Journal of Tuberculosis Research
Vol.03 No.02(2015), Article ID:57310,8 pages
10.4236/jtr.2015.32005
Comparison of the Sensitivities of Different PCR Assays for the Detection of Mycobacterium tuberculosis Complex Isolates from Five Regions of Cameroon
Fidelis Cho-Ngwa1, Irene A. Anyangwe1, Emiliene T. Berinyuy1, Henry D. Meriki1, Jean-Paul Assam-Assam2, Vincent P. Titanji1
1Biotechnology Unit, Faculty of Science, University of Buea, Buea, Cameroon
2Laboratory of Tuberculosis Research, Biotechnology Centre of Nkolbisson, Faculty of Science, University of Yaoundé I, Yaoundé, Cameroon
Email: fidelis.ngwa@fulbrightmail.org
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 25 April 2015; accepted 18 June 2015; published 23 June 2015
ABSTRACT
The accurate diagnosis of tuberculosis caused by members of the Mycobacterium tuberculosis complex (MTBC) has remained a major challenge in clinical laboratories world-wide. Several studies have evaluated the use of highly specific in-house PCR assays targeting the IS6110, hupB, rpoB, oxyR, and IS1081 genes in the detection of MTBC species with reports on variable sensitivities depending on the geographical sourcing of isolates. In the present investigations, we evaluated the sensitivities of these PCR assays on 125 MTBC cultured isolates from five (West, Centre, Littoral, North West and South West) of the ten regions of Cameroon. Of this number, 124 (99.2%), 117 (93.6%), 123 (99.1%), 119 (95.2%) and 118 (94.4%) were positive by the IS6110, hupB, rpoB, oxyR, and IS1081-based PCR assays respectively. A total of 110 (88%) of the cultured isolates were also identified as MTBC by standard biochemical tests. Of this number, 109 (99.1%), 104 (94.5%), 109 (99.1%), 106 (96.4%) and 104 (94.5%) were positive in the IS6110, hupB, rpoB, oxyR, and IS1081-based PCR assays respectively. Concordant PCR results were obtained for 108 of the 125 samples. The 15 isolates that were negative biochemically scored sensitivities ranging from 100% (for the IS6110 assay) to 86.7% (for the hupB and oxyR assay). The combination of the IS6110 assay, which turned out to be the most sensitive, and each of the other assays gave 100% sensitivity. We conclude that the combined targeting of the IS6110 and rpoB genes is likely to yield the most sensitive PCR procedure for the diagnosis of MTBC infection in the five regions of Cameroon.
Keywords:
PCR-Assays, Sensitivities, MTB Complex

1. Introduction
The causative agents of tuberculosis (TB) are members of the Mycobacterium tuberculosis complex (MTBC). The group includes M. tuberculosis (Mtb), M. africanum, M. bovis, M. canetti, and M. microti. They share a > 99% similarity at the nucleotide level. Mtb is the major cause of TB in humans. While TB due to MTBC strains is the most common mycobacterial infection in developing countries, many non-tuberculous mycobacteria (NTM) are also of medical relevance, especially for immuno-compromised patients [1] . Approximately a third of the world’s population is latently infected with MTBC, of which 5% - 10% of people not co-infected with HIV may ultimately develop active [2] . An estimated 9.4 million new cases of active TB and 1.7 million deaths were recorded in 2009 alone [2] . TB remains an important public health challenge in the 10 regions of Cameroon, a country with over 20 million inhabitants, where the prevalence of the disease is estimated to be 237 per 100,000 inhabitants, and mortality rates, 29 per 100,000 [3] .
The rapid diagnosis of active TB is required for rapid initiation of the right therapy to avoid the devastating effects of late-stage infections, and for the isolation of patients to avoid the spread of disease. The detection and identification of Mtb through culture is presently the gold standard for diagnosis. In clinical laboratories in Cameroon, TB diagnosis is principally by microscopy and chest X-ray analysis. These tests suffer seriously from low sensitivity and specificity. As a result, many patients with pulmonary TB (PTB) are often left untreated. A battery of other tests based on bacterial growth, phenotypic and biochemical properties have also been frequently used in more specialized laboratories in Cameroon and elsewhere to identify MTBC in clinical specimens. However, these tests are time-consuming, laborious, inaccurate, and not always reproducible [4] . Alternative rapid detection and identification methods, including the polymerase chain reaction (PCR), have more recently been developed and evaluated for clinical and field use [5] -[7] .
