International Journal of Organic Chemistry
Vol. 2 No. 4 (2012) , Article ID: 25860 , 4 pages DOI:10.4236/ijoc.2012.24045
Ti(IV) Chloride-Promoted Diastereoselective Conjugate Addition of 1-Enoyl-5-Substituted Hydantoins with Allyltrimethylsilane
Department of Applied Chemistry, Faculty of Engineering, Kanagawa Institute of Technology, Atsugi, Japan
Email: *yamagu@chem.kanagawa-it.ac.jp
Received August 22, 2012; revised October 10, 2012; accepted October 22, 2012
Keywords: Hydantoin; Conjugate Addition; Lewis Acid; Allyltrimethylsilane
ABSTRACT
Diastereoselective conjugate addition of 1-enoyl-5-substituted hydantoins with allyltrimethylsilane in the presence of Ti(IV) chloride proceeded to give the corresponding allyl adducts in high yield and high diastereoselectivity. In order to determine the absolute configuration on the β-position of the acyl group, the hydantoin was removed by hydrolysis of the allyl adducts with a base to give the corresponding carboxylic acid. It was found that the absolute configuration was S on the basis of specific rotation.
1. Introduction
Chiral auxiliaries are one of the most useful tools to synthesize optically active compounds. Among the many types of chiral auxiliaries, 5-membered heterocycles containing nitrogen atom(s), such as 2-oxazolidinone or 2-imidazolidinone, are the most popular [1]. Optically active hydantoins, which resemble 2-oxazolidinone or 2-imidazolidinone in structure, were easily prepared from amino acid amides without racemization [2] and they play a role as a chiral auxiliary [3]. We have reported high diastereoselective conjugate addition of 1-enoyl-5-substituted hydantoin with a nucleophile, e.g., diethylaluminum chloride or dialkylcuprate reagent [3]. Generally, the diastereoselectivity at the β-position of the acyl group can be lower than that at the β-position of the acyl group (Scheme 1) [4,5] in the synthesis of optically active compounds via the use of a chiral auxiliary.
Therefore, development of a new method and a new chiral auxiliary is required. In this paper, we examined Ti(IV) chloride-promoted diastereoselective conjugate addition of 1-enoyl-5-substituted hydantoins with allyltrimethylsilane and found that optically active hydantoins are effective chiral auxiliaries for inducing a chiral center at the β-position of the acyl group.
2. Results and Discussion
The results in the diastereoselective conjugate addition of 1-enoyl-5-substituted hydantoins 1 with allyltrimethylsilane (All-TMS) are shown in Table 1 and Scheme 2. Ti(IV) chloride was a suitable promoter for the conjugate addition of 1a with All-TMS, but Sn(IV) chloride was not (entries 1 and 2). However, the diastereomeric excess (d.e.) of the allyl adduct 2a was not determined by 1H NMR or HPLC analysis. The diastereoselectivity of 2a could be successful determined from the HPLC analysis of 3a, which was obtained from the catalytic hydrogenation of 2a (Scheme 3). Conjugate additions of 1-enoylhydantoin 1b and 1c, which were prepared from Tryptophan and Valine, respectively, were also examined. Unfortunately, the d.e.s of both allyl adducts were lower (entries 3 and 4).
In addition, 1-cinnamoylhydantoin 1d was used as a substrate. Since the steric hindrance of the phenyl group at the cinnamoyl group is relative higher than that of the methyl group at the crotonyl group, the reactivity of 1d was expected to decrease relative to that of 1a. In fact, a reaction temperature of −50˚C was necessary to complete the reaction, however, their chemical yield and d.e. were similar (entry 5). Under the conditions of a higher reaction temperature at room temperature, the chemical yield and d.e. decreased (entry 6).
