International Journal of Organic Chemistry
Vol. 2 No. 3 (2012) , Article ID: 22735 , 10 pages DOI:10.4236/ijoc.2012.23027
Synthesis and Solvatochromic Behavior of Hexaphenylbenzenes and Indeno[1,2-b]fluorene Derivatives with Hydroxy Groups
Department of Chemistry, Interdisciplinary Graduate School of Faculty of Science and Engineering, Shimane University, Matsue, Japan
Email: *iyamaguchi@riko.shimane-u.ac.jp
Received July 3, 2012; revised August 14, 2012; accepted August 26, 2012
Keywords: Hexaphenylbenzene; Indeno[1,2-b]fluorene; Donor Number; Photoluminescence; Solvatochromism
ABSTRACT
Hexakis(4-methoxyphenyl)benzene (HPB-OMe(1)) and hexakis(2,6-dimethyl-4-methoxy phenyl)benzene (HPB-OMe(2)) were synthesized via organometallic complex catalysis. The treatment of HPB-OMe(1) with FeCl3 caused cyclodehydrogenation at two positions to yield an oligophenylene with an indeno[1,2-b]fluorene structure (IF-OMe). Deprotection of the methoxy groups of these compounds was conducted by treatment with BBr3. Deprotonation of the OH groups of HPB-OH(1), HPB-OH(2), and IF-OH through treatment with NaH caused a bathochromic shift in the absorption and photoluminescence (PL) peaks. The bathochromic shift of the deprotonated species increased with the donor number (DN) of the solvents. These observations can be explained as the consequence of intramolecular charge transfer (ICT) from the ONa groups to the inner benzene rings.
1. Introduction
Oligo(p-phenylene)s (OPs) are an important class of π- conjugated oligomer [1-18]. We have recently reported on oligo(p-phenylene) compounds with an OH group located at either one end or at both ends, namely, OPP(n) -OH (where n is the number of benzene rings) and HOArPh(n)-OH species (Ar = 9,9-dihexylfluorene -2,7-diyl and 2,5-dioctyloxybenzene-1,4-diyl), respectively [19, 20]. These OPP(n)-OH and HO-ArPh(n)-OH compounds exhibited significant solvatochromism where deprotonation of the OH groups, when treated with NaH, caused a bathochromic shift of λmax that increased with the DN of the solvent. The solvatochromism exhibited by OPP(n) -ONa and NaO-ArPh(n)-ONa was attributed to an ICT from the sodium phenoxy group(s) to the adjacent rings [19,20]. The degree of bathochromic shift in the deprotonated species increased with an increase in the chain length that corresponds to the expansion of the π-conjugation system. However, ICT behavior in branched oligophenylenes containing OH groups remains unclear.
Hexaphenylbenzenes (HPBs) are an important class of aromatic compounds in the field of materials science, acting as precursors for graphite-like, dendritic, or photoconductive polycyclic aromatic hydrocarbons [21,22]. HPBs also serve as guest-inclusion organic crystals directed to organic zeolites [23,24]. In this study, the optical properties of HPB-OH compounds were investigated before and after deprotonation of the OH groups. The π-conjugated system of the HPBs is comparatively small because of the presence of steric hindrance between the phenyl groups. We herein therefore also studied an OP which has its OH groups substituted with a planar indeno[1,2-b]fluorene structure (IF-OH), synthesized by the cyclodehydrogenation of HPB-OH, and subsequently investigated its solvatochromic behavior. IFs have attracted considerable attention because they can be useful materials for electroluminescence and photovoltaic devices [25,26]. These applications are based on the fact that IFs have a more extended coplanar, fused structure, which thus enables extended π-conjugation and improves their carrier mobilities compared to simple fluorene derivatives. The large carrier mobility in IFs is suited to the development of new solvatochromic materials based on ICT. The investigation into the optical properties of HPB-OH and IF-OH will afford information pertinent to the development of new solvatochromic materials. It is noteworthy that IF-OH is expected to be a useful starting material for the synthesis of new IFs through reactions using the OH groups. To the best of our knowledge, this is the first example of IFs with reactive groups.
We herein report on the synthesis of HPBs and IF with OH groups and their optical properties before and after the deprotonation of the OH groups.
2. Experimental
2.1. General
Solvents were dried, distilled, and stored under nitrogen. Reagents were purchased and used without further purification. Reactions were carried out with standard Schlenk techniques under nitrogen.