Several authors have proposed TB tests based on in-house PCR. The latter is technically faster, less laborious and perhaps cheaper than both culture and biochemical techniques. However, the choice of the most favourable target sequence is conflict-ridden since results from different geographical regions are at odds concerning sensitivity. This is most likely the consequence of genetic variations within the bacteria from one region to another.
Hitherto, there has been no study evaluating the sensitivities of popular in-house PCR assays based on the IS6110 [8] , IS1081 [9] , rpoB [10] , oxyR [10] and hupB [11] genes on isolates or clinical specimens from Cameroon. All these assays scored 100% specificity each when they were developed and evaluated. In the present study, we evaluated these PCR assays in order to guide the selection of the most sensitive one or combination in the PCR-based diagnosis of TB in the most economically vibrant regions of Cameroon.
2. Methods
2.1. Study Design and Study Population
This was a cross-sectional survey in which samples were collected between January and June 2009. The study population comprised of suspected PTB patients visiting five specialized centres, one in each region, for the detection and treatment of TB. These were the Regional Hospital Limbe (for the South West Region), L’Hôpital de District de Bonassama (for the Littoral Region), L’Hôpital Regional de Bafoussam (for the West Region), L´Hôpital Jamot Yaoundé (for the Centre Region) and the Regional Hospital Bamenda (for the North West Region).
2.2. Data and Sample Collection
Information on the history of previous TB treatment, age, sex and region of residence of each TB patient were recorded through the administration of a questionnaire. One early morning sputum specimen was collected from smear positive TB patients.
2.3. Bacterial Isolation and Identification
Lowenstein Jensen (LJ) medium supplemented with glycerol was prepared according to the manufacturer’s instructions (Liofilchem, Italy). Sample decontamination was performed following standard procedures [4] . Briefly, for every 2 ml of sputum sample, 3 ml of decontaminating solution (SDS, 3%; NaOH, 1%) were added and vortexed in a closed tube. After incubation at room temperature for 30 minutes, neutralization solution (phosphoric acid, 0.567%; bromothymol blue, 0.4%) was added drop wise, until a light green colour appeared, indicating the neutralization of pH. The samples were centrifuged for 30 minutes at 3000 g. Then 3 ml of distilled water was added to the sediment and inoculated on LJ medium in duplicate. Cultures were incubated at 37˚C and examined weekly for a maximum duration of 8 weeks. Identification of Mycobacterium tuberculosis complex was based on microscopy of Ziehl-Neelsen stained smears, colony morphology, nitrate reduction, niacin accumulation, catalase activity at 25˚C and 68˚C [12] . A positive control (Mtb H37Rv strain) and negative control (sterile distilled water) were included in the test and all negative tests were repeated for confirmation.
2.4. Genomic DNA Extraction and PCR Amplification
The extraction of mycobacterial DNA from the heat-killed cells was performed essentially as described by Wilson [13] . DNA pellet was dried at room temperature, re-dissolved in TE buffer (pH 8.0) and stored at 4˚C for immediate use or at −20˚C for longer term storage. The DNA was analysed by 0.5% standard agarose gel electrophoresis for verification of purity. Genomic DNA of the isolates were analysed by PCR using previously published and specific primers for the identification of the following gene targets: hupB, oxyR, rpoB, IS6110, and IS1081 genes (Table 1). The primers were synthesized and purified commercially (SIGMA, Germany). The 25 μl PCR reaction mixture consisted of 12.5 µl of RedTaq PCR SuperMix (SIGMA, Germany), 1 µl of purified bacterial DNA or 5 µl of bacterial thermolysate and each of the two flanking primers at a final concentration of 0.5 µM. A positive control (DNA from Mtb H37Rv strain) and two negative controls were also included in each assay. The negative control was made up of: one with PCR water replacing template DNA (negative control 1), and another with template DNA but no primers (negative control 2). Samples showing negative results were re-tested with variations in amount of template DNA. Both the purified DNA and the thermolysates of 30 isolates selected at random were tested in all the PCR assays to assess the effect of DNA isolation method on the sensitivity of PCR assays. A G-STORM thermocycler (Gene Technologies, UK) was used for all the PCR amplifications.