Wu et al. reported a similar conjugate addition of 1- cinnamoyl-4-substituted 2-oxazolidinone with allylsilane in the presence of Ti(IV) chloride at 25˚C [6]. However, the present conjugate addition using 1-cinnamoylhydantoin proceeded under a lower temperature of −50˚C. Comparing the two reaction temperatures above, the re-
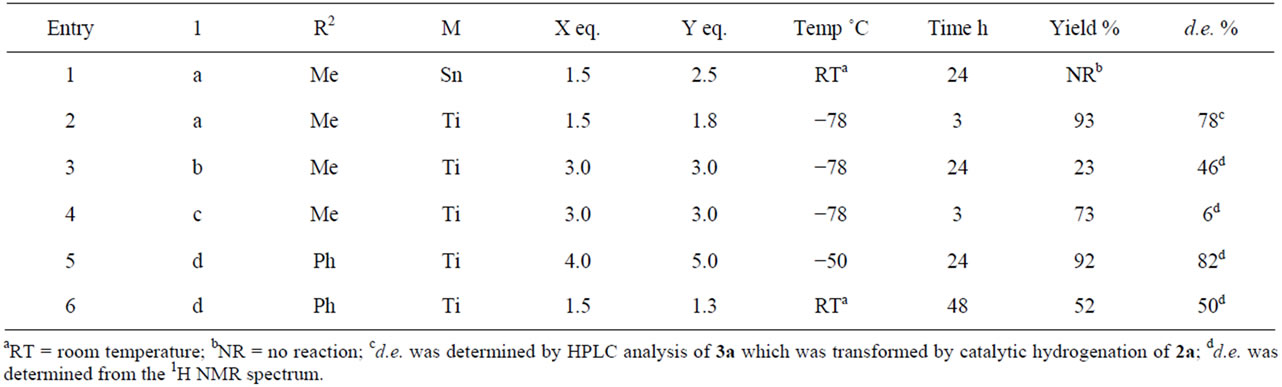
Table 1. Lewis acid-promoted diastereoselective conjugate addition of 1 with All-TMS.
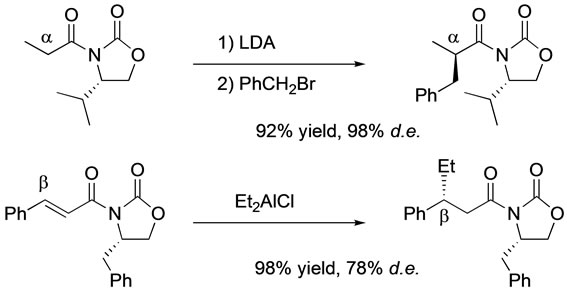
Scheme 1. Comparison of diastereoselectivities.
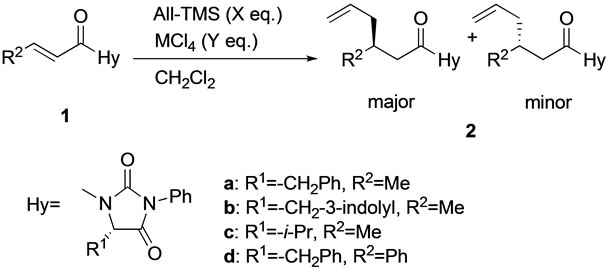
Scheme 2. Ti(IV) chloride-promoted diastereoselective conjugate addition of 1-enoyl-5-substituted hydantoins with AllTMS.
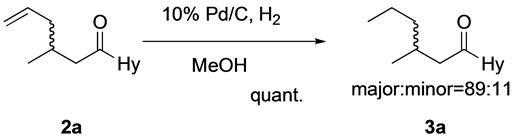
Scheme 3. Catalytic hydrogenation of 2a to 3a.
activity of hydantoin would be higher than that of 2-oxazolidinone in the conjugate addition.
In order to determine the absolute configuration of the β-position of the acyl group, hydantoin was removed from the adduct to give the corresponding carboxylic acid [7]. Allin and co-workers reported that the specific rotation of (3R)-3-phenyl-hex-5-enoic acid was 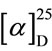 - 20.0˚ (CH2Cl2), c 1.27) [8]. The specific rotation of 4, which was cleaved from 2d, was
- 20.0˚ (CH2Cl2), c 1.27) [8]. The specific rotation of 4, which was cleaved from 2d, was 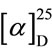 + 27.6˚ (CH2Cl2, c 0.59). Comparison between the specific rotations, the absolute configuration of 4 was S (Scheme 4). The above result suggested that the structure of intermediate 5 in the transition state would be a bicoordinated model, and thus allylsilane attacks the enoyl group from the Si-face (Figure 1).