IR and NMR spectra were recorded on a JASCO FT/IR-660 PLUS spectrophotometer and a JEOL AL-400 spectrometer, respectively. Elemental analysis was performed on a Yanagimoto MT-5 CHN corder. UV-Vis and PL spectra were obtained by a JASCO V-560 spectrometer and a JASCO FP-6200 spectrofluorometer, respectively. Quantum yields were calculated by using a diluted ethanol solution of 7-dimethylamino-4-methylcoumarin as the standard.
2.2. Synthesis
2.2.1. Synthesis of 1
4-Bromoanisole (1.4 g, 7.5 mmol), bis(tributylstannyl) acetylene (2.4 g, 3.9 mmol), and Pd(PPh3)4 (0.50 g, 0.43 mmol) were dissolved in 20 ml of dry toluene under N2. After the reaction solution was refluxed for 48 h, the solvent was removed under vacuum. The resulting solid was purified by column chromatography (eluent = CHCl3) and recrystallized from chloroform. The yellow crystals were collected by filtration, washed with cold methanol, and dried under vacuum to give 1 as a yellow solid (0.38 g, 43%). 1H NMR (400 MHz, CDCl3): δ 7.45 (d, J = 8.8 Hz, 4H), 7.26 (d, J = 8.8 Hz, 4H), 3.83 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 156.8, 140.3, 133.6, 132.5, 112.2, 55.0. Anal Calcd for C16H14O2: C, 80.65; H, 5.92. Found: C, 80.88; H, 6.10.
2.2.2. Synthesis of 2
2 was synthesized by the reaction of 2,5-dimethyl-4- bromoanisole with bis(tributylstannyl)acetylene analogously.
Data of 2: Yield = 53%. 1H NMR (400 MHz, CDCl3): δ 7.18 (s, 4H), 3.72 (s, 6H), 2.27 (s, 12H). 13C NMR (100 MHz, CDCl3): δ 157.2, 132.0, 131.1, 118.8, 88.2, 59.8, 16.0. Anal Calcd for C20H22O2: C, 81.60; H, 7.53. Found: C, 81.44; H, 7.36.
2.2.3. Synthesis of HPB-OMe(1)
Dicobalt octacarbonyl (0.068 g, 0.20 mmol) and 1 (0.38 g, 1.6 mmol) were dissolved in 20 ml of dry toluene under N2. After the reaction solution was refluxed for 24 h, the solvent was removed under vacuum. The resulting solid was purified by silica gel column chromatography (eluent = CHCl3), washed with methanol, and dried under vacuum to give HPB-OMe(1) as a light brown solid (0.30 g, 79%). 1H NMR (400 MHz, CDCl3): δ 6.67 (d, J = 8.8 Hz, 12H), 6.42 (d, J = 8.8 Hz, 12H), 3.63 (s, 18H). 13C NMR (100 MHz, DMSO-d6): δ 156.7, 140.2, 133.5, 132.4, 112.1, 54.9. IR (KBr, cm–1): 3008, 2957, 2835, 1611, 1577, 1518, 1464, 1419, 1397, 1287, 1244, 1178, 1109, 1033, 834, 804, 551. Anal Calcd for C48H42O6·0.25H2O: C, C, 80.14; H, 5.96. Found: C, 80.09; H, 5.99. Mp = 378˚C.
2.2.4. Synthesis of HPB-OMe(2)
HPB-OMe(2) was synthesized using a procedure similar to that used for HPB-OMe(1) the analogous method.
Data of HPB-OMe(2): Yield = 65%. 1H NMR (400 MHz, CDCl3): δ 6.41 (s, 12H), 3.50 (s, 18H), 1.93 (s, 36H). 13C NMR (100 MHz, DMSO-d6): δ 154.1, 139.6, 136.5, 132.2, 127.8, 59.5, 15.6. IR (KBr, cm–1): 2935, 1487, 1219, 1126, 1016, 867, 644. Anal Calcd for C60H66O6·0.5H2O: C, 80.77; H, 7.57. Found: C, 80.74; H, 7.14. Mp = 254˚C - 255˚C.