The PCR cyclic conditions were as follows: For the IS6110 assay, initial denaturation at 95˚C for 5 min, then 30 cycles at 95˚C for 1 min, 63˚C for 1 min, and 72˚C for 1 min, with a final elongation step at 72˚C for 7 min. For IS1081 assay, initial denaturation at 95˚C for 5 min, then 30 cycles at 94˚C for 1 min, 1 min at 68˚C and 2 min at 72˚C, with a final elongation step at 72˚C for 7 min. For rpoB assay, initial denaturation at 95˚C for 5 min, then 30 cycles at 94˚C for 1 min, 1 min at 68˚C and 1 min at 72˚C, with a final elongation step at 72˚C for 10

Table 1. Primers used in the study.
min. For hupB assay, initial denaturation at 94˚C for 10 min, then 35 cycles at 94˚C for 1 min, 1 min at 60˚C and 1 min at 72˚C, with a final elongation step at 72˚C for 10 min. For oxyR assay, initial denaturation at 95˚C for 5 min, then 30 cycles at 94˚C for 1 min , 1 min at 68˚C and 1 min at 72˚C, with a final elongation step at 72˚C for 7 min. The PCR products were analysed by standard agarose gel electrophoresis on a 2% gel. The 1-kb-plus (1 g/µ l) DNA ladder (Invitrogen, USA) was electrophoresed in parallel with the PCR products.
2.5. Ethical Consideration and Consent
Ethical clearance for this study was obtained from the Cameroon National Ethics Committee in Yaoundé and administrative authorization obtained from the various establishments where this study was conducted. Each participant ≥21 signed a consent form and those <21 an assent was obtained from patients guardians.
2.6. Statistical Analysis
Statistical calculations were carried out using SPSS 18.0 (Statistical Package for the Social Sciences, Chicago, Illinois). The diagnostic sensitivity of each PCR assay was calculated conventionally. The Chi-Square test was used to determine statistically significant differences between the assay sensitivities. A p-value of < 0.05 was considered statistically significant.
3. Results
3.1. Sample Population Distribution
The 384 smear positive sputum samples collected were distributed as follows: 62 from the Northwest region, 61 from the Littoral region, 114 from the South West region, 74 from the Central region, 73 from the West region. However, only 130 culture positive samples were included in the analysis. The participants were aged between 6 and 67 years old with mean age of 29.2 years old. Of the 130 culture positive samples, 71 were male (54.6%) and 59 (45.38%) were female (Table 2).
3.2. Identification of Mycobacterial Isolates and DNA Preparation
Initial presumptive distinction of MTBC from NTM was obtained by direct observation of colony aspect (morphology and pigmentation) and presence of serpentine-like cords on Ziehl-Neelsen stained smears obtained from cultures. All isolates retained were also non-pigmented, had rough colonies and were slow growers (greater than 7 days), indicative of MTBC isolates.
Of the 130 isolates of MTBC recovered at random from the growing cultures: 25 were from the centre region; 20 from Littoral; 20 from the North West; 38 from the South West and 27 from the West Region. DNA was successfully purified from 125 of the 130 samples using the cetyltrimethylammonium bromide (CTAB) standard method. Figure 1 depicts a typical electrophoretic analysis of the purified DNA samples. The 5 negative samples
Table 2. Demographic characteristics of the 130 study participants used for the analysis.
Figure 1. Agarose gel electrophoresis of DNA isolated from the 125 samples using the standard CTAB method.
were therefore excluded from the PCR analyses. The DNA molecules from culture thermolysates of all samples were also analysed by agarose gel electrophoresis. A typical electrophoregram is shown in Figure 2. A total of 110 (88%) of the 125 samples were positive in all 3 biochemical tests and were thus confirmed as belonging to the MTBC.
