Journal of Encapsulation and Adsorption Sciences
Vol.07 No.01(2017), Article ID:74415,30 pages
10.4236/jeas.2017.71002
Microencapsulation by Spray Drying of Vitamin A Palmitate from Oil to Powder and Its Application in Topical Delivery System
Avinash B. Gangurde, Purnima D. Amin
Department of Pharmaceutical Sciences and Technology, Institute of Chemical Technology, Mumbai, India

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: September 28, 2016; Accepted: February 24, 2017; Published: February 27, 2017
ABSTRACT
Vitamin A palmitate (VAP) contains retinol and palmitic acid which is easily absorbed by body and widely used in skin care products. But, it is a hydrophobic and oxidation sensitive molecule which undergoes rapid degradation especially in an aqueous environment. The purpose of this study was to prepare microcapsules of VAP using combination maltodextrin and modified starches. Emulsion of VAP was prepared using cremophore RH 40 with Tween 80 in a homogenizer and formed emulsion was spray-dried. The spray process was optimized using a central composite design for two variables to obtain microcapsules with desirable characteristics. Microcapsules containing 30% of VAP were produced using different concentration of wall materials. The prepared microcapsules were evaluated for their physical, morphological, in-vitro drug release and SEM study. The results showed that obtained microcapsules are nearly spherical in shape with a particle size ranged from 1 to 12 μm. The drug content and encapsulation efficiency (53% - 63%) of different batches were found within acceptable range. These stabilized drug loaded microcapsules were incorporated into silicone cream based formulation for convenient topical application and evaluated for its physicochemical parameters. The drug release study showed 80.18% to 83.43% of drug release from VAP microcapsules while topical formulations prepared by VAP microcapsules showed 67.09% to 71.45% drug release at the end of 24 hrs. The formulations were kept for 3 months stability study as per ICH guidelines and found to be stable.
Keywords:
Vitamin A Palmitate, Microencapsulation, Spray Drying, Starch Derivatives, Topical Delivery

1. Introduction
Vitamin A is the fat-soluble polyunsaturated hydrocarbon vitamin including retinoids and carotenoids [1] . Vitamin A obtained from plant source is a carotenoid that your body can transform into a retinol, while the vitamin A from animal sources is already in a form of retinol which is easily absorbed by your body [2] . Vitamin A palmitate (VAP) is the ester of retinol and palmitic acid which is available in oily or dry forms. Figure 1 showed VAP chemical structure [3] .
Sufficient amount of vitamin A is important in childhood development because of its role in ocular health, immune system and nutritional development. It has to be provided by food or dietary supplements because, it cannot be synthesized in humans [1] [4] .
Recommended dietary allowance (RDA) for adults is 900 micrograms daily (3000 IU) for men and 700 micrograms daily (2300 IU) for women [5] [6] . Supplementation with large, pharmaceutically administered doses of vitamin A can substantially reduce the incidence and severity of some infectious diseases. Oxygen accelerates photo-catalyzed degradation of retinoids under light or chemically generated free radicals [7] . Also, the degradation of VAP in aqueous solution is rapid, and the solubility of retinoids in aqueous solvents is poor because of their low polarity [8] . Therefore, there is a need to encapsulate VAP in a form that protects them from chemical degradation during storage, to solubilize it in aqueous systems and also does not adversely affect product quality [9] .
Dispersibility and stability can be improved by encapsulating VAP oil in powder microcapsules by using suitable barrier component [8] [10] . This barrier used for encapsulation may protect against light, oxygen, water, protect it from other ingredients or give control diffusion. It is generally made of compounds with chains to create a network, with hydrophilic and hydrophobic properties. Physical properties of microcapsules may also depend on the properties of the wall material. The wall materials used for stabilization may require specific stability properties during heating or freezing processes, and for an optimal release of the encapsulated active component [11] .
Different techniques were used for encapsulation, among which spray drying of a formulated emulsion/suspension/solution is the most widely used technique for preparation of microcapsules where the active component is well dispersed in the matrix [12] . The wall material acts as a stabilizer in the case of emulsion and also it is able to disperse again in water. The fraction of oil on the surface

Figure 1. Structure of Vitamin A palmitate.
of powder showed it is unprotected against oxidation and it does not seem to be significant to increase its shelf life [13] .
For spray drying process, carbohydrates, gums, semisynthetic cellulose derivatives and synthetic polymers are the commonly used carriers [14] [15] [16] . Maltodextrin is a hydrolyzed starch having multiple functions including film formation properties, binding ability, reduction of oxygen permeability of wall matrix. However, it has a low glass transition temperature and low emulsifying capacity to agglomeration or caking of micro particulate powders [17] [18] . It affects release of encapsulated actives and degradation/oxidation process may occur during the storage period. To increase efficiency of microencapsulation combination of wall materials is often used [19] .
In this research work combination of maltodextrin with modified starches such as Hi cap 100, which have good emulsifying capacity helped in increasing stability of active. The modified form very stable oil-in-water emulsions and is recommended as a total replacement for expensive encapsulating agents such as gum arabic and gelatin [20] . The characteristics of the VAP microcapsules loaded with combination of maltodextrin and modified starch matrix prepared by using spray drying technique.
VAP is also a constituent of some topically applied skin care products. After its absorption into the skin, retinyl palmitate is converted to retinol, and ultimately to retinoic acid. It increases moisturization of skin, decreases skin wrinkling and also acts as antioxidant when applied topically [21] [22] .
This study investigates the formulation and characterization of powder microcapsules of VAP. The microcapsules were incorporated into silicone cream based formulation for convenient topical application.
2. Materials and Methods
Vitamin A palmitate was donated from Piramal enterprises Ltd. BASF Ltd. (Batch no.: MH0033) (India). The surfactants used were PEG-40 hydrogenated castor oil (Mw = 2589) (CremophorRH40) kindly donated by BASF (Ludwigshafen Germany) and Polyoxymethyl sorbitan monooleate (Mw = 1310 g/mol) (Tween 80) purchased from SD Fine chemicals (India). Maltodextrin and HICAP 100 (Batch no. DC16611) were purchased from Gujrat ambuja exports ltd. (India) and Ingredion Ltd. (CP ingredients India Pvt. Ltd.) respectively. Silicone cream base were procured from DOW corning Ltd. Mumbai. All other chemicals were of HPLC grade and were purchased from Merck (Darmstadt, Germany).
2.1. Determination of VAP Solubility in Fixed Oils
Solubility of VAP in three different fixed oils namely olive oil, coconut oil and hydrogenated castor oil was initially investigated. Excess amount of VAP was added to 2 g of each of the oils and the mixtures were stirred using magnetic stirrer and covered with aluminum foil to protect it from light. Aliquots were taken to determine the amount of VAP until it reaches the equilibrium solubility.
2.2. Preparation of VAP Emulsion
A series of emulsions were prepared with fixed concentration of VAP (6%w/w), Cremophore RH 40 (4%w/w), and Tween 80 (0.2% w/w) as emulsifier (lipid phase). And varying the concentration of maltodextrin and OSA modified starch in combination (Aqueous phase). Aqueous phase were prepared by dissolving OSA modified starches or maltodextrin in distilled water at 70˚C under gentle stirring for 30 min to enhance hydration then cooled to room temperature. The lipid phase was prepared by mixing VAP with Cremophore RH 40 and heated to 60˚C - 65˚C also added Tween 80. The aqueous phase containing combination of OSA modified starch and maltodextrin were also heated to 60˚C - 65˚C and added very slowly to lipid phase with thorough stirring using a rotor-stator homogenizer (Ultra-turrax IKA T18 Basic, Wilmington, NC, USA) operated at 10,000 rpm for 30 min at room temperature. As a result of hydration, the solution thickens, with the viscosity attaining a maximum after about half of the water has been added. Further addition of water then decreases the viscosity again. If the first half of the water is added too quickly, the solution can become opalescent. Alternatively, the warm mixture of the VAP and Cremophore RH 40 can be slowly stirred into the water, which results in a lower increase in intermediate viscosity. The composition of emulsions was showed in Table 1.
2.3. Emulsion Stability
Immediately after the emulsion preparation, 25 mL aliquots of each sample were transferred to graduated cylinders, 25 mL, sealed, stored at room temperature for one day, and the volume of the aqueous phase measured after 24 hours. The stability was measured by % of separation, expressed as:
 (1)
(1)
where, Ho represents the emulsion initial height and H1 is the upper phase height.
2.4. Particle Characterization
The mean particle diameter, particle size distribution, and electrical charge of mixed micelles were determined. The mean particle diameter and particle size