2.2.5. Synthesis of HPB-OH(1)
After a dichloromethane solution (5 ml) of BBr3 (0.34 ml, 3.6 mmol) was added dropwise to a dichloromethane solution (20 ml) of HPB-OMe(1) (0.096 g, 0.13 mmol), the reaction solution was stirred at 20˚C for 30 h and quenched with water. The resulting precipitate was collected by filtration and dried under vacuum to give HPB-OH(1) as a pink solid (0.047 g, 55%). 1H NMR (400 MHz, CDCl3): δ 8.83 (s, 6H), 6.53 (d, J = 8.0 Hz, 12H), 6.22 (d, J = 8.0 Hz, 12H). 13C NMR (125 MHz, CD3OD): δ 155.4, 141.8, 134.3, 133.7, 114.5. IR (KBr, cm–1): 3531, 3340, 3033, 1612, 1518, 1436, 1401, 1261, 1236, 1173, 1101, 1015, 826, 548. Anal Calcd for C42H30O6·0.5H2O: C, 78.86; H, 4.88. Found: C, 78.79; H, 4.72. Mp > 400˚C.
2.2.6. Synthesis of HPB-OH(2)
HPB-OH(2) was synthesized using a procedure similar to that used for HPB-OH(1) the analogous method.
Data of HPB-OH(2): Yield = 91%. 1H NMR (400 MHz, DMSO-d6): δ 7.55 (s, 6H), 6.42 (s, 12H), 1.83 (s, 36H). 13C NMR (100 MHz, DMSO-d6): δ 150.0, 140.4, 132.8, 131.7, 122.1, 16.9. IR (KBr, cm–1): 3576, 3036, 2967, 2917, 2861, 1604, 1490, 1420, 1396, 1310, 1190, 1127, 1022, 943, 868, 730. Anal Calcd for C54H54O6·0.2H2O: C, 80.81; H, 6.83. Found: C, 80.63; H, 6.97. Mp > 400˚C.
2.2.7. Synthesis of IF-OMe
After a dichloromethane solution (300 ml) of HPBOMe(1) (82 mg, 0.12 mmol) (N2 bubbled before use) was added to dry nitromethane solution (7 ml; N2 bubbled before use) of FeCl3 (0.37 g, 2.3 mmol) at 0˚C, the reaction solution was stirred at 20˚C for 1.5 h. The reaction solution was added to water (50 ml), extracted with dichloromethane, and dried over magnesium sulfate. After the solvent was removed under vacuum, the resulting solid was washed with methanol, collected filtration, and dried under vacuum to give IF-OMe as a light brown solid (57 mg, 72%). 1H NMR (400 MHz, CDCl3): δ 7.18 (d, J = 8.4 Hz, 4H), 6.90 (d, J = 8.0 Hz, 4H), 6.51 - 6.57 (m, 8H), 6.23 (d, J = 8.8 Hz, 2H), 6.14 (d, J = 9.6 Hz, 4H), 3.88 (s, 6H), 3.68 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 185.9, 159.9, 159.5, 149.7, 144.2, 142.3, 138.9, 134.7, 134.1, 130.8, 129.0, 128.3, 124.4, 114.9, 113.6, 109.2, 56.6, 55.5, 55.4. IR (KBr, cm–1): 2936, 2836, 1661, 1604, 1521, 1496, 1457, 1436, 1247, 1174, 1024, 861, 833. Anal Calcd for C46H34O6·H2O: C, 78.84; H, 5.18. Found: C, 78.55; H, 5.07. Mp = 399˚C - 403˚C.
2.2.8. Synthesis of IF-OH
After a dichloromethane solution (2 ml) of BBr3 (0.21 ml, 2.2 mmol) was added to a dichloromethane solution (13 ml) of IF-OMe (0.057 g, 0.083 mmol), the reaction solution was stirred at 20˚C for 30 h and quenched with water. The resulting precipitate was collected by filtration and dried under vacuum to give IF-OH as a green solid (0.030 g, 58%). 1H NMR (400 MHz, DMSO-d6): δ 9.61 (s, 2H), 9.56 (s, 2H), 7.05 (d, J = 8.0 Hz, 4H), 6.66 (d, J = 8.0 Hz, 4H), 6.45 (d, J = 8.4 Hz, 2H), 6.35 (s, 2H), 6.08 (d, J = 8.4 Hz, 2H), 6.00 (d, J = 9.6 Hz, 4H). 13C NMR (125 MHz, DMSO-d6): δ 184.2, 157.0, 156.9, 149.7, 143.8, 141.9, 137.9, 133.9, 130.3, 127.8, 127.7, 126.5, 123.4, 115.2, 114.3, 110.3, 55.8.IR (KBr, cm–1): 3301, 1652, 1608, 1524, 1434, 1364, 1236, 1173, 1106, 868, 839. Anal Calcd for C42H26O6·H2O: C, 78.25; H, 4.38. Found: C, 78.11; H, 4.65. Mp > 400˚C.
