013) , Article ID: 27879 , 7 pages DOI:10.4236/ojpc.2013.31002
Density Functional Theory Studies on the Addition and Abstraction Reactions of OH Radical with Benzoate Anion
Department of Environmental Science and Technology, Faculty of Engineering, Shinshu University, Nagano, Japan
Email: *ntanaka@shinshu-u.ac.jp
Received November 28, 2012; revised December 29, 2012; accepted January 27, 2013
Keywords: DFT Calculation; Benzoate Anion; H-Atom Abstraction; OH Addition; Transition State Theory
ABSTRACT
The addition and abstraction reactions of OH radical with benzoate anion are investigated by density functional theory calculations that include solvent effects using UB3LYP, UCAM-B3LYP, UmPW1PW91 and UM06-2X functionals with the 6-311++G(2d,2p) basis set. Geometry optimizations of the reactants, products and transition state species are performed for the possible reaction paths. For the addition reactions, those targeting the ipso-, ortho-, metaand para-carbons are predicted to be exoergic. The H-atom abstraction reactions from ortho, meta and para positions are also predicted to be exoergic. On the basis of the rate constants calculated by means of the transition state theory, the H-atom abstraction reaction from the ortho position is determined to be the favored path followed by the ortho OH addition reaction.
1. Introduction
Sodium salicylate is known as a good phosphor under the x-ray and vacuum ultraviolet excitation [1]. Since its proposal as a sensitive dosimeter [2], the salicylate has been also utilized as a probe to measure OH radical generation in aqueous solutions where salicylate anion is generated by the reaction of benzoate anion with OH radical. This method is widely used in radiochemical, sonochemical and photocatalytic reactions [3] as well as the method using the 2-hydroxyterephthalate dianion [4]. Peräkylä et al. [5] have theoretically studied the reactivity of benzoate towards OH addition reaction at the ROHF/6-31G(d) and ROMP2/6-31G(d) levels including solvent effects on the basis of the qualitative frontier molecular orbital theory and compared the stability of the isomeric OH adducts of benzoate. They revealed the correlation between the kinetic reactivity and the thermodynamic stabilities of the addition products. However, no theoretical study of the detailed kinetics and thermochemistry of the reactions has been reported. As concerns the water environmental chemistry, the OH radical initiated reactions of aromatic compounds have been studied [6,7]. The rate constants, the branching ratios and product distribution were in good agreement with the experimental values. In the present study, we have focused on the kinetics and thermochemistry of the primary steps; namely, OH radical addition to the benzoate anion and H-atom abstraction from the benzoate anion (Scheme 1).
2. Computational Methods
The equilibrium geometries of the reactants, transition states, products and complexes were optimized using the density functional theory (DFT) method. Hybrid GGA functionals B3LYP [8] and mPW1PW91 [9], hybrid meta-GGA functional M06-2X [10] and range-separated hybrid GGA functional CAM-B3LYP [11] were employed with the 6-311++G(2d,2p) basis set. As compared with the standard B3LYP functional, the mPW1PW91 and CAM-B3LYP improve long-range behavior and the M06-2X functional has been recommended for thermochemistry and kinetics by the authors. Harmonic vibrational frequencies were calculated to confirm the predicted structures as local minima or transition states (one imaginary frequency) and elucidate zero-point vibra-

Scheme 1. Possible reaction paths for benzoate anion + OH reaction.
tional energy corrections (ZPE). The obtained transition states were confirmed as those connecting the investigational species by a calculation of the subsequent intrinsic reaction coordinates. The solvation effects were included using a polarizable continuum method (IEF-PCM). All calculations were performed using Gaussian 09 [12]. The spin-squared values were checked and the deviations from the doublet value of
The rate constants for the addition and abstraction reactions were estimated using the conventional thermodynamic formulation of the transition state theory. The rate constant k is given by
 (1)
(1)
where kB is the Boltzmann constant, T is the temperature, h is Planck’s constant, ΔG≠ is the Gibbs free energy of activation and κ is the transmission coefficient. The tunneling effect was approximated using the Wigner correction [13].
3. Results and Discussion
3.1. OH Addition Reaction
There are five possible sites for OH addition, i.e. carbonyl, ipso, ortho, meta and para carbons of benzoate anion. The optimized structures of benzoate anion and addition products for the addition reaction are shown in Figure 1. For the carbonyl, ipso, meta and para addition products, respectively two minima were found on the CCOH torsion potentials, where the O-H bonds point toward or away from the ring. For the ortho addition product, two conformers are present, where the O-H bond points toward the ring or toward the COO group to form the H-bond. Some optimized geometrical parameters are also given in Figure 1.