Table 1. Formulation composition of VAP microcapsules.
distribution were measured using the laser light diffraction instrument (Malvern Instruments, Mastersizer 2000, UK). This instrument calculates the particle size of oil droplets in a medium by the scattering pattern of a traversing laser light. The electrical charge (ζ-potential) was determined using electrophoretic mobility measurements (Particulate system, Nano-Plus, Malvern Instruments). This instrument determines the sign and magnitude of the particles charge by measuring the direction and velocity of their movement when they are placed in a capillary test tube between two electrodes. Samples were equilibrated for 1 min inside the instrument before data were collected over at least 10 sequential readings and analyzed using the Smoluchowski’s model.
2.5. Shear Viscosity Measurement
The shear viscosity of samples was measured using a rheoplus instrument (Anton Parr, Rheoplus/32). A cone and plate geometry consisting of a measuring system with spindle number of CP50-2-SN15926 (diameter = 0.209 mm) was used for measurement of viscosity. The samples were loaded into the rheometer measurement plate and allowed to equilibrate at 25˚C for 5 min before beginning all experiments. Shear viscosity (h) measurements were carried out at different shear rates (0.01 to 100 s−1), and all oil phases and aqueous phases tested were found to be Newtonian. We therefore only reported the shear viscosity measurements at a fixed shear rate (10 s−1).
2.6. Interfacial Tension Measurement
The interfacial tension was measured at oil-water interfaces using a drop shape analysis instrument Rame-hart automated dispensing system (Model no. 100-22). The aqueous phases were prepared by mixing appropriate amounts of water, cosolvents and wall materials whereas oil phases were prepared by mixing appropriate amounts of VAP and Cremophore RH 40. The oil phase was injected into the aqueous phase after the equilibrium (more than 5 min.) and then the interfacial tension was determined by drop shape analysis method.
2.7. Statistical Analysis
All measurements were performed on to three freshly prepared samples (i.e., new samples were prepared for each series of experiments). The final values were reported as mean of the three measurements + standard deviation (SD) calculated from these values.
2.8. Spray-Drying Process
Homogenized microemulsions were spray-dried in a Mini spray dryer JISL equipped with a co-current nozzle atomizer. Inlet and outlet temperatures were 110˚C - 130˚C and 55˚C - 60˚C respectively, with a feed rate of 1 - 5 ml/min atomization air pressure 2-3 kg/cm2 and aspiration rate 40% - 45%. Obtained microcapsules were sealed into amber colored glass bottles (50 mL) and then put in plastic bag and kept into a desiccator at room temperature for further analysis. Composition for microcapsule preparation was showed in Table 1.
2.9. Determination of Drug Content
Accurate weight (10 mg) of VA-loaded microcapsules was dissolved and diluted with isopropanol to an appropriate concentration for determination of drug content of the microcapsules. The obtained solutions were measured for its absorbance (in triplicate) using ultraviolet (UV) spectrophotometer (Shimadzu UV-1650, Tokyo, Japan) at 325 nm using isopropanol as solvent. The amounts of drug content were calculated by comparing the measured absorbance values with those in the standard curve [23] .
Validation of the method was performed to ensure that the calibration curve between 6 and 12 μg/ml of VAP in isopropanol solutions was in the linearity range (r2 > 0.999) and the measured absorbance was in the range of 0.2 - 0.8.
2.10. Encapsulation Efficiency
The microcapsules equivalent to 100 mg of VAP were accurately weighed. For, determination of encapsulation efficiency microcapsules powder was dissolved in cyclohexane (5 ml) in volumetric flask and the volume was made with 0.1 N HCl. This solution was then filtered through whatmann filter paper No. 44. After suitable dilution, the absorbance was measured at 325 nm using UV spectrophotometer and the percentage of drug entrapped was calculated Equation (2). The drug entrapment study was conducted in triplicate.
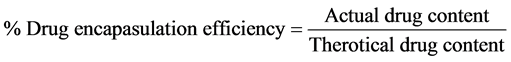 (2)
(2)
2.11. Moisture Uptake and Stability Studies
Moisture uptake by prepared microcapsules was studied using Moisture balance MB 50C (Citizen, India). Moisture content was calculated as percentage. After moisture content analysis microcapsules were placed in crucible at accelerated conditions of temperature 40˚C ± 2˚C and humidity 75% ± 5% RH in an environmental test chamber for 24 h (Thermo lab, India). These samples were then analyzed for drug content by UV spectroscopy. This method is useful to determine the effect of moisture on degradation of drug and prepared microcapsules systems [24] (need to add reference).
2.12. Design of Experiment (DOE)
The design of the experiment was done by software (Design Expert®, Version 9.0.4.1, Stat-Ease, system techno craft services pvt. Ltd. Ser. No. 5401-1156- 2569-7020 Inc., USA). The key elements of the study were the moisture content, encapsulation efficiency, drug content and moisture uptake stability study.
In the preliminary study, the composition of the emulsion to be spray dried (the quantity of VAP, Cremophore RH 40 oil, Tween 80 and combinations of wall materials ratio) and the process parameters (inlet air temperature, aspiration, feed rate, and solid concentration) were studied by single factor design. Based on the results of preliminary study, the major factors were found to be the feed rate and solid concentration.
Two-factor central composite design (CCD) with five center points was used to find the optimum condition. The factors and levels were shown in Table 2 and Table 3. The best-fitting mathematical model was selected based on the F-value provided by analysis of variance (ANOVA). The optimum values of the process variables were obtained from the model. The reproducibility of the process was studied on the optimized process by triplicate.
2.13. Flow Properties
The prepared microcapsules were evaluated for the parameters like bulk density, tapped density, compressibility index (Carr’s index), angle of repose, and Hausner’s ratio.
2.14. Solubility
The powder solubility in water was determined by dispersing the VAP microcapsules in water solution (0.3% w/v) and then gently stirred until solid solubilization. The microcapsules powder was considered soluble when the time of solubilization was not greater than 5 min. The time required to completely solubilize microcapsules was recorded [25] .
Table 2. Factors and levels.
Table 3. Experimental runs.
2.15. Contact Angle Measurement
To investigate the behavior of water in contact with the VAP microcapsules surfaces, changes in the contact angle of water droplets were measured. For the contact angle measurement, G10 Contact angle meter (KRUSS, Germany) was used. Static contact angle method was adopted to measure it. A solid disc of VAP microcapsules with flat surface was prepared by spray drying technique. A single drop of distilled water was deposited on the flat disc surface and the contact angle was measured immediately and after 60 s of equilibrium. In order to minimize the evaporation of water, contact-angle measurements were carried out in an atmosphere with saturated relative humidity. Measurements were carried out in triplicate for each sample.
2.16. Solid State Characterization of Formulations
The solid VAP microcapsules were characterized by particle size analysis and morphological characterization was done using scanning electron microscopy (SEM).
2.16.1. Particle Size Analysis
The microcapsules were evaluated for their particle size distribution using the laser light diffraction instrument (Malvern Instruments, Mastersizer 2000 Ver. 5.30.010, UK). The microcapsules were fed into the sample micro feeder. This technique measures the size of particles dispersed in a medium by the scattering pattern of a traversing laser light. Before analysis, microcapsules were suspended in ethanol and submitted to an ultrasound during 1.50 min. During the analysis (in triplicates), the samples were maintained at constant agitation.
All samples were analyzed 5 times, and average of results were taken. The characterization of particles size distribution was analyzed by D10, D50 and D90 of the microcapsule size. The average of d (0.1), d (0.5), and d (0.9) values is taken as mean diameter.
The size uniformity and dispersity of the microcapsules were represented by span value. The narrow size distribution was indicated by small span value. Span value was measured from the following Equation (3):
 (3)
(3)
2.16.2. Scanning Electron Microscopy (SEM)
To evaluate physical surface and morphology of microcapsules like size and shape was analyzed using scanning electron microscope (XL 30 Model, JEOL 5400, Japan). Sample was prepared by affixing double-sided carbon tape on aluminum stubs over which sample of VE microcapsules were placed. The radiation of the platinum plasma beam (JFC-1600) was bombarded on aluminum stubs to make 2 mm coating thickness above the sample for 25 min. These prepared coated stubs were then placed in the vacuum chamber of a scanning electron microscopy (SEM) and adjusted to maximum magnification to obtain excellent quality scanning images. Then, those samples were observed for morphological characterization using a gaseous secondary electron detector (working pressure: 0.8 Torr, acceleration voltage: 20.00 kV). SEM images were obtained at a maximum and visible magnification to understand the surface morphology VAP microcapsules.