3. Results and Discussion
3.1. Synthesis
Indeno[1,2-b]fluorene derivatives were synthesized by the methods shown in Scheme 1. The 1:2 Stille coupling reacon of bis(tributylstannyl)acetylene with 4-bromoanisole and 4-bromo-2,5-dimethylanisole yielded bis(4-methoxyenyl)acetylene (1) and bis(3,5-dimethyl-4-ethoxyphenyl) cetylene (2), respectively. The Co2(CO)8-catalyzed cyclotrimerization of 1 and 2 yielded HPB-OMe(1) and HPBOMe(2) in 79% and 65% yields, respectively. The treatment of HPB-OMe(1) with FeCl3 caused oxidative cyclodehydrogenation to yield IF-OMe. It has been rerted that the treatment of HPBs with FeCl3 can cause cyclodehydrogenation to yield compounds with an inno[1,2-b]fluorene structure [27]. The deprotection of the OMe groups of HPB-OMe(1), HPB-OMe(2), and IF-Me with BBr3 resulted in the production of HPB-OH(1), HPB-H(2), and IF-OH, respectively.
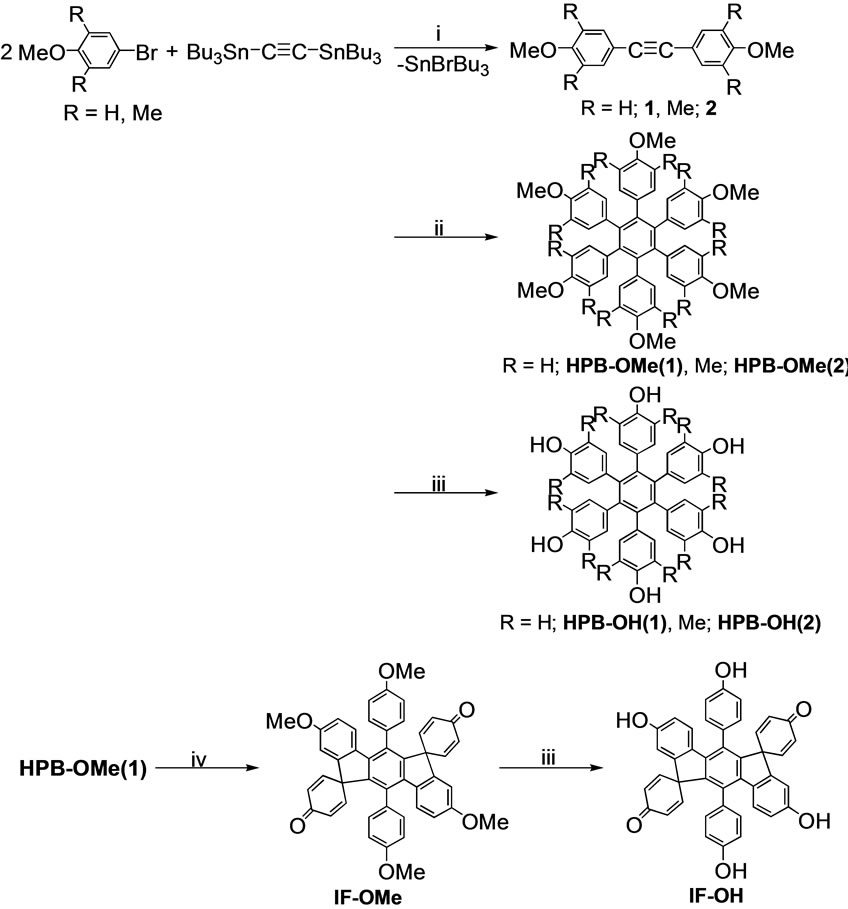
Scheme 1. Synthesis of HPBs and oligophenylenes with an indeno[1,2-b]fluorene structure. i) Pd(PPh3)4, reflux, toluene, 48 h; ii) Co2(CO)8, toluene, reflux, 24 h; iii) BBr3, CH2Cl2, 20 ˚C, 30 h; iv) FeCl3, CH3NO2, CH2Cl2, 1.5 h.