Table 1 lists the relative electronic energies, including the zero-point vibrational energies (ΔrE), relative enthalpies at 298 K (ΔrH) and relative Gibbs energies at 298 K (ΔrG). The ΔrG for the carbonyl addition reactions are calculated to be positive while others to be negative. The spin density of the carbonyl addition product is localized on the oxygen atoms of the OH added COO group, whereas those of the other products are delocalized to gain the resonance stabilization energies. The ortho1 addition product was calculated to be the thermodynamiccally most stable one due to an intramolecular hydrogen bond with CO···HO distances in the range of 1.707 (UmPW1PW91) to 1.826 (UM06-2X) Å. The ipso1 addition product also forms an intramolecular hydrogen bond with CO···HO distances in the range of 1.758 (UmPW1PW91) to 1.816 (UM06-2X) Å.
Optimized structures of the transition states for the OH
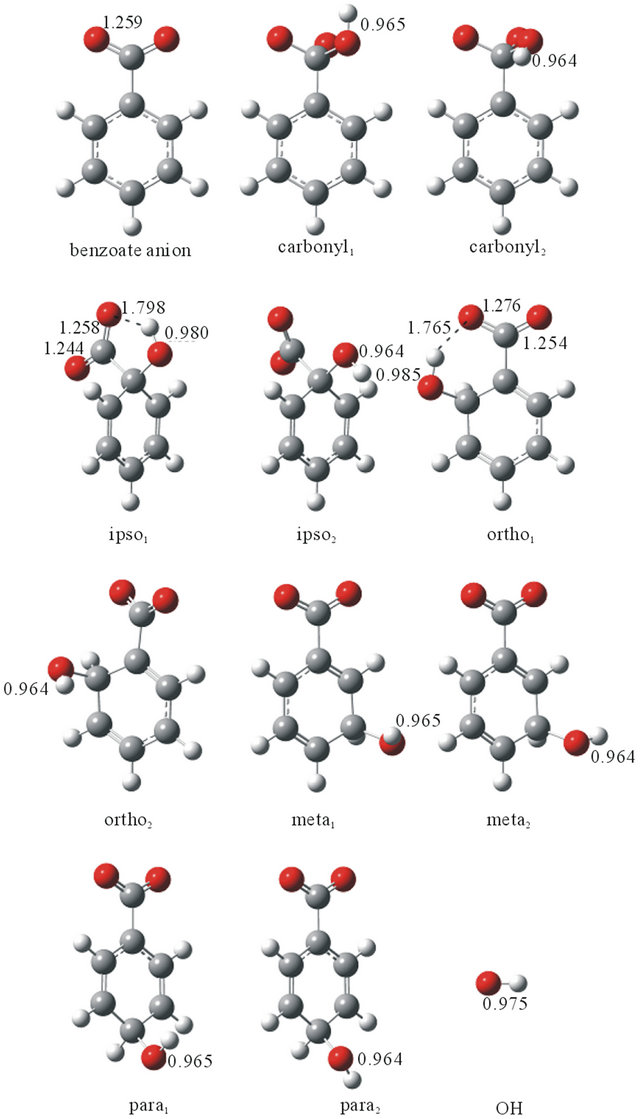
Figure 1. Optimized structures of the reactants and OH addition products calculated at the UB3LYP/6-311++G (2d,2p) level of theory.
addition reactions and complexes calculated at the UB3LYP/6-311++G(2d,2p) level of theory are shown in Figure 2. Transition states TScarbonyl1, TScarbonyl2, TSipso1, TSipso2, TSortho1, TSortho2, TSmeta and TSpara correspond to the formation of addition products carbonyl1, carbonyl2, ipso1, ipso2, ortho1, ortho2, meta1 and para1, respectively. IRC calculations revealed that transition states TSortho1 and TSipso1 are connected with complex1, and TSortho2, TSmeta and TSpara are connected with complex2, complex3 and complex4, respectively.
The imaginary frequencies and relative energies of the transition states and distance ratios of rC···OH/rC–OH are summarized in Table 2. The C···OH distances in the transition states for the ipso, ortho, meta and para addition reactions are elongated by over 40% as compared with those of the addition products, indicative of an early transition state. On the contrary, in the transition states
Table 1. Thermochemical parameters (kcal∙mol−1) of addition products.