2.17. Development of Semi-Solid Dosage Form for Topical Delivery of VAP
2.17.1. Preparation of Semi-Solid Dosage Forms
Semi-solid formulations of VAP were prepared according to the composition given below in Table 4 using aqueous silicone cream base. VAP oil and VAP microcapsules incorporated into the formulation at 0.3% concentration of active VAP. To obtain homogenous mass all the actives were triturated by using geometrical dilution procedure.
2.17.2. Cream Enriched with VAP Microcapsules
Creams were prepared using silicone cream base. For the preparation of cream, glycerol, SLS and microcapsules of VAP were weighed accurately and dispersed in water [26] . The dispersion obtained was, then stirred in high shear homogenizer (IKA T18 Digital Ultra-turrax) (7000 rpm). The clear solution of actives was added in small aliquots to silicone cream base with constant agitation. The cream was stirred till cream of desired uniformity and consistency was obtained.
2.17.3. Conventional Cream (Neat VAP)
Conventional cream of a Vitamin A was prepared by incorporating the VAP in silicone cream base at the same concentration as microcapsule enriched cream. The VAP was dispersed in part of water containing sodium lauryl sulphate and glycerol [22] . The part containing VAP was added to the silicone cream base and the cream was stirred till desired uniformity and consistency was obtained.
2.17.4. Evaluation of Topical Cream Formulations
1) Organoleptic properties
The physical appearance of formulations was checked visually (color, odor
Table 4. Composition of topical formulations of VAP cream.
and homogeneity) while greasiness was assessed by application into the skin surface. In addition, the pH of the VAP cream formulations was determined using a Eutech digital pH meter.
2) Rheological properties
The viscosity of VAP cream was measured on a Brookfield viscometer RV model. T-bar spindle (C) was used for measurement of viscosity and viscometer was operated at 0.5, 1, 2.5 and 5 rpm. A graph of shear rate vs. shearing stress (viscosity X r.p.m.) was plotted which indicate rheology of VAP topical formulation [27] .
3) Texture profile
The spreadability of VAP topical formulation was analyzed using texture analyzer Brookfield engineering labs which consist of a set of matched male and female Perspex cones. CT3 with 100 gm load cell was used for measurement of spreadability. For measurement of the spreadability of the VAP cream parameters was maintained as follows:
Test type: compression, pre-test speed: 1.0 mm/s, target value and target force: 5.0 mm and 6.80 gm respectively.
4) Stability under centrifugation
The stability of VAP cream against aggregation of emulsified oil particles (i.e. creaming at the top of the sample) was evaluated. The VAP cream was evaluated for aggregation of emulsified oil particles i.e. creaming at the top of the sample. The centrifugation tubes filled with 10 ml of cream were centrifuged at 5000 rpm for 30 min. and evaluated for phase separation. This test is used for evaluation of VAP cream under accelerated deterioration and high stress conditions [26] [28] .
5) Stability under thermal cycling
The prepared VAP cream was stored in wide mouth plastic containers made up of Low Density Polyethylene (LDPE) and subjected to refrigerated temperature (2˚C - 8˚C) for 24 hrs. Followed by exposure to 30˚C/60% RH and 40˚C/75% RH accelerated storage conditions for 24 hrs. This cycle was repeated three times and any change in the color, viscosity was recorded.
6) In vitro drug release of VAP cream
In vitro drug release studies were performed using Franz glass diffusion cells. It contains 0.785 cm2 diffusion area and 10 ml volume of receptor chamber. For permeation study, Dow Corning 7-4107 Silicone elastomer was used as semi- permeable membrane for 24 hrs. Phosphate buffer pH 7.4 containing 3% polysorbate 80 was used as receptor fluid. A weighed quantity of VAP cream formulations were placed evenly on the surface of the membrane and immersed slightly in receptor fluid which was continuously stirred with magnetic stirrer and maintained at temperature 37˚C ± 0.2˚C. Samples of 1 ml were collected over a period of 24 hrs. The withdrawn samples were diluted suitably and VAP content was estimated spectrophotometrically at 325 nm [22] .
2.18. In-Vitro Release Profile of VAP Microcapsules
The dissolution study of prepared microcapsules was carried out by using eight station USP apparatus II (Electro lab, TDT-08L, India) in 900 ml phosphate buffer pH 7.4 at a temperature of 37˚C ± 0.5˚C at 75 rpm. A 5 gm of powdered microcapsules were introduced into the dissolution medium [29] . And dissolution study was carried out over a period of 24 hr. A 5 ml of aliquots sample were withdrawn and same amount replaced by fresh medium to maintain sink condition. The withdrawn samples were analyzed through UV-Visible spectrophotometer at 325 nm. All studies were carried out in triplicates.
2.19. Antioxidant Assay
Free radical scavenging activity of VAP microcapsules and topical formulations of VAP was determined by stable 2, 2-diphenyl-1-picrylhydrazyl (DPPH) radical. Methanolic solutions (1 ml) of different formulations containing 1, 2, 3, 4 and 5 μg/mL concentration of actives were added to 1 ml methanolic solution of DPPH (2 mg/ml). Ascorbic acid was taken as reference compound. The absorbance is measured at 517 nm after 30 min. The results were evaluated as scavenging percentage of radical [30] .
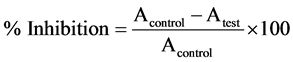 (4)
(4)
whereas, A = absorbance; Acontrol = absorbance of blank solution Atest = absorbance of the reference standard.
The percentages of inhibition were plotted against the concentration for each sample, and the concentration that corresponds to 50% inhibition of DPPH reduction was reported as the IC50 value [31] .
2.20. Stability Study
Stability study was conducted by placing powder microcapsule samples in closed glass ambered color vials and topical cream formulation of VAP in wide mouth plastic containers made up of low density polyethylene. Both, the samples were stored in controlled temperature environments inside stability chamber with RH of 75% and 40˚C temperature and 65% and 30˚C temperature. Microcapsule samples were reevaluated for physical appearance, moisture content, and drug content while the VAP cream formulation were reevaluated for appearance, pH and drug content after 1, 2 and 3 months storage.
3. Results and Discussion
Vitamin A supplementation via foods and/or pharmaceuticals has been widely used for prevention of diseases and ensures healthy childhood development in developing countries [1] . However, utilization of VAP is limited due to its poor dispersibility in aqueous systems and its vulnerability to degradation during processing, transport and storage.
Preparation of VAP microcapsules with combination of carbohydrate materials is a promising way to improve solubility and stability.
In the present study, we investigated the impact of different combination ratios of carbohydrate materials such as maltodextrin and modified starch (OSA starch) upon stabilization of VAP in the solid phase, dissolution, and solubilization in aqueous media. Also, the prepared microcapsules were incorporated into semi-solid dosage formulation (silicone cream base) for convenient topical application.
3.1. Preparation of VAP Encapsulated Microcapsules
In the present study, VAP microcapsules were prepared by spray drying technique for delivery of VAP by topical route. In order to prepare stabilized VAP microcapsule combinations of natural ingredients such as maltodextrin and starch derivative (Hi cap100) were used in different concentrations as wall material for microencapsulation.
The loading capacity of VAP was targeted by preparing emulsions containing a 30% of VAP within the oil phase. However, it is difficult to homogenize alone due to its relatively high viscosity and its oily nature. Therefore, for the development of emulsion a right blend of low and high HLB (hydrophile-lipophile balance) surfactant is necessary for the formation of stable emulsion [32] [33] .
Initially, the solubility of VAP in three different fixed oils namely olive oil, coconut oil and hydrogenated castor oil was investigated. VAP was found to be very soluble in all the investigated fixed oils, but its solubility in hydrogenated castor oil (1.24 g/1g) was found to be higher than olive oil and coconut oil as showed in Table 5. Therefore, hydrogenated castor oil (Cremophore RH 40) was chosen to formulate VAP microcapsules.
Polyoxyl 40 Hydrogenated Castor Oil (Cremophore RH 40) acts as nonionic solubilizer and Tween 80 acts as emulsifier. Addition of surfactant is necessary for preparation of stable emulsion. Also, the emulsion may become unstable when it is highly diluted because the surfactant concentration may then fall below the critical micelle concentration (CMC), so some of the surfactant leaves the droplet surfaces, eventually leading to aggregation.
A series of oil-in-water emulsions with oil phase containing VAP, Cremophore RH 40, Tween 80 and wall materials containing different composition of combined wall materials was prepared as shown in Table 1. The emulsions were produced using the high shear homogenizer (IKA T18 Digital Ultra-turrax) (10,000 rpm).
All emulsions prepared with combination of starch derivatives showed 100% stability for 24 hours, with no phase separation.
The electrical characteristics of the emulsion were determined which showed that negative charge was formed on the micelles which can be attributed to the presence of anionic carboxylic groups (-COO). The prepared emulsion showed
Table 5. Solubility of VAP in different oils.