The structures of the newly synthesized compounds were determined by 1H and 13C NMR spectroscopy and elemental analysis. The solubilities of the obtained compounds are summarized in Table 1. HPB-OMe(1) and HPB-OMe(2) were soluble in polar organic solvents such as 1,4-dioxane, tetrahydrofuran (THF), N,N-imethylformamide (DMF) and dimethyl sulfoxide (DMSO) as well as in less polar organic solvents such as chloroform and dichloromethane. However, HPB-OH(1) and HPB-OH(2) were soluble in DMF and DMSO but insoluble in dichloromethane and chloroform because of the presence of hydrophilic OH groups. IF-OMe was soluble in dichloromethane and 1,4-dioxane but insoluble in polar organic solvents such as DMF and DMSO, whereas IF-OH was soluble in the polar organic solvents because of the presence of the OH groups but insoluble in less polar organic solvents.
3.2. IR and NMR Spectra
Figure 1 shows the IR spectra of HPB-OMe(1), HPBH(1), HPB-OH(2), and IF-OH The main features of the IR spectra of HPB-OMe(1) and HPB-OMe(2) were identical, with absorption bands resulting from C-O stretching, the presence of a phenyl ring, and out-oflane C-H bending vibrations of p-phenylene observed at approximately 1244 cm–1, 1517 cm–1, and 804 cm–1, respectively. Similarly, the main features of the IR spectra of HPB-OH(1) and HPB-OH(2) were identical, except for the absorption resulting from the O-H stretching, with the absorption bands resulting from C-O stretching, the presence of a phenyl ring, and out-of-plane C-H bending vibrations of p-phenylene observed at approximately 1261 cm–1, 1518 cm–1, and 826 cm–1, respectively. The IR spectrum of HPB-OH(1) exhibited a strong sharp absorption due to the hydrogen bonding free OH group at 3576 cm–1 and a broad absorption due to the hydrogen bonding OH group at around 3340 cm–1. In contrast, while the IR spectrum of HPB-OH(2) exhibited a strong sharp absorption due to the hydrogen bonding free OH group at 3576 cm–1, no absorption was observed for the hydrogen bonding OH group. The inhibition of intermolecular hydrogen bonding in HPB-OH(2) is attributed to
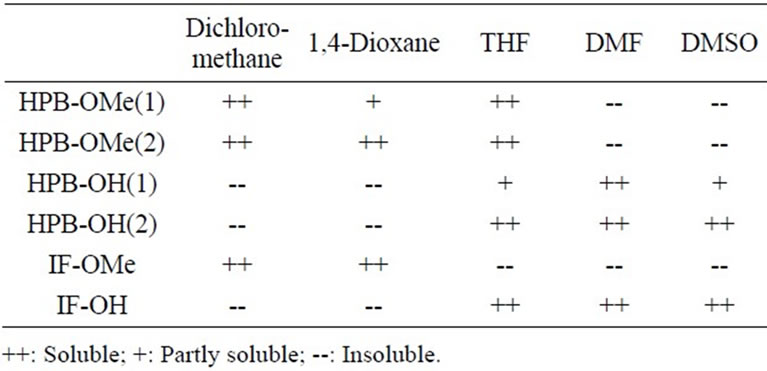
Table 1. Solubility of the obtained compounds.
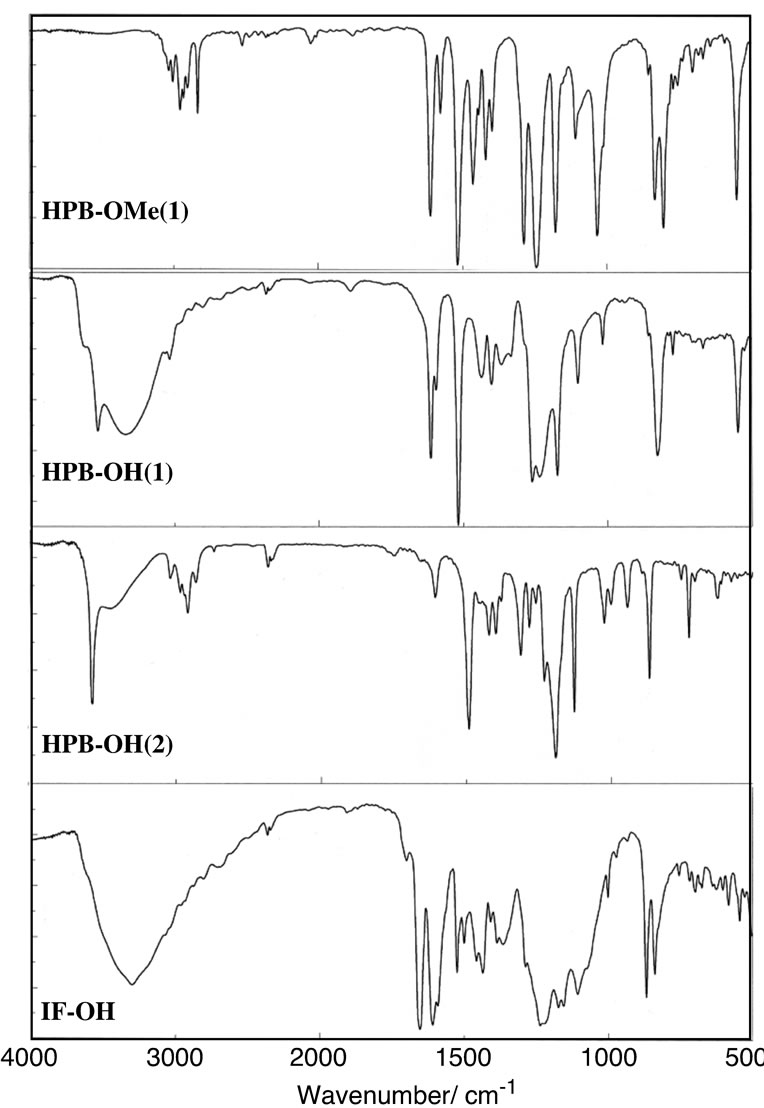
Figure 1. IR spectra of HPB-OMe(1), HPB-OH(1), HPBOH(2), and IF-OH.