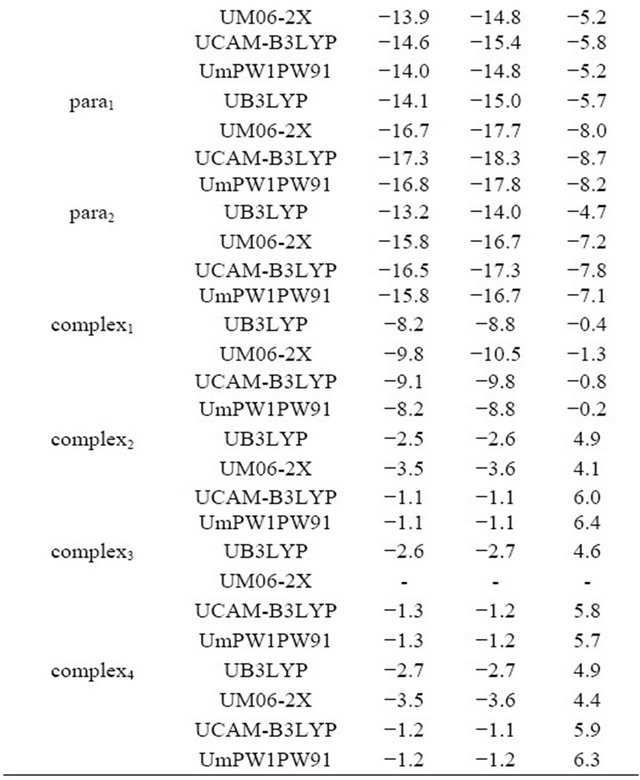
Figure 2. Optimized structures of the transition states for OH addition reaction and complexes calculated at the UB3LYP/6-311++G(2d,2p) level of theory.
for the carbonyl addition reaction, the C···OH distances are approximately 20% longer than the equilibrium distances in the addition products, indicative of a late transition state. The ΔG≠ value for the addition of OH to carbonyl carbon is found to be much higher than those of others. The ΔG≠ values for the formation of ortho1 and para1 are comparable and the lowest, though it depends on the functionals. Therefore, ortho and para positions should be the kinetically most favored sites for addition of OH radical. In complex1, the O-H bond length is elongated by 4.0% (UB3LYP) to 4.6% (UM06-2X) as compared with that of the bare OH radical. The CO bond interacting with the OH radical is also elongated while the other CO bond is contracted as compared with that of the benzoate anion. The ΔrG values for the formation of the complex1 are calculated to a range from −1.3 (UM06-2X)
Table 2. Calculated imaginary frequencies, CO distance ratios and thermochemical parameters (kcal∙mol−1) of the transition states and rate constants for addition reactions.
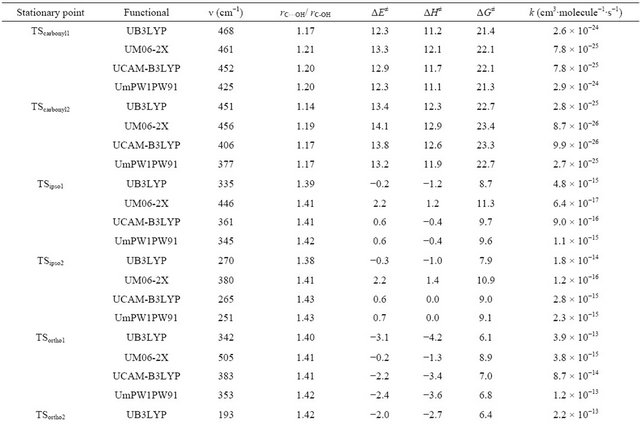
to −0.4 kcal∙mol−1 (UB3LYP). The ΔrH values for the formation of the complex2, complex3 and complex4 are calculated to be negative while ΔrG values to be positive. Therefore, at 298 K the formation of complex2, complex3 and complex4 are negligible. Complex3 was unable to be located on the potential energy surfaces at the UM06-2X level.
The functional dependence of ΔrG shows a similar trend for each stationary point. The value of ΔrG increases in the order of UmPW1PW91 ≈ UM06-2X ≈ UCAM-B3LYP < UB3LYP for the products and ΔG≠, in the order of UB3LYP < UmPW1PW91 ≈ UCAMB3LYP < UM06-2X.
The calculated rate constants for OH addition to the carbonyl, ipso, ortho, meta and para carbons are listed in Table 2. The rate constants for the carbonyl addition are much lower than those for others owing to a large barrier height. Total rate constants for the addi tion reaction were obtained to be 5.2 × 10−12, 1.1 × 10−12, 7.3 × 10−13 and 3.2 × 10−14 cm3∙mol−1∙s−1 using UB3LYP, UmPW1- PW91, UCAM-B3LYP and UM06-2X functionals, respectively. The UB3LYP value is slightly smaller than the experimental one, 9.8 × 10−12 cm3∙mol−1∙s−1 [14].