+ 27.6˚ (CH2Cl2, c 0.59). Comparison between the specific rotations, the absolute configuration of 4 was S (Scheme 4). The above result suggested that the structure of intermediate 5 in the transition state would be a bicoordinated model, and thus allylsilane attacks the enoyl group from the Si-face (Figure 1).
3. Experimental
General procedure for preparation of 1: To a DMF solution of hydantoin, acid anhydride and N,N-diisopropylethylamine was added a catalytic amount of N,Ndimethylaminopyridine at room temperature and the reaction mixture was stirred for over night. DMF was removed under reduced pressure and the residue was extracted with EtOAc. The organic layer was washed with 5% citric acid aq., 5% NaHCO3 aq., and brine. The organic layer was dried with Na2SO4 and was concentrated under reduced pressure. The residue was purified by preparative TLC on silica-gel and 1 was given.
(S)-5-benzyl-1-crotonyl-3-phenylhydantoin: mp 85˚C.  + 233.7˚ (CHCl3, c1.0). IR (KBr) 1791, 1733, 1686, 1639 cm−1. 1H NMR (CDCl3, 400 MHz) δ 1.98 (3H, dd, J = 1.4 and 6.4 Hz), 3.35 (1H, dd, J = 2.3 and 13.7 Hz), 3.67 (1H, dd, J = 5.0 and 13.7 Hz), 5.02 (1H, dd, J = 2.3 and 5.0 Hz), 6.84-6.90 (2H, m), 7.02 - 7.10 (2H, m), 7.13 - 7.40 (8H, m). 13C NMR (CDCl3, 400 MHz) δ 18.63, 34.80, 59.58, 123.12, 126.44, 127.67, 128.61, 128.92, 129.10, 129.78, 130.25, 133.37, 147.72, 152.43, 164.20, 169.65.
+ 233.7˚ (CHCl3, c1.0). IR (KBr) 1791, 1733, 1686, 1639 cm−1. 1H NMR (CDCl3, 400 MHz) δ 1.98 (3H, dd, J = 1.4 and 6.4 Hz), 3.35 (1H, dd, J = 2.3 and 13.7 Hz), 3.67 (1H, dd, J = 5.0 and 13.7 Hz), 5.02 (1H, dd, J = 2.3 and 5.0 Hz), 6.84-6.90 (2H, m), 7.02 - 7.10 (2H, m), 7.13 - 7.40 (8H, m). 13C NMR (CDCl3, 400 MHz) δ 18.63, 34.80, 59.58, 123.12, 126.44, 127.67, 128.61, 128.92, 129.10, 129.78, 130.25, 133.37, 147.72, 152.43, 164.20, 169.65.