Figure 2. Zeta potential of VE emulsion determined by using nano-plus zeta sizer.
ζ-potential (mv) is in the range of −15.98 to −16.74 mv as shown Figure 2.
The rheological behavior of emulsions was evaluated by determination of shear viscosity at 25˚C as showed in Figure 3. The experimental data showed that emulsion showed Newtonian flow over the shear rate, according to which viscosity is constant with shear rate. The shear viscosity of emulsions was increased with increasing proportion of Hi cap 100 in wall material systems. The shear viscosity measurement at fixed shear rate (10−1) was found to be 1.431, 1.656 and 1.770 for VA1, VA2 and VA3 formulations respectively as shown in Figure 3.
The interfacial tension plays an important role in the formation of small droplets during homogenization, which is proportional to the force required to deform and disrupt droplets within the homogenization zone [34] . Therefore, we measured the interfacial tension at the oil-water interface of oil phases with different composition as showed in Figure 4. The interfacial tension was increased by increasing concentration of Hicap 100 in the aqueous phase changing from 23.6 mNm−1 to 29.2 mNm−1. This study indicates that the free energy required to increase the interfacial area by increasing amount of Hi cap 100 in aqueous phase which is having higher emulsifying capacity.
3.2. Optimization of Process Parameters
To optimize the levels of solid concentration and inlet air temperature statistically designed experiments were performed using central composite design (CCD). A high correlation coefficient explained the goodness of fit of the experimental data in the response surface models of moisture content, encapsulation efficiency, drug content and moisture uptake drug content stability study.
The different graphs of 4 responses were developed as a function of the two independent variables (levels of solid concentration and inlet air temperature) according to their significance to the response.
The graphs for moisture content, encapsulation efficiency, drug content and moisture uptake drug content stability study value as a function of levels of solid concentration and inlet air temperature are showed in Figure 5. The optimized