3.2. H-Atom Abstraction Reaction
Optimized structures of the relevant stationary points for H-atom abstraction from the benzoate anion by the OH radical calculated at the UB3LYP/6-311++G(2d, 2p) level of theory are shown in Figure 3. Table 3 lists the ΔE≠, ΔH≠ and ΔG≠ values for the H-atom abstraction reactions. For H-atom abstractions from the ortho and meta positions, the transition states were optimized by starting from the three initial geometries where the OH molecular axis was perpendicular to the molecular plane of the benzoate anion or the OH molecular axis was in the plane of the benzoate anion with either the H-atom or O-atom of the OH radical pointing to the carbonyl oxygen atom. For H-atom abstraction from the para position, the calculation was started from the two initial geometries where the OH molecular axis was perpendicular to or in the molecular plane of the benzoate anion. TSab_ortho1 was calculated to be planar with the OH···OC distance in the range from 1.650 (mPW1PW91) to 1.704 (B3LYP) Å. TSab_ortho2 was calculated to be nonplanar with the O-H bond pointing away from the carbonyl group. In the TS structures for the meta and para H-atom abstraction reactions, the OH molecular axis was perpendicular to the molecular plane of the benzoate anion. The breaking C-H bonds are elongated by about 17, 14, 12 and 13% as compared with the equilibrium C-H bond of the benzoate anion while the forming O-H bonds are elongated by about 28, 35, 37 and 35% as compared with the O-H bond length of H2O for TSab_ortho1, TSab_ortho2, TSab_meta and TSab_para, respectively for the UB3LYP calculation. The reactant-like geometry is characteristic of an early transition state. The ΔG≠ values of TSab_ortho1 are smaller than those of TSab_ortho2 owing to hydrogen bonding stabilization.
The calculated rate constants for H-atom abstraction are listed in Table 3. The rate constant for the abstraction reaction via TSab_ortho1 is one to three orders of magnitude larger than those for the other abstraction reactions. Total rate constants for the abstraction reaction were obtained to be 7.0 × 10−12, 4.0 × 10−12, 1.4 × 10−12 and 1.3 × 10−13 cm3 mol−1 s−1 using UB3LYP, UmPW1- PW91, UCAM-B3LYP and UM06-2X functionals, respectively. Figure 4 gives the potential energy profile for the reaction pathway. The product distribution was obtained for the UB3LYP calculations as follows: carbonyladdition:ipso-addi-tion:ortho-addition:meta-addition:para-addition:ortho-abstraction:meta-abstraction:para-abstraction = 0.0:0.4:19.9:8.6: 13.7:50.7:4.7:2.1. Comparing the rate constants for the addition reactions versus the abstraction reaction, it can be concluded that the latter is the slightly favored reaction channel for the reaction of OH radical with the benzoate anion.
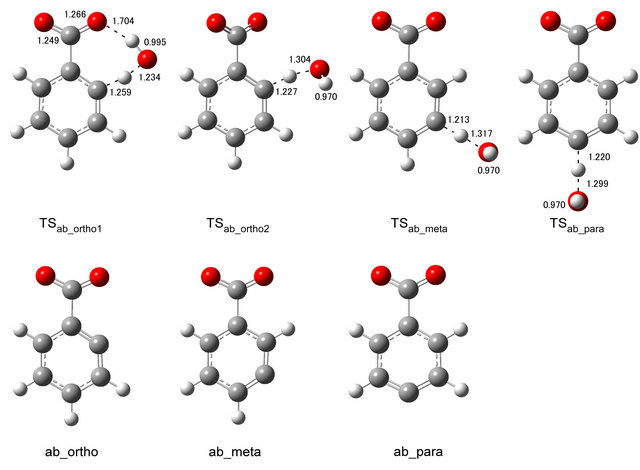
Figure 3. Optimized structures of the transition states for H-atom abstraction reaction and abstraction products calculated at the UB3LYP/6-311++G(2d,2p) level of theory.
Table 3. Calculated imaginary frequencies, ratios of HO and CH distances, and thermochemical parameters (kcal∙mol−1) of the transition states and rate constants for abstraction reactions.

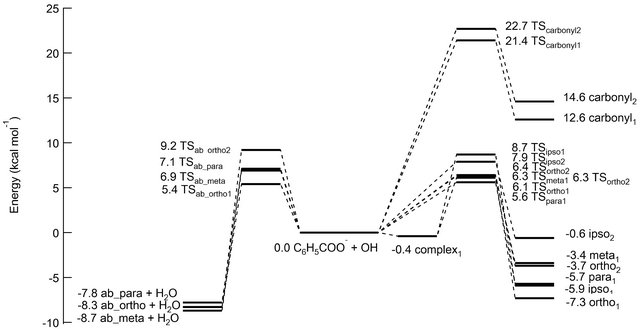
Figure 4. Potential energy diagram for the addition and H-atom abstraction reactions of OH + benzoate anion calculated at the UB3LYP/6-311++G(2d,2p) level of theory.