the presence of the methyl groups at the 2- and 6-positions in HPB-OH(2). The IR spectrum of IF-OMe exhibited absorption bands corresponding to C=O and C-O stretching vibrations and out-of-plane C-H bending vibrations of p-phenylene, observed at 1661 cm–1, 1246 cm–1, and 833 cm–1, respectively. IF-OH exhibited absorption peaks corresponding to hydrogen bonding O-H stretching vibrations in the range 2500 - 3300 cm–1, and peaks corresponding to C=O and C-O stretching vibrations and out-of-plane C-H bending vibrations of pphenylene at 1652 cm–1, 1236 cm–1, and 868 cm–1, respectively. The fact that the band corresponding to the C=O stretching vibrations of IF-OH was observed at a shorter wavenumber than that for IF-OMe is attributed to the intermolecular hydrogen bonding via the OH group.
The 1H NMR spectra of HPB-OMe(1) and HPBMe(2) exhibited a peak corresponding to the methoxy H-atoms at δ 3.63 and 3.50, respectively, and that of IF-OMe(1) exhibited two peaks corresponding to the methoxy H-atoms at δ 3.68 and 3.88. These peaks disappeared in the 1H NMR spectra of the corresponding deprotected species, thus suggesting that the deprotection reaction proceeded to completion. The peaks corresponding to the phenyl H-atoms of HPB-OMe(1) and HPB-OMe(2) were observed at higher magnetic field positions than those of 1 and 2. These observations may be due to the presence of the ring current effect in HPB-OMe(1) and HPB-OMe(2). The peaks corresponding to the OH groups of HPB-OH(1) and HPB-OH(2) were observed at δ 8.83 and 7.56, respectively, whereas those of IF-OH were observed at δ 9.56 and 9.61. The fact that the peak corresponding to the OH groups of HPB-OH(2) was observed at a higher magnetic field position than that of HPB-OH(1) is because of the presence of the electron-donating methyl groups adjacent to the OH groups in HPB-OH(2).
Deprotonation of the OH groups of HPB-OH(1), HPBOH(2), and IF-OH was carried out through treatment with an excess amount of NaH in DMSO-d6. The result is that the signal corresponding to the OH group disappeared from the 1H NMR spectra for solutions of HBCOH, HPB-OH(1), HPB-OH(2), and IF-OH(1) in the presence of NaH, indicating that the deprotonation proceeded quantitatively.