(S)-1-crotonyl-5-(3-indolylmethyl)-3-phenylhydantoin: mp 85˚C. 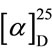 + 172.3˚ (CHCl3, c1.0). IR (KBr) 1791, 1731, 1684, 1635 cm−1. 1H NMR (CDCl3, 400 MHz) δ 1.97 (3H, dd, J = 1.8 and 6.9 Hz), 3.62 (1H, dd, J = 2.3 and 14.7 Hz), 3.85 (1H, dd, J = 5.0 and 14.7 Hz), 5.03 (1H, dd, J = 2.3 and 5.0 Hz), 6.55 - 6.65 (2H, m), 6.89 (1H, s), 7.05 (1H, dt, J = 0.9 and 8.2), 7.10 - 7.20 (2H, m), 7.20 - 7.35 (5H, m), 7.51 (1H, dd, J = 0.9 and 8.2 Hz), 8.31 (1H, s). 13C NMR (CDCl3, 400 MHz) δ 18.62, 25.27, 59.59, 107.64, 111.05, 119.20, 120.03, 122.42, 123.39,
+ 172.3˚ (CHCl3, c1.0). IR (KBr) 1791, 1731, 1684, 1635 cm−1. 1H NMR (CDCl3, 400 MHz) δ 1.97 (3H, dd, J = 1.8 and 6.9 Hz), 3.62 (1H, dd, J = 2.3 and 14.7 Hz), 3.85 (1H, dd, J = 5.0 and 14.7 Hz), 5.03 (1H, dd, J = 2.3 and 5.0 Hz), 6.55 - 6.65 (2H, m), 6.89 (1H, s), 7.05 (1H, dt, J = 0.9 and 8.2), 7.10 - 7.20 (2H, m), 7.20 - 7.35 (5H, m), 7.51 (1H, dd, J = 0.9 and 8.2 Hz), 8.31 (1H, s). 13C NMR (CDCl3, 400 MHz) δ 18.62, 25.27, 59.59, 107.64, 111.05, 119.20, 120.03, 122.42, 123.39,
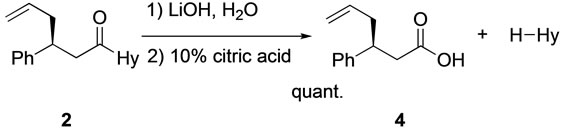
Scheme 4. Hydrolysis of 2 to carboxylic acid 4 and hydantoin.
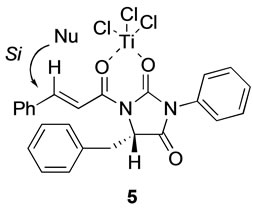
Figure 1. Conformation of intermediate 5 in the transition state.
123.75, 126.44, 127.36, 128.77, 128.97, 130.27, 135.89, 147.33, 152.70, 164.32, 170.58.
(S)-1-crotonyl-5-isopropyl-3-phenylhydantoin: oil. 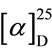 + 19.7˚ (CHCl3, c 0.27). IR (neat) 1790, 1732, 1688, 1637 cm−1. 1H NMR (CDCl3, 400 MHz) δ 0.97 (3H, d, J = 6.9 Hz), 1.26 (3H, d, J = 6.9 Hz), 1.97 (3H, dd, J = 6.4 and 0.9 Hz), 2.66 (1H, dsept, J = 6.9 and 3.2 Hz), 4.70 (1H, d, J = 3.2 Hz), 7.20 (1H, dq, J = 15.1 and 6.4 Hz), 7.30 (1H, dq, J = 15.1 and 0.9 Hz), 7.33 (2H, dt, J = 7.3 and 2.7 Hz), 7.41 (1H, dt, J = 7.3 and 2.3 Hz), 7.48 (2H, tt, J = 7.3 and 2.3 Hz). 13C NMR (CDCl3, 400 MHz) δ 15.64, 18.06, 18.58, 29.70, 63.02, 123.41, 126.47, 128.90, 129.22, 130.60, 147.39, 153.10, 164.07, 169.32.
+ 19.7˚ (CHCl3, c 0.27). IR (neat) 1790, 1732, 1688, 1637 cm−1. 1H NMR (CDCl3, 400 MHz) δ 0.97 (3H, d, J = 6.9 Hz), 1.26 (3H, d, J = 6.9 Hz), 1.97 (3H, dd, J = 6.4 and 0.9 Hz), 2.66 (1H, dsept, J = 6.9 and 3.2 Hz), 4.70 (1H, d, J = 3.2 Hz), 7.20 (1H, dq, J = 15.1 and 6.4 Hz), 7.30 (1H, dq, J = 15.1 and 0.9 Hz), 7.33 (2H, dt, J = 7.3 and 2.7 Hz), 7.41 (1H, dt, J = 7.3 and 2.3 Hz), 7.48 (2H, tt, J = 7.3 and 2.3 Hz). 13C NMR (CDCl3, 400 MHz) δ 15.64, 18.06, 18.58, 29.70, 63.02, 123.41, 126.47, 128.90, 129.22, 130.60, 147.39, 153.10, 164.07, 169.32.