4. Conclusion
The kinetics and thermochemistry of the addition and abstraction reactions of OH radical with benzoate anion have been investigated using DFT. On the basis of the rate constants calculated by means of the transition state theory, the H-atom abstraction reaction from the ortho position is determined to be the favored path followed by the ortho OH addition reaction. Hydrogen bonds present in the transition states influence the reaction rates.
REFERENCES
- C. B. Zurro, “A Broad-Band Plasma Radiation Detector with Spatial-Resolution Based on the Optical-Scanning of the Fluorescence of a Phosphor,” Review of Scientific Instruments, Vol. 62, No. 1, 1991, pp. 118-123. doi:10.1063/1.1142318
- W. A. Armstrong and D. W. Grant, “A Highly Sensitive Chemical Dosimeter for Ionizing Radiation,” Nature, Vol. 182, No. 4637, 1958, p. 747.
- G. L. Newton and J. R. Milligan, “Fluorescence Detection of Hydroxyl Radicals,” Radiation Physics and Chemistry, Vol. 75, No. 4, 2006, pp. 473-478.
- S. E. Page, W. A. Arnold and K. McNeill, “Terephthalate as a Probe for Photochemically Generated Hydroxyl Radical,” Journal of Environmental Monitoring, Vol. 12, No. 9, 2010, pp. 1658-1665. doi:10.1039/c0em00160k
- M. Peräkylä and T. A. Pakkanen, “Ab Initio Molecular Orbital Study on Reactivity of Phenol, Biphenyl, Benzoate and p-Hydroxybenzoate in the ·OH Addition Reaction and Stability of the Corresponding ·H and ·OH Cyclohexadienyl Adducts,” Journal of the Chemical SocietyPerkin Transactions 2, No. 7, 1995, pp. 1405-1410. doi:10.1039/P29950001405
- M. Kılıç, G. Koçtürk, N. San and Z. Çınar, “A Model for Prediction of Product Distributions for the Reactions of Phenol Derivatives with Hydroxyl Radicals,” Chemosphere, Vol. 69, No. 9, 2007, pp. 1396-1408. doi:10.1016/j.chemosphere.2007.05.002
- A. Hatipoglu, D. Vione, Y. Yalçın, C. Minero and Z. Çınar, “Photo-Oxidative Degradation of Toluene in Aqueous Media by Hydroxyl Radicals,” Journal of Photochemistry and Photobiology A: Chemistry, Vol. 215, No. 1, 2010, pp. 59-68. doi:10.1016/j.jphotochem.2010.07.021
- C. Lee, W. Yang and R. G. Parr, “Development of the Colic-Salvetti Correlation-Energy Formula into a Functional of the Electron Density,” Physical Review B, Vol. 37, No. 2, 1988, pp. 785-789. doi:10.1103/PhysRevB.37.785
- C. Adamo and V. Barone, “Exchange Functionals with Improved Long-Range Behavior and Adiabatic Connection Methods without Adjustable Parameters: The mPW and mPW1PW Models,” The Journal of Chemical Physics, Vol. 108, No. 2, 1998, pp. 664-675. doi:10.1063/1.475428
- D. G. Truhlar and Y. Zhao, “The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals,” Theoretical Chemistry Accounts, Vol. 120, No. 1-3, 2008, pp. 215-241. doi:10.1007/s00214-007-0310-x
- T. Yanai, D. P. Tew and N. C. Handy, “A New Hybrid Exchange-Correlation Functional Using the CoulombAttenuating Method (CAM-B3LYP),” Chemical Physics Letters, Vol. 393, No. 1-3, 2004, pp. 51-57. doi.org/10.1016/j.cplett.2004.06.011
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery, J. A., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, “Gaussian 09, Revision B.01,” Gaussian, Inc., Wallingford CT, 2009.
- E. Wigner, “Crossing of Potential Thresholds in Chemical Reactions,” Zeitschrift für Physikalische Chemie, Vol. B19, 1932, pp. 203-216.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, “Critical Review of Rate Constants for Reations of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (·OH/·O−) in Aqueous Solution,” Journal of Physical and Chemical Reference Data, Vol. 17, No. 2, 1988, pp. 513-886. doi:10.1063/1.555805
NOTES
*Corresponding author.