Figure 3. Shear viscosity as function of shear rate.

Figure 4. Interfacial tensions of different formulations between oil phase (30 % VE acetate: 20% cremophore RH 40 oil) and aqueous phase containing different combinations of wall materials VA1, VA2 and VA3.
formulation was obtained by superimposing graphical methods which gives the optimum area [35] .
3.2.1. Moisture Content and Encapsulation Efficiency
The coefficients of the first order terms variables showed that the moisture content decreased and encapsulation efficiency increased with the increase in inlet air temperature and decreasing solid concentration. Inlet air temperature was found to have maximum influence on moisture content followed by solid concentration. The generation of high hot air during drying which might have trapped the moist air that was found in the fed product and it might have reduced the moisture content and increased encapsulation efficiency to a greater extent [36] . The moisture content was decreased by increasing the inlet air temperature during spray drying [37] .
The linear model was a statistically significant model (p = 0.0005). The mathematical model (a polynomial equation) which was obtained by the software (Design-Expert®) was described below:
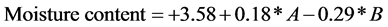
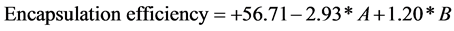
where, A solid concentration, B inlet temp
3.2.2. Drug Content and Moisture Uptake Stability Study (45˚C/75%)
The coefficients of the first order terms for variables indicated that drug content and moisture uptake stability drug content of the VAP microcapsules increased significantly by reducing the solid concentration and increasing inlet air temperature.
The Quadratic model was a statistically significant model (p = 0.0005). The mathematical model (a polynomial equation) which was obtained by the software (Design-Expert®) was described below:


where, A solid concentration, B inlet temp.
ANOVA shown in Table 6 indicated that the effect of the corresponding factors on the response was significant. The response surface plots were showed in Figure 5. The R2 value of all variables indicates that the model was in good fit with the R.S.D. value and CV. An optimization procedure was used to obtain the levels of solid concentration and inlet temperatures at which all the variables are in acceptable range was obtained. The optimum levels for all the variables were given in Table 6.
The VAP microcapsules under the optimum condition were prepared in triplicate. The observed value agreed with the predicted value which was showed in
Table 6. Significant level of powder response.


Figure 5. Effect of solid concentration and inlet temperature on different physico-chemical properties of the prepared microcapsules: (A) moisture content; (B) Encapsulation efficiency; (C) Drug content (D) Moisture uptake stability (40˚C/75%).
Table 7. This proved the validity of the model. The reproducibility was proved by the low S.D.
After, optimization of processing variables for spray drying process the microcapsules were evaluated for flow and compaction behavior of VAP microcapsules. It was found that VAP microcapsules has poor flow properties and needs manual tapping to make it freely flowable and also has poor compaction behavior because of fluffy nature of microcapsules as showed in Table 8.
Drug content of VAP microcapsules were determined by UV spectrophotometric method. Table 8 showed that VA1 to VA3 were in the range 28% - 30% which is implied that encapsulation could produce good reproducibility of drug content. The encapsulation efficiency of VAP microcapsules varied between 59% - 64% as showed in Table 8. Results demonstrate that increasing concentration of maltodextrin increases encapsulation efficiency of VAP microcapsules.
The morphological study of VAP microcapsules showed that the uniform and regular shape of particles. The aspect ratio of microcapsules varied from 1.03 to 1.08 which indicates almost spherical shape of microcapsules while the wall materials used for encapsulation showed irregular and non-uniform shape of particles with aspect ratio varied from 1.40 to 1.56.
3.3. Solubility and Contact Angle Measurement
The powder microcapsules were studied for time required to solubilize the microcapsules in water. The solubilization time required to solubilize the microcapsules was ranged from 3.3 to 4.5 min. Table 9 which indicated reducing concentration of maltodextrin decreases solubility of powder microcapsules [25] . To check wettability changes through surface modification contact angle measurement of VAP microcapsules were done. The time evolution of the water contact angle of VAP microcapsule films is shown in Table 9. The contact angle of all
Table 7. Predicted and observed values.
Table 8. Evaluation of micro capsules for their flow properties, assay, aspect ratio and encapsulation efficiency.
Table 9. Solubility and contact angle measurement of VAP microcapsules prepared by spray drying (n = 3, mean ± SD).
Table 10. Moisture content and drug content of encapsulated VE microcapsules after accelerated temperature conditions.
VAP microcapsule is in the range of 15˚ - 25˚ which showed higher wettability of microcapsules. Conversion of VAP oil into the solid state leads to decrease in contact angle which showed higher wettability of microcapsules. Therefore, VAP microcapsule films are hydrophilic in nature with lower contact angle.
3.4. Moisture Uptake and Stability Studies
To check hygroscopic nature of microcapsules moisture uptake study was conducted. VAP microcapsules were subjected to accelerate conditions of temperature and humidity and it shows that there is no significant change in weight of the microcapsules.
The amount of moisture absorption is directly proportional to the amount of hygroscopic surface area on the encapsulated particles (Ref. need to add). Therefore, it indicates the intimacy of mixing of oil with wall materials in encapsulated VAP. The Moisture content in encapsulated VAP was found to be in the range of 3.45% to 3.53% as showed in Table 10 which is negligible and has no degradation effect on drug efficacy. The drug content analysis was carried out after treatment and observed to be in the range of 27.53% to 29.32% analyzed by UV-spectrophotometric method.
3.5. Solid State Characterization
3.5.1. Particle Size Distribution of Microcapsules
The particle mean diameters ranged between 5.039 μm and 7.712 μm. The microcapsules produced from VA3 containing higher concentration of Hi-Cap 100 showed greater size, probably due their higher emulsion viscosity. Table 11 showed the particle size distribution of microcapsules produced with different proportion of maltodextrin and Hi-Cap 100 wall materials combination.
Particle size analysis showed a multi-modal particle size distribution. All prepared VAP microcapsules showed the narrowest size distribution with lower span value which showed regular particle size and shape Figure 6.
3.5.2. Scanning Electron Microscopy (SEM)
The surface analysis of the microcapsule particles obtained with combination of different proportion of encapsulating agents was performed using SEM allowing three-dimensional characteristics to be visualized. Figure 7 shows the photomicrographs of the VAP microcapsules obtained using the combination of maltodextrin and Hi-Cap 100 carrier agents.
Table 11. Volume weighted mean particle size, the d (0.1), d (0.5), d (0.9), and span values of VAP microcapsules determined by laser diffractometry.