(S)-5-benzyl-1-cinnnamoyl-3-phenylhydantoin: mp 129˚C - 130˚C. 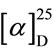 + 233.7˚ (CHCl3, c1.0). IR (KBr) 1786, 1726, 1679, 1618 cm−1. 1H NMR (CDCl3, 400 MHz) δ 3.40 (1H, dd, J = 2.8 and 13.7 Hz), 3.74 (1H, dd, J = 5.0 and 13.7 Hz), 5.11 (1H, dd, J = 2.8 and 5.0 Hz), 6.85 - 6.95 (2H, m), 7.05 - 7.15 (2H, m), 7.25 - 7.30 (3H, m), 7.35 - 7.45 (6H, m), 7.60 - 7.65 (2H, m), 7.88 (1H, d, J = 16.0 Hz), 8.00 (1H, d, J = 16.0 Hz). 13C NMR (CDCl3, 400 MHz) δ 34.91, 59.82, 118.20, 126.52, 27.77, 128.69, 128.72, 128.93, 129.07, 129.22, 129.87, 130.27, 130.91, 133.38, 134.41, 147.13, 152.69, 164.51, 169.66.
+ 233.7˚ (CHCl3, c1.0). IR (KBr) 1786, 1726, 1679, 1618 cm−1. 1H NMR (CDCl3, 400 MHz) δ 3.40 (1H, dd, J = 2.8 and 13.7 Hz), 3.74 (1H, dd, J = 5.0 and 13.7 Hz), 5.11 (1H, dd, J = 2.8 and 5.0 Hz), 6.85 - 6.95 (2H, m), 7.05 - 7.15 (2H, m), 7.25 - 7.30 (3H, m), 7.35 - 7.45 (6H, m), 7.60 - 7.65 (2H, m), 7.88 (1H, d, J = 16.0 Hz), 8.00 (1H, d, J = 16.0 Hz). 13C NMR (CDCl3, 400 MHz) δ 34.91, 59.82, 118.20, 126.52, 27.77, 128.69, 128.72, 128.93, 129.07, 129.22, 129.87, 130.27, 130.91, 133.38, 134.41, 147.13, 152.69, 164.51, 169.66.
General procedure for conjugate addition of 1 with All-TMS: To a CH2Cl2 solution of 1 was added Ti(IV) chloride under nitrogen. The mixture was stirred for 10 min. All-TMS was added to the reaction mixture. The reaction was quenched by adding saturated NH4Cl solution, and the organic materials were extracted with CH2Cl2. The CH2Cl2 layer was washed with brine, and dried over Na2SO4. After removal of solvent under reduced pressure, the residue was purified by silica-gel column chromatography, and the allyl adduct was isolated.
Allyl adduct 2a (signals of the major adduct were showed only): 1H NMR (CDCl3, 300 MHz) δ 1.03 (3H, d, J = 7.2 Hz), 2.05 - 2.25 (3H, m), 2.79 (1H, dd, J = 16.7 and 7.5 Hz), 2.93 (1H, dd, J = 16.7 and 5.1 Hz), 3.37 (1H, dd, J = 13.9 and 2.7 Hz), 3.66 (1H, dd, J = 13.9 and 5.1 Hz), 4.95 - 5.10 (3H, m), 5.70 - 5.90 (1H, m), 6.85 - 6.95 (2H, m), 7.05 - 7.10 (2H, m), 7.20 - 7.45 (6H, m).