3.3. Performance of PCR Assays
Five (5) of the 30 purified DNA samples (16.7%) that were included in a comparative analysis of DNA isolation methods were negative by all PCR assays, whereas none of the thermolysate preparations was negative. This was most likely due to the sharing of template DNA during the rather harsh purification process. Due to this high performance of the thermolysates, they were used in all the PCR reactions. However, a purified DNA sample was also tested in a situation where the thermolysate yielded a negative result, for the purpose of confirmation.
Of the 125 isolates tested, 124 (99.2%), 117 (93.6%), 123 (99.1%), 119 (95.2%) and 118 (94.4%) were positive by the IS6110, hupB, rpoB, oxyR, and IS1081-based PCR assays respectively (Figure 3). Of the 110 isolates biochemically identified as MTBC, 109 (99.1%), 104 (94.5%), 109 (99.1%), 106 (96.4%) and 104 (94.5%) were positive in the IS6110, hupB, rpoB, oxyR, and IS1081-based PCR assays respectively (Figure 3). Concordant PCR results were obtained for 108 of the 125 samples. The 15 isolates that could not be identified biochemically scored sensitivities ranging from 100% (for the IS6110 assay) to 86.7% (for the hupB and oxyR assays) (Figure 3). Twelve (12) of these samples (80%) tested positive (concordantly) by all the PCR assays. Thus, as expected, the PCR was more sensitive than the biochemical tests. There was, however, no statistically significant difference (p > 0.05) between sensitivities of all the different PCR assays. None of the PCR assays was 100% sensitive, but the combined (duplex PCR) or consecutive use of the IS6110 assay and each of the other assays increased the sensitivity to 100% for all the combinations (Figure 3). Table 3 shows the interrelationship of isolates that were not identified by PCR but were identified by biochemical tests as MTBC isolates.
No size polymorphisms were observed for all the PCR products and all the sizes obtained were as expected for each of the assays: 123bp for IS6110, 645bp for hupB, 473bp for oxyR, 235bp for rpoB and 306 bp for IS1081. Figure 4 typifies the PCR results obtained with the different PCR assays. The lone isolate that was negative by the IS6110 assay was positive in the biochemical tests, while the lone isolate that was negative by the catalase test at 22˚C was positive in all the PCR assays. No single isolate was negative by all the PCR assays.
4. Discussion
We evaluated the sensitivities of five popular and highly specific diagnostic PCR assays for TB targeting the IS6110, hupB, rpoB, oxyR, and IS1081 genes of MTBC using cultured bacterial isolates from 5 of the 10 regions of Cameroon as sources of template DNA. The objective was to determine which assay or combination of primer pairs would be the most sensitive in detecting or diagnosing the strains of MTBC circulating in these most socio-economically active regions of Cameroon. The various assays reliably detected the MTBC genes of the isolates with high sensitivities ranging from 93.6% to 99.2%. No significant statistical difference (p > 0.05) was, however, found between the different PCR assays.
The highest sensitivity (99.2%) was obtained with the IS6110-based assay. This high performance could have
Figure 2. A typical agarose gel after electrophoretic analysis of thermolysate DNA.