Figure 6. Particle size distribution of VAP microcapsules (A) VA1, (B) VA2, (C) VA3.

Figure 7. SEM micrographs of (A) VA1 (B) VA2 & (C) VA3 prepared by combination of maltodextrin and Hi-Cap 100 at different concentrations.
Microcapsules obtained with combination of encapsulating agents VA1 to VA3 exhibited irregular surfaces of angular shapes with several indentations. The formation of these indentations obtained by spray drying technique on the surface of microcapsules is usually attributed to particle shrinkage due to the drastic loss of moisture followed by cooling [38] .
The agglomeration of smallest particles between themselves and to the larger particles was also observed in the microcapsules. Microcapsules with rough surfaces or dents can have problems in their flow properties, but they do not affect stability [39] .
The microcapsules prepared by spray drying technique are spherical in shape but they have irregularities on their outer surface.
3.6. Development of Semi-Solid Dosage for Topical Delivery of VAP
VAP is widely used in dermatological and cosmetic preparation as active component. It involves in regulation of epidermal cell growth, participates in collagen synthesis process, inhibits final step of keratinization and enhances glycosaminoglycan synthesis and essential in reproduction of basal membrane cells. It also stimulates skin cell proliferation, which eventually causes the epidermis to thicken, resulting in a stronger skin barrier function. Also it improves skin appearance and its elasticity [3] .
The importance of VAP to skin has inspired development of topical drug delivery.
The recommended concentration of VAP 1.7 million IU/g is 0.05% - 0.3%.
3.7. Evaluation of Semi-Solid Topical Cream Formulations
All the topical formulations of VAP were white in color and found to be smooth, free from grittiness on application and homogenous in nature. The drug concentration of VAP in dermatological bases was found to be in the range of 94% - 99% (Table 12). It reveals that the method used was suitable for preparation of topical dosage forms. There is no degradation of the active during the preparation as indicated by drug content. The pH of the VAP formulations was in the range of 7.1 - 7.7 which is suitable for skin pH, indicated suitability of VAP formulation when applied to skin.
The shear viscosity of VAP topical formulations was evaluated at 25˚C as showed in Figure 8. The shear viscosity measurement of topical cream formulation at fixed shear rate 5 rpm was in the range of (6.00 - 6.70) × 106 centipoise. The viscosity of VAP cream formulations was ranged from (6.24 - 6.72) × 106
Table 12. Evaluation of topical formulations of VAP for their drug content and texture studies [Hardness (gm) and Hardness work done (mJ)].

Figure 8. Viscosities of VAP topical formulations (Neat VAP, VAPT1, VAPT2, VAPT3) as a function of shear rate.
spreadability using texture analyzer which consists of a set of matched male and centipoise at 5 rpm. The VAP topical formulations were evaluated for their female perspex cones. The graph of Time (seconds) vs. Load (gm.) Figure 9 indicates the maximum force value which is a measure of the firmness of cream at specified area and depth. Also, the energy required to deform the cream formulations which is the hardness work done. Lower the firmness and hardness work done value indicates a more spreadable sample [26] . All the topical cream formulations showed lower firmness and hardness work done which indicates that VAP topical formulations were more spreadable as showed in Table 12.
The topical formulations of VAP were also evaluated for their stability under centrifugation and thermal cycling. It indicates that no creaming or phase separations were observed under centrifugation and no change in viscosity and color was observed under thermal cycling for VAP cream formulations [28] .
3.8. In-Vitro Release Study
The in vitro release profile of VAP microcapsules was showed in Figure 10. In the initial 2 hrs. the drug release was less than 20% this may be because of slow dissolution of drug from the wall material. After 2 hrs. the drug release rate was increased with time until 8 hrs. Comparing the drug release from semi-solid topical formulation containing VAP microcapsules was slower from the cream as compared with VAP microcapsules. As can be seen in this Figure 10 80.18% to 83.43% of drug release from all VAP microcapsules (VA1, VA2 and VA3) while semi-solid topical formulations prepared by VAP microcapsules showed 67.09% to 71.45% drug release at the end of 24 hrs. Neat VAP topical formulation of cream showed 85.09% drug release in 8 hrs.
Drug release from all the formulations containing VAP microcapsule powder and topical formulation followed the first order release kinetics. Higher correlation, as indicated by R2 value was observed for Higuchi matrix release kinetics in all the selected formulation, suggesting diffusion as a probable prominent mechanism of drug release.
3.9. Stability Studies
VAP microcapsules prepared by combination of starch derivatives packed in ambered colored glass bottles exhibited no significant change in moisture content (which is in the range of 4.30% to 4.35% at 30˚C/65% and 4.41% to 4.46% at 40˚C/75%) and drug content (which is in the range of 27.03% to 28.13% at 30˚C/65% and 27.01% to 27.19% at 40˚C/75%) after 3 months storage at accelerated conditions. Topical formulations of VAP indicated no evidence of phase separation, change in color, consistency of formulation and development of disagreeable odor during stability study; and, also showed no significant difference in drug content (which is in the range of 93.23 to 93.51 at 30˚C/65% and 94.12% to 94.27% at 40˚C/75%), and pH of the formulations after 3 months storage at accelerated storage conditions.

Figure 9. Texture profile of (A) Neat VAP, (B) VA1, (C) VA2, (D) VA3.

Figure 10. In vitro release profile of (A) VAP powder microcapsules VA1, VA2, VA3 (B) VAP topical formulation Neat VAP, VAPT1, VAPT2, VAPT3.
3.10. Antioxidant Activity
Antioxidant activity of VAP is characterized by a stable free radical by virtue of the delocalization of the electron over the molecule, the delocalization gives rise to the deep violet color, characterized by an absorption in methanol solution centered at about 527 nm. When a solution of DPPH is mixed with VAP formulations it can donate a hydrogen atom, then this gives rise to the reduced form with the loss of this violet color which is characterized by absorbance at 517 nm [40] . Antioxidant DPPH radical scavenging ability of VAP formulations showed in Figure 11 as decrease in absorbance with concentration of antioxidant at 517 nm. VAP formulations exhibited a comparable antioxidant activity (p ˂ 0.5) with ascorbic acid as the standard at various concentrations (1, 2, 3, 4 and 5 μg/ml). Results showed that the VAP formulations have a significant free radical scavenging activity belonging to its hydrogen-donating ability. The comparison of the antioxidant activity of VAP formulations and ascorbic acid as % inhibition of DPPH scavenging activity (IC50) are shown in Figure 12.

Figure 11. Reduction of oxidizing ability of DPPH by VAP microcapsules, VAP topical formulations and standard ascorbic acid (as function of decrease in absorbance).