Allyl adduct 2b: 1H NMR (CDCl3, 400MHz) δ 1.03 (2.19H, d, J = 6.4 Hz, the major adduct signal), 1.07 (0.81H, d, J = 6.9 Hz, the minor adduct signal), 2.00 - 2.30 (3H, m), 2.64 (0.27H, dd, J = 16.9 and 7.3 Hz, the minor adduct signal), 2.79 (0.73H, dd, J = 16.9 and 6.4 Hz, the major adduct signal), 2.84 (0.73H, dd, J = 16.9 and 6.9 Hz, the major adduct signal), 2.99 (0.27H, dd, J = 16.9 and 5.5 Hz, the minor adduct signal), 3.59 (1H, dd, J = 14.2 and 2.3 Hz), 3.85 (1H, dd, J = 14.2 and 5.0 Hz), 5.00 - 5.10 (3H, m), 5.70 - 5.90 (1H, m), 6.50 - 6.60 (2H, m), 6.92 (1H, d, J = 2.8 Hz), 7.07 (1H, td, J = 6.9 and 0.9 Hz), 7.19 (1H, td, J = 8.2 and 0.9 Hz), 7.25 - 7.30 (3H, m), 7.33 (1H, d, J = 8.2 Hz), 7.50 (1H, d, J = 8.2 Hz), 8.10 (1H, s).
Allyl adduct 2c: 1H NMR (CDCl3, 400 MHz) δ 0.95 - 1.05 (3H, m), 1.25 (3H, d, J = 6.9 Hz), 1.95 - 2.30 (3H, m), 2.55 - 2.65 (1H, m), 2.71 (1H, dd, J = 16.0 and 8.2 Hz, the minor adduct signal), 2.92 (1H, dd, J = 16.0 and 6.4 Hz, the major adduct signal), 2.96 (1H, dd, J = 16.0 and 7.3 Hz, the major adduct signal), 3.14 (1H, dd, J = 16.0 and 5.0 Hz, the minor adduct signal), 4.65 (1H, d, J = 3.2 Hz), 4.95 - 5.10 (2H, m), 5.70 - 5.85 (1H, m), 7.30 - 7.35 (2H, m), 7.30 - 7.35 (3H, m).
Allyl adduct 2d: 1H NMR (CDCl3, 400 MHz) δ 2.47 (2H, t, J = 6.9 Hz), 3.17 (1H, dd, J = 16.5 and 5.0 Hz), 3.29 (1H, dd, J = 13.7 and 2.8 Hz), 3.35 - 3.50 (1H, m), 3.51 (1H, dd, J = 16.5 and 8.7 Hz), 3.59 (1H, dd, J = 13.7 and 5.0 Hz), 4.79 (0.91H, dd, J = 5.0 and 2.5 Hz), 4.88 (0.09H, dd, J = 5.0 and 2.3 Hz), 4.95 - 5.10 (2H, m), 5.65 - 5.80 (1H, m), 6.57 (0.18H, d, J = 6.9 Hz), 6.80 - 6.85 (1.82H, m), 6.95 - 7.05 (1.82H, m), 7.09 (0.18H, t, J = 7.3 Hz), 7.15 - 7.40 (11H, m).
Catalytic hydrogenation of 2a: To a methanol (80 mL) and a small amount of CH2Cl2 solution of 2a was added 10% Pd/C as catalyst under hydrogen, and the reaction mixture was stirred for 2 days. After removal of the catalyst by Celite filtration, the organic layer was concentrated under reduced pressure and 3a was given.
3a (signals of the major adduct were showed only): 1H NMR (CDCl3, 300 MHz) δ 0.93 (3H, t, J = 6.6 Hz), 1.02 (3H, d, J = 6.6 Hz), 1.25 - 1.45 (4H, m), 2.00 - 2.10 (1H, m), 2.75 (1H, dd, J = 16.4 and 8.1 Hz), 2.93 (1H, dd, J = 16.4 and 5.4 Hz), 3.37 (1H, dd, J = 13.9 and 2.7 Hz), 3.66 (1H, dd, J = 13.9 and 4.9 Hz), 4.79 (1H, dd, J = 4.9 and 2.7 Hz), 6.85 - 6.95 (2H, m), 7.05 - 7.10 (2H, m), 7.25 - 7.40 (6H, m).