3.3. UV-Vis Absorption and Solvatochromism
Figure 2 shows the UV-Vis spectra of HPB-OMe(1), HPB-OMe(2), HPB-OH(1), HPB-OH(2), IF-OMe, IFOH, and their deprotonated species in organic solvents. The optical data of these compounds are summarized in Tables 2-4. The THF solutions of HPB-OH(1) and HPB-OH(2) each exhibited three absorption peaks. It has been reported that m-terphenyl can be regarded as a combination of two molecules of biphenyl of the C1 group [28]. The observation of three absorption peaks in HPB-OH(1) probably derives from the assumption that it can be regarded as a combination of three molecules of dihydroxy-o-, m-, and p-terphenyl. o-, m-, and p-terphenyls exhibit an absorption peak at 260 nm, 240 nm, and 290 nm, respectively. These wavelengths are consistent with those observed for the THF solution of HPB-OH(1). The absorption peaks of HPB-OH(2) are observed at shorter wavelengths than those of HPBOH(1). This observation is attributed to the larger bond twisting between the 2,6-dimethyl-4-methoxyphenyl ring and the central benzene ring in HPB-OH(1) compared with that between the 4-methoxyphenyl ring and the central benzene ring in HPB-OH(2). As shown in Figure 2iii, the UV-Vis spectra of IF-OMe and IF-OH can be divided into two clearly distinguishable parts. These two regions appear to be separate states of electronic transition, shown in Figure 3. The absorption peak at 238 nm is probably a result of the electronic transition directed along the a and b axes, while those in the range of 310 - 344 nm are likely due to the electronic transition directed along the c, d, and e axes, respectively, as shown in Figure 4. The models of the electronic transition axes were proposed in the case of branched oligophenylenes. In CH2Cl2 and 1,4-dioxane, the λmax values of IF-OMe were longer than those of HPB-OMe(1) because of the presence of the planar indeno[1,2-b]fluorene structure in IF-OMe.
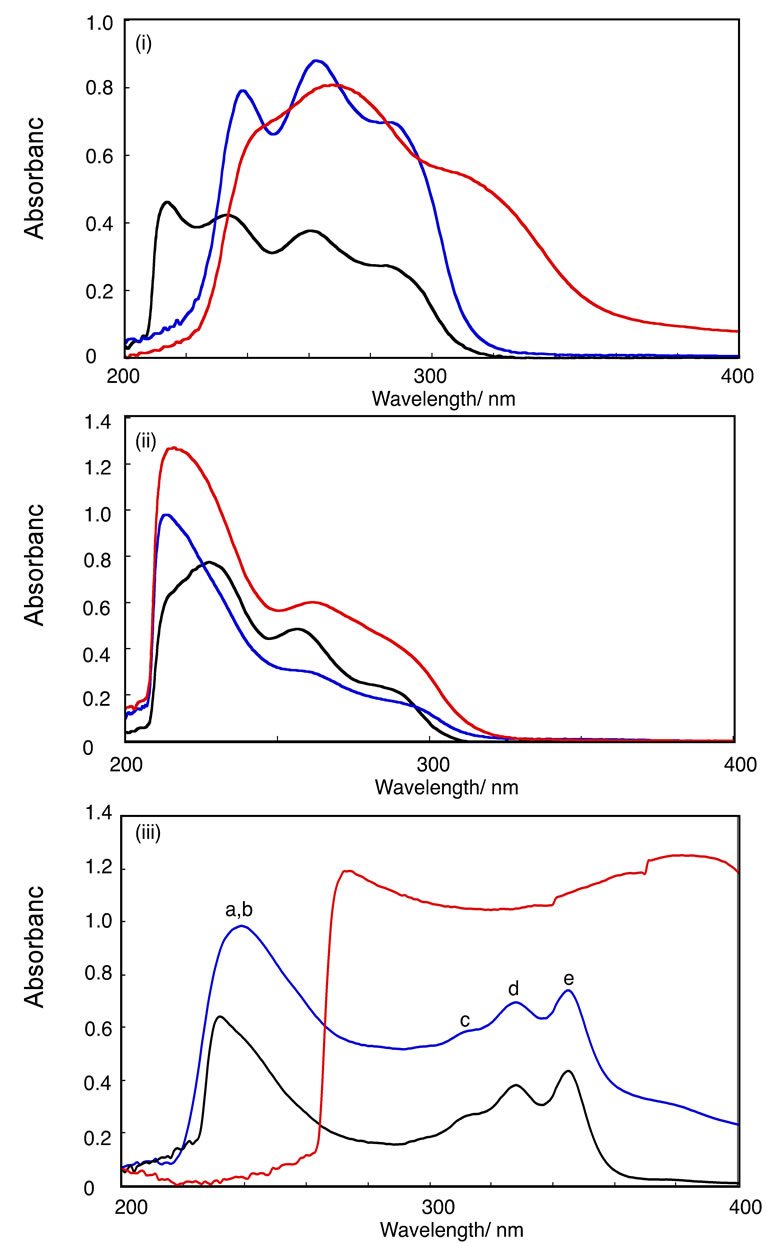
Figure 2. UV-Vis spectra of HPB-OMe(1) (i; black curve), HPB-OMe(2) (ii; black curve), HPB-OH(1) (i; blue curve), HPB-OH(2) (ii; blue curve), and their deprotonated species (i, ii, iii; red curves) in THF, IF-OMe in dichloromethane (iii; black curve), and IF-OH (iii; blue curve) and IF-ONa (iii; red curve) in DMSO.