Figure 12. % Inhibition of anti-oxidant activity of DPPH by VAP microcapsules, VAP topical formulations and standard ascorbic acid.
Results suggested that VAP retains its antioxidant activity as complex with combination of starch derivatives and in topical cream based formulations. All, formulations showed significant in vitro free radical scavenging activity.
4. Conclusion
The results obtained from this study revealed that using combination of wall materials such as maltodextrin and Hi cap 100 can be an economically viable alternative over expensive encapsulating agents such as gum arabic and gelatin. These starches forms very stable oil-in-water emulsions and helps in increasing stability of VAP. The spray drying technique used for micro encapsulation of VAP was optimized using a response surface methodology (RSM) for two variables such as solid concentration and inlet temperature to obtain microcapsules with desirable characteristics. The drug content and encapsulation efficiency of microcapsules was found to be in the range of 29.24% to 30.86% and 59% - 63% respectively. Particle size analysis showed a multi-modal particle size distribution for all samples, along with a main mode in intermediate diameters range (5.039 to 7.712 μm). The morphological study showed spherical shape of microcapsules with some irregularity on the outer surface. The topical formulation of VAP containing moisturizing cream base developed using simple cold processing technique was non-sticky and easily water washable. The drug release study showed 80.18% to 83.43% of drug release from all VAP microcapsules while topical formulations prepared by VAP microcapsules showed 67.09% to 71.45% drug release at the end of 24 hrs. All formulations showed significant in vitro antioxidant activity and were found to be stable for 3 months at accelerated conditions.
Acknowledgements
The authors are thankful to Piramal Pvt Ltd., and Ingredion Pvt. Ltd. India for providing the gift sample of VAP acetate and Hicap 100. The authors are also thankful to UGC-SAP-2801-PH for providing the research fellowship and Institute of Chemical Technology, Mumbai, India, for providing all facilities and guidance.
Conflict of Interest
This article does not contain any studies with human and animal subjects performed by any of the authors. All authors (AB Gangurde and PD Amin) declare that they have no conflict of interest.
Cite this paper
Gangurde, A.B. and Amin, P.D. (2017) Microencapsulation by Spray Drying of Vitamin A Palmitate from Oil to Powder and Its Application in Topical Delivery System. Journal of Encapsulation and Adsorption Sciences, 7, 10- 39. https://doi.org/10.4236/jeas.2017.71002
References
- 1. Loveday, S.M. and Singh, H. (2008) Recent Advances in Technologies for Vitamin A Protection in Foods. Trends in Food Science & Technology, 19, 657-668.
https://doi.org/10.1016/j.tifs.2008.08.002 - 2. West, C.E., Eilander, A. and van Lieshout, M. (2002) Consequences of Revised Estimates of Carotenoid Bioefficacy for Dietary Control of Vitamin A Deficiency in Developing Countries. The Journal of Nutrition, 132, 2920S-2926S.
- 3. Moyano, M. and Segall, A. (2011) Vitamin A Palmitate and-Lipoic Acid Stability in o/w Emulsions for Cosmetic Application. Journal of Cosmetic Science, 62, 405-415.
- 4. Sauvant, P., Cansell, M., Sassi, A.H. and Atgié, C. (2012) Vitamin A Enrichment: Caution with Encapsulation Strategies Used for Food Applications. Food Research International, 46, 469-479.
https://doi.org/10.1016/j.foodres.2011.09.025 - 5. Promislow, J.H., Goodman-Gruen, D., Slymen, D.J. and Barrett-Connor, E. (2002) Retinol Intake and Bone Mineral Density in the Elderly: The Rancho Bernardo Study. Journal of Bone and Mineral Research, 17, 1349-1358.
https://doi.org/10.1359/jbmr.2002.17.8.1349 - 6. Feskanich, D., Singh, V., Willett, W.C. and Colditz, G.A. (2002) Vitamin A Intake and Hip Fractures among Postmenopausal Women. JAMA, 287, 47-54.
https://doi.org/10.1001/jama.287.1.47 - 7. Failloux, N., Bonnet, I., Perrier, E. and Baron, M.H. (2004) Effects of Light, Oxygen and Concentration on Vitamin A1. Journal of Raman Spectroscopy, 35, 140-147.
https://doi.org/10.1002/jrs.1116 - 8. Semenova, E.M., Cooper, A., Wilson, C.G. and Converse, C.A. (2002) Stabilization of All-Trans-Retinol by Cyclodextrins: A Comparative Study Using HPLC and Fluorescence Spectroscopy. Journal of Inclusion Phenomena and Macrocyclic Chemistry, 44, 155-158.
https://doi.org/10.1023/A:1023042612880 - 9. Lesmes, U. and McClements, D.J. (2009) Structure-Function Relationships to Guide Rational Design and Fabrication of Particulate Food Delivery Systems. Trends in Food Science & Technology, 20, 448-457.
https://doi.org/10.1016/j.tifs.2009.05.006 - 10. Fuchs, M., Turchiuli, C., Bohin, M., Cuvelier, M., Ordonnaud, C., Peyrat-Maillard, M., et al. (2006) Encapsulation of Oil in Powder Using Spray Drying and Fluidised Bed Agglomeration. Journal of Food Engineering, 75, 27-35.
https://doi.org/10.1016/j.jfoodeng.2005.03.047 - 11. Sinton, R. (1998) TasteTech—The Taste of Things to Come. Nutrition & Food Science, 98, 227-230.
https://doi.org/10.1108/00346659810215172 - 12. Shahidi, F. and Han, X.Q. (1993) Encapsulation of Food Ingredients. Critical Reviews in Food Science & Nutrition, 33, 501-547.
https://doi.org/10.1080/10408399309527645 - 13. Buffo, R. and Reineccius, G. (2000) Optimization of Gum Acacia/Modified Starch/ Maltodextrin Blends for the Spray Drying of Flavors. Perfumer & Flavorist, 25, 45-54.
- 14. Chiu, Y., Chiu, C., Chien, J., Ho, G., Yang, J. and Chen, B. (2007) Encapsulation of Lycopene Extract from Tomato Pulp Waste with Gelatin and Poly(γ-Glutamic Acid) as Carrier. Journal of Agricultural and Food Chemistry, 55, 5123-5130.
https://doi.org/10.1021/jf0700069 - 15. Ersus, S. and Yurdagel, U. (2007) Microencapsulation of Anthocyanin Pigments of Black Carrot (Daucus carota L.) by Spray Drier. Journal of Food Engineering, 80, 805-812.