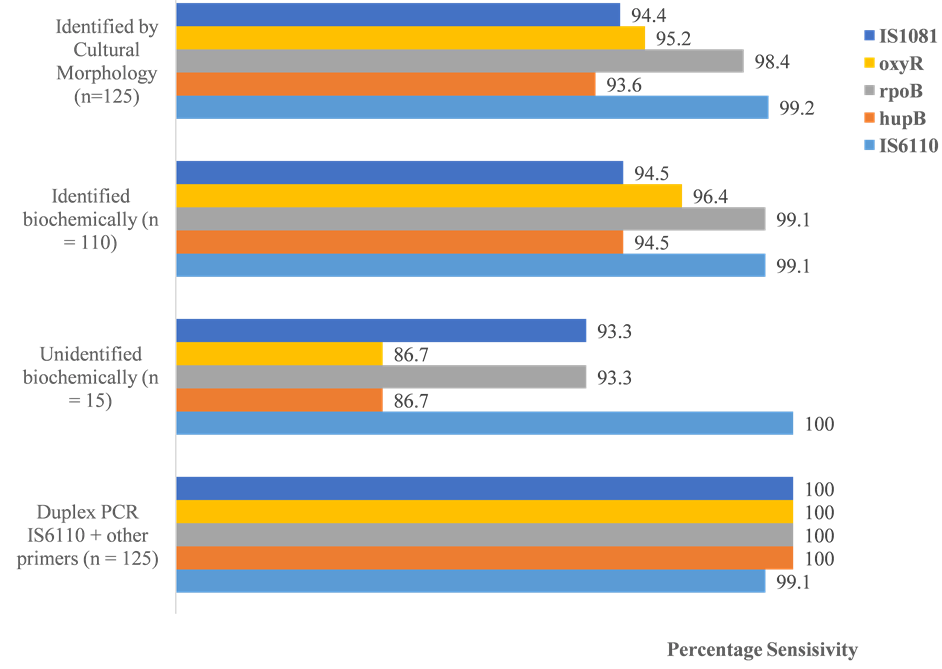
Figure 3. Comparison of the sensitivities of cultural identification, biochemical methods and PCR assays.
been due to the frequent occurrence of multiple copies of the IS6110 sequence in MTBC genomes. This result was in line with previous reports in which 100% sensitivity and specificity were obtained for this gene [8] [14] . One sample was negative in the IS6110 assay. This negativity was possibly due to a mutation in the target sequence, rather than to PCR inhibitors since this same isolate DC108, gave positive PCR results in all the other PCR assays (Table 3). At least one study has reported the absence of the IS6110 sequence in some MTBC strains [15] .
The rpoB-based assay also gave a high positivity rate of 98.4%. This assay has also yielded 100% sensitivity and specificity in a previous study [10] . Although the target site for PCR amplification contains frequent muta-
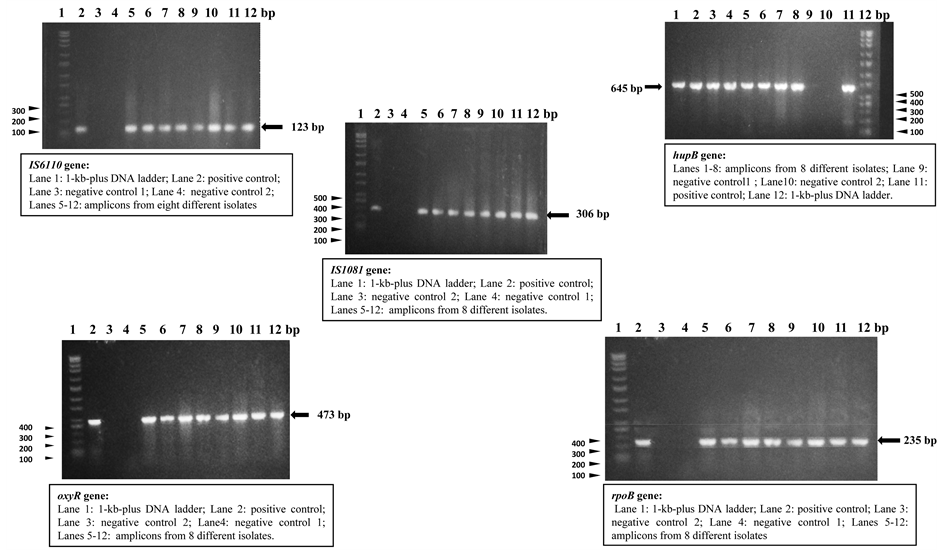
Figure 4. Agarose gel electrophoretic analysis results obtained with the different PCR assays.