Hydrolysis of the allyl adduct into 4 [7]: To a solution of LiOH and H2O2 was added a solution (THF and H2O) of 4 at 0˚C. The reaction mixture was stirred for 2 h. Na2SO3 was added to the reaction mixture at 0˚C, and it was stirred for 30 min. After removal of the solvent under reduced pressure, the residue was aciditified by adding 10% citric acid solution. The organic materials were extracted with CH2Cl2. The organic layer was dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by silica-gel preparative TLC and 4 was obtained as oil.
4: 1H NMR (CDCl3, 400 MHz) δ 2.37 (2H, t, J = 7.3 Hz), 2.57 (1H, dd, J = 16.0 and 8.2 Hz), 2.69 (1H, dd, J = 16.0 and 6.4 Hz), 3.17 (1H, quint, J = 7.3 Hz), 4.90 - 5.05 (2H, m), 5.63 (1H, ddt, J = 16.9, 10.0, and 7.3 Hz), 7.10 - 7.30 (5H, m), 8.65 (1H, brs).
4. Conclusion
We showed that hydantoin is more reactive than conventional 2-oxazolidinone chiral auxiliaries. Further studies on the utilizations of hydantoin as a unique chiral auxiliary are now in progress.
REFERENCES
- R. M. Coates and S. E. Denmark, “Handbook of Reagents for Organic Synthesis: Reagents, Auxiliaries, and Catalysts for C-C Bond Formation,” John Willey & Sons Ltd., Chichester, 1999.
- J. Yamaguchi, M. Harada, T. Kondo, T. Noda and T. Suyama, “A Facile Method for Preparation of Optically Active Hydantoin,” Chemistry Letters, Vol. 32, No. 4, 2003, pp. 372-373. doi:10.1246/cl.2003.372
- J. Yamaguchi, M. Harada, T. Narushima, A. Saitoh, K. Nozaki and T. Suyama, “Diastereoselective Conjugate Addition of 1-(α,β-Unsaturated acyl)hydantoin with Nucleophiles,” Tetrahedron Letters, Vol. 46, No. 38, 2005, pp. 6411-6415. doi:10.1016/j.tetlet.2005.07.116
- D. A. Evans, M. D. Ennis and D. J. Mathre, “Asymmetric Alkylation Reaction of Chiral Imide Enolate. A Practical Approach to the Enantioselective Synthesis of α-Substituted Carboxylic Acid Derivatives,” Journal of the American Chemical Society, Vol. 104, No. 6, 1982, pp. 1737-1739. doi:10.1021/ja00370a050
- K. Rück and H. Kunz, “Stereoselective Conjugate Addition of Organoaluminum Chloride to α,β-Unsaturated Carboxylic Acid Derivatives,” Synthesis, Vol. 1993, No. 10, 1993, pp. 1018-1028. doi:10.1055/s-1993-25990
- M.-J. Wu, C.-C. Wu and P.-C. Lee, “Lewis Acid Promoted Asymmetric 1,4-Addition of Allyltrimethylsilane to Chiral α,β-Unsaturated N-Acylamides,” Tetrahedron Letters, Vol. 33, No. 18, 1992, pp. 2547-2548. doi:10.1016/S0040-4039(00)92238-X
- J. R. Harding, R. A. Hughes, N. M. Kelly, A. Sutherland and C. L. Willis, “Synthesis of Isotopically Labelled L-aAmino Acid with an Asymmetric Centre at C-3,” Journal of the Chemical Society, Perkin Transactions 1, Vol. 2000, 2000, pp. 3406-3416. doi:10.1039/b005172l
- S. M. Allin, M. S. Essat, C. H. Pita, R. D. Baird, V. McKee, M. Elsegood, M. Edgar, D. M. Andrews, P. Shah and I. Aspinall, “Applications of the Amino-Cope Rearrengement: Synthesis of Tetrahydropyran, d-Lactone and Piperidine Targets,” Organic & Biomolecular Chemistry, Vol. 3, 2005, pp. 809-815. doi:10.1039/b416179c
NOTES
*Corresponding author.