https://doi.org/10.1016/j.jfoodeng.2006.07.009 - 16. Tuyen, C.K., Nguyen, M.H. and Roach, P.D. (2010) Effects of Spray Drying Conditions on the Physicochemical and Antioxidant Properties of the Gac (Momordica cochinchinensis) Fruit Aril Powder. Journal of Food Engineering, 98, 385-392.
https://doi.org/10.1016/j.jfoodeng.2010.01.016 - 17. Reineccius, G.A. (1989) Flavor Encapsulation. Food Reviews International, 5, 147-176.
https://doi.org/10.1080/87559128909540848 - 18. Drusch, S., Serfert, Y., Van Den Heuvel, A. and Schwarz, K. (2006) Physicochemical Characterization and Oxidative Stability of Fish Oil Encapsulated in an Amorphous Matrix Containing Trehalose. Food Research International, 39, 807-815.
https://doi.org/10.1016/j.foodres.2006.03.003 - 19. Kolanowski, W., Laufenberg, G. and Kunz, B. (2004) Fish Oil Stabilisation by Microencapsulation with Modified Cellulose. International Journal of Food Sciences and Nutrition, 55, 333-343.
https://doi.org/10.1080/09637480410001725157 - 20. Balasubramani, P., Palaniswamy, P., Visvanathan, R., Thirupathi, V., Subbarayan, A. and Maran, J.P. (2015) Microencapsulation of Garlic Oleoresin Using Maltodextrin as Wall Material by Spray Drying Technology. International Journal of Biological Macromolecules, 72, 210-217.
https://doi.org/10.1016/j.ijbiomac.2014.08.011 - 21. Desorgher, M. and Desorgher, S. (2002) The Nutrient Needs of the Flautist—Vitamin A. Autism, Pigments, and the Immune System.
- 22. Pople, P.V. and Singh, K.K. (2006) Development and Evaluation of Topical Formulation Containing Solid Lipid Nanoparticles of Vitamin A. AAPS PharmSciTech, 7, E63-E69.
https://doi.org/10.1208/pt070491 - 23. Junyaprasert, V.B., Mitrevej, A., Sinchaipanid, N., Boonme, P. and Wurster, D.E. (2001) Effect of Process Variables on the Microencapsulation of Vitamin A Palmitate by Gelatin-Acacia Coacervation. Drug Development and Industrial Pharmacy, 27, 561-566.
https://doi.org/10.1081/DDC-100105181 - 24. Fule, R., Meer, T., Amin, P., Dhamecha, D. and Ghadlinge, S. (2014) Preparation and Characterisation of Lornoxicam Solid Dispersion Systems Using Hot Melt Extrusion Technique. Journal of Pharmaceutical Investigation, 44, 41-59.
https://doi.org/10.1007/s40005-013-0099-7 - 25. Wang, Y., Lu, Z., Lv, F. and Bie, X. (2009) Study on Microencapsulation of Curcumin Pigments by Spray Drying. European Food Research and Technology, 229, 391-396.
https://doi.org/10.1007/s00217-009-1064-6 - 26. Patil, A.K., Gangurde, A.B. and Amin, P.D. (2014) Comparative Study of Different Formulations of Aceclofenac as a Topical Drug Delivery System and It’s in Vitro and in Vivo Characterization. International Journal of Pharmaceutical Sciences and Research, 5, 3401.
- 27. Kamble, S., Udapurkar, P., Nakhat, P., Yeole, P. and Biyani, K. (2011) Development and Evaluation of Sorbitan Monostearate Organogels as a Topical Delivery System for Aceclofenac. Indian Journal of Pharmaceutical Education and Research, 45, 65-70.
- 28. Segall, A., Sosa, M., Alami, A., Enero, C., Hormaechea, F., Pizzorno, M., et al. (2004) Stability Study of Lipoic Acid in the Presence of Vitamins A and E in o/w Emulsions for Cosmetic Application. Journal of Cosmetic Science, 55, 449-462.
- 29. Wakode, R. and Bajaj, A. (2011) Gelatin Microspheres for Topical Delivery of Vitamin A Palmitate. NHSM Journal of Pharmacy and Healthcare Management, 2, 61-65.
- 30. Nenadis, N. and Tsimidou, M. (2002) Observations on the Estimation of Scavenging Activity of Phenolic Compounds Using Rapid 1,1-Diphenyl-2-picrylhydrazyl (DPPH•) Tests. Journal of the American Oil Chemists’ Society, 79, 1191-1195.
https://doi.org/10.1007/s11746-002-0626-z - 31. Erasto, P., Bojase-Moleta, G. and Majinda, R.R. (2004) Antimicrobial and Antioxidant Flavonoids from the Root Wood of Bolusanthus speciosus. Phytochemistry, 65, 875-880.
https://doi.org/10.1016/j.phytochem.2004.02.011 - 32. Pouton, C.W. (2000) Lipid Formulations for Oral Administration of Drugs: Non-Emulsifying, Self-Emulsifying and “Self-Microemulsifying” Drug Delivery Systems. European Journal of Pharmaceutical Sciences, 11, S93-S98.
https://doi.org/10.1016/s0928-0987(00)00167-6 - 33. Craig, D., Barker, S., Banning, D. and Booth, S. (1995) An Investigation into the Mechanisms of Self-Emulsification Using Particle Size Analysis and Low Frequency Dielectric Spectroscopy. International Journal of Pharmaceutics, 114, 103-110.
https://doi.org/10.1016/0378-5173(94)00222-Q - 34. Yang, Y. and McClements, D.J. (2013) Encapsulation of Vitamin E in Edible Emulsions Fabricated Using a Natural Surfactant. Food Hydrocolloids, 30, 712-720.
https://doi.org/10.1016/j.foodhyd.2012.09.003 - 35. Patil, V., Chauhan, A.K. and Singh, R.P. (2014) Optimization of the Spray-Drying Process for Developing Guava Powder Using Response Surface Methodology. Powder Technology, 253, 230-236.
https://doi.org/10.1016/j.powtec.2013.11.033 - 36. Jaya, S. and Das, H. (2004) Effect of Maltodextrin, Glycerol Monostearate and Tricalcium Phosphate on Vacuum Dried Mango Powder Properties. Journal of Food Engineering, 63, 125-134.
https://doi.org/10.1016/S0260-8774(03)00135-3 - 37. Loh, S.K., Man, C., Yaakob, B., Tan, C.P., Osman, A. and Hamid, N.S.A. (2005) Process Optimisation of Encapsulated Pandan (Pandanus amaryllifolius) Powder Using Spray-Drying Method. Journal of the Science of Food and Agriculture, 85, 1999-2004.
https://doi.org/10.1002/jsfa.2169 - 38. Silva, P.I., Stringheta, P.C., Teófilo, R.F. and de Oliveira, I.R.N. (2013) Parameter Optimization for Spray-Drying Microencapsulation of Jaboticaba (Myrciaria jaboticaba) Peel Extracts Using Simultaneous Analysis of Responses. Journal of Food Engineering, 117, 538-544.
https://doi.org/10.1016/j.jfoodeng.2012.08.039 - 39. Tonon, R.V. (2009) Secagem por atomização do suco de açaí: Influência das variáveis de processo, qualidade e estabilidade do produto. Universidade Estadual de Campinas, Campinas, Doutorado em Engenharia de Alimentos.
- 40. Molyneux, P. (2004) The Use of the Stable Free Radical Diphenylpicrylhydrazyl (DPPH) for Estimating Antioxidant Activity. Songklanakarin Journal of Science and Technology, 26, 211-219.