Table 3. Interrelationships between 15 samples that were both culture and biochemically positive, but negative by PCR assays.
tion sites for rifampicin resistance, the intra-species variation of MTBC rpoB sequences was reported to be less than 1%. The sequence is thus highly conserved [16] . One of the two samples that gave a negative result with this assay was also negative in the biochemical tests, while the other was positive for all the other PCR assays. Thus, when all the assays were limited to samples that were positive by both the culture and biochemical tests, the sensitivity of the rpoB-based assay was equal to that of the IS6110 assay (99.1%). Additionally, the rpoB test was able to detect what the IS6110 test could not detect. The former test could therefore be a good substitute or preferably, a complement to the IS6110 test. The IS1081-based assay was the least sensitive. In one study using infected human blood samples, the sensitivity of the IS1081 PCR assay was only 43.75% [9] . However, when the latter assay was applied in the identification of M. bovis culture isolates, it identified 27 out of 27 (100%) of the isolates [17] . This indicates that although the assay was not very sensitive in the present study, it could nonetheless be a sensitive assay in the detection of M. bovis. The IS1081 gene is also known to exist in multiple copies in the MTBC genomes. On the other hand, the hupB and oxyR genes have been shown in previous studies to have sensitivities and specificities of 100% each, in the detection of MTBC strains [10] [11] . The combination of the IS6110 assay, which turned out to be the most sensitive, and each of the other assays gave 100% sensitivity (Figure 3). This strategy is preferable since genetic variants are possible.
Genetic differences have been observed among various MTBC isolates from across the world at many loci. These studies uniformly show that distinct geographic regions of the world have variable distributions of lineages, with certain regions having a major lineage that is a minor contributor elsewhere [18] . The consideration of variability is noteworthy especially in the design of drugs, vaccines and commercial diagnostic assays. False- negative results on PCR or other amplification assays can also occur when primer sequences are not properly complementary to the target hybridization sites. It may therefore be necessary to subsequently sequence the relevant genetic regions of the samples that are negative in PCR in the present study to verify this possibility.
DNA purification to eliminate polymerase inhibitors has been recognized to be one of the most important steps in sputum-based PCR diagnosis of pulmonary TB. In this study template DNA from each isolate was prepared by two different methods from all isolates: a standard CTAB method, yielding purified DNA [13] and a simple heat treatment method yielding culture thermolysates containing unpurified DNA. Five (5) of the 30 samples in which purified DNA served as template were negative in all PCR assays (83.33% positivity), while 100% sensitivity was obtained from thermolysate samples. The procedure for purification of mycobacterial DNA is very lengthy and involves a great deal of heating and harsh treatment of samples. This can greatly compromise the integrity of the DNA molecules [19] . It is therefore interesting that 5 of the purified DNA samples give negative PCR results. In clinical samples, however, there are usually numerous PCR inhibitors compared to cultured isolates, hence the isolation of bacterial cells for thermolysate preparation or the careful purification of DNA prior to PCR testing becomes necessary.
The major limitation was the absence of a perfect standard test to completely identify members of MTBC. Culture and biochemical tests (as an adjunct test) were the reference standard used in this study but they have been shown to have low sensitivity.
In conclusion, the combined targeting of the IS6110 and rpoB genes should yield a highly sensitive and specific PCR procedure for the diagnosis of TB in the five regions of Cameroon.
Acknowledgements
This research was funded by Biosciences Eastern and Central Africa Network (BecANet), a network of the New Partnership for Africa’s Development (NEPAD). The authors thank all the staff at the local, regional and district hospitals for interviewing patients and collecting sputum samples. We also thank the staff of the Centre Pasteur du Cameroun for their technical assistance during the bacteriological works.
References
- Katoch, V.M. (2004) Infections Due to Non-Tuberculous Mycobacteria (NTM). Indian Journal of Medical Research, 120, 290-304.
- World Health Organisation (2010) Tuberculosis, WHO Fact Sheet No. 104, November 2010. http://www.Who.Int/Mediacentre/Factsheets/Fs104/En/
- World Health Organisation (2008) Global Tuberculosis Control: Surveillance, Planning, Financing: WHO Report 393.
- Kent, P.T. and Kubica, G.P. (1985) Public Health Mycobacteriology: A Guide to the Level III Laboratory. USDHHS. Centres for Disease Control, Atlanta, 159-184.
- Katoch, V.M. (2003) Advances in Molecular Diagnosis of Tuberculosis. Medical Journal Armed Forces of India, 59, 182-186. http://dx.doi.org/10.1016/S0377-1237(03)80001-1
- Katoch, V.M. (2004) Newer Diagnostic Techniques for Tuberculosis. Indian Journal of Medical Research, 120, 418- 428.
- Balasingham, S.V., Davidsen, T., Szpinda, I., Frye, S.A. and Tønjum, T. (2009) Molecular Diagnostics in Tuberculosis. Molecular Diagnosis and Therapy, 13, 137-151. http://dx.doi.org/10.1007/BF03256322
- Eisenach, K.D., Cave, M.D., Bates, J.H. and Crawford, J.T. (1990) Polymerase Chain Reaction Amplification of a Repetitive DNA Sequence Specific for Mycobacterium tuberculosis. Journal of Infectious Diseases, 161, 977-981. http://dx.doi.org/10.1093/infdis/161.5.977
- Niyaz, A., Ashok, K.M., Utpal, M., Virender, K.B. and Sunita, G. (1998) PCR-Based Rapid Detection of Mycobacterium tuberculosis in Blood from Immuno-Competent Patients with Pulmonary Tuberculosis. Journal of Clinical Microbiology, 36, 3094-3095.
- Mokaddas, E. and Ahmad, S. (2007) Development and Evaluation of a Multiplex PCR for Rapid Detection and Differentiation of Mycobacterium tuberculosis Complex Members from Non-Tuberculous Mycobacteria. Japanese Journal of Infectious Disease, 60, 140-144.
- Prabhakar, S., Mishra, A., Singhal, A., Katoch, V.M., Thakral, S.S., Tyagi, J.S. and Prasad, H.K. (2004) Use of the Hupb Gene Encoding a Histone-Like Protein of Mycobacterium tuberculosis as a Target for Detection and Differentiation of M. tuberculosis and M. Bovis. Journal Clinical Microbiology, 42, 2724-2732. http://dx.doi.org/10.1128/JCM.42.6.2724-2732.2004
- International Union against Tuberculosis and Lung Disease (1998) The Public Health Service National Tuberculosis Reference Laboratory and the National Laboratory Network: Minimum Requirements, Role and Operation in a Low- Income Country. Paris, 62-71.
- Wilson, K. (1990) Preparation of Genomic DNA from Bacteria. In: Ausbel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A., et al., Eds., Current Protocols in Molecular Biology, Volume 1, Green and Wiley- Interscience, New York, 241-242.
- Ogusku, M.M. and Salem, J.I. (2004) Analysis of Different Primers Used in the PCR Method: Diagnosis of Tuberculosis in the State of Amazonas. Jornal Brasileiro De Pneumologia, 30, 343-349.
- Van Soolingen, D., De Haas, P.W., Hermans, P.W., Groenen, P.M.A. and Van Embden, J.D.A. (1993) Comparison of Various Repetitive DNA Elements as Genetic Markers for Strain Differentiation and Epidemiology of Mycobacterium tuberculosis. Journal of Clinical Microbiology, 31, 1987-1995.
- Kim, B.J., Lee, S.H., Lyu, M.A., Kim, S.J., Bai, G.H., et al. (1999) Identification of Mycobacterial Species by Comparative Sequence Analysis of the RNA Polymerase Gene (Rpob). Journal of Clinical Microbiology, 37, 1714-1720.
- Nassar, A.F., Miyashiro, S., Oliveira, C.G., Pacheco, W.A. and Ogata, R.A. (2007) Isolation and Identification of Bovine Tuberculosis in a Brazilian Herd (São Paulo). Memórias do Instituto Oswaldo Cruz, 102, 639-642. http://dx.doi.org/10.1590/S0074-02762007005000073
- Kato-Maeda, M., Bifani, P.J., Krieswirth, B.N. and Small, P.M. (2001) The Nature and Consequence of Genetic Variability in Mycobacterium tuberculosis. Journal of Clinical Investigation, 107, 533-537. http://dx.doi.org/10.1172/JCI11426
- Bemer-Melchior, P. and Drugeon, H. (1999) Inactivation of Mycobacterium tuberculosis for DNA Typing Analysis. Journal of Clinical Microbiology, 37, 2350-2351.