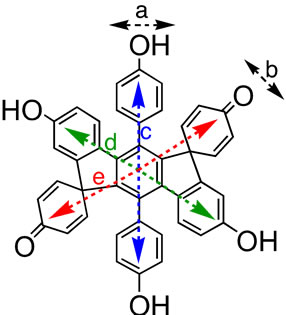
Figure 3. Electronic transition directions in IF-OH.
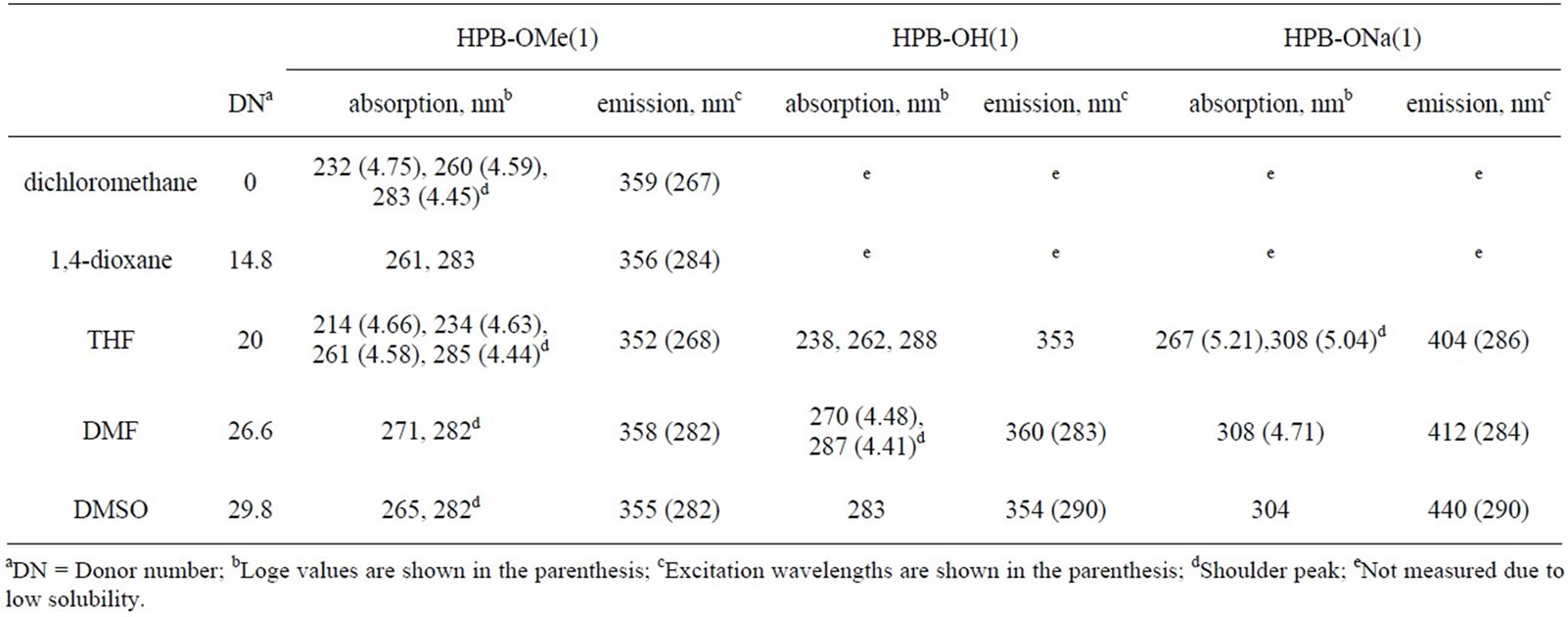
Table 2. Optical data of HPB-OMe(1), HPB-OH(1), and HPB-ONa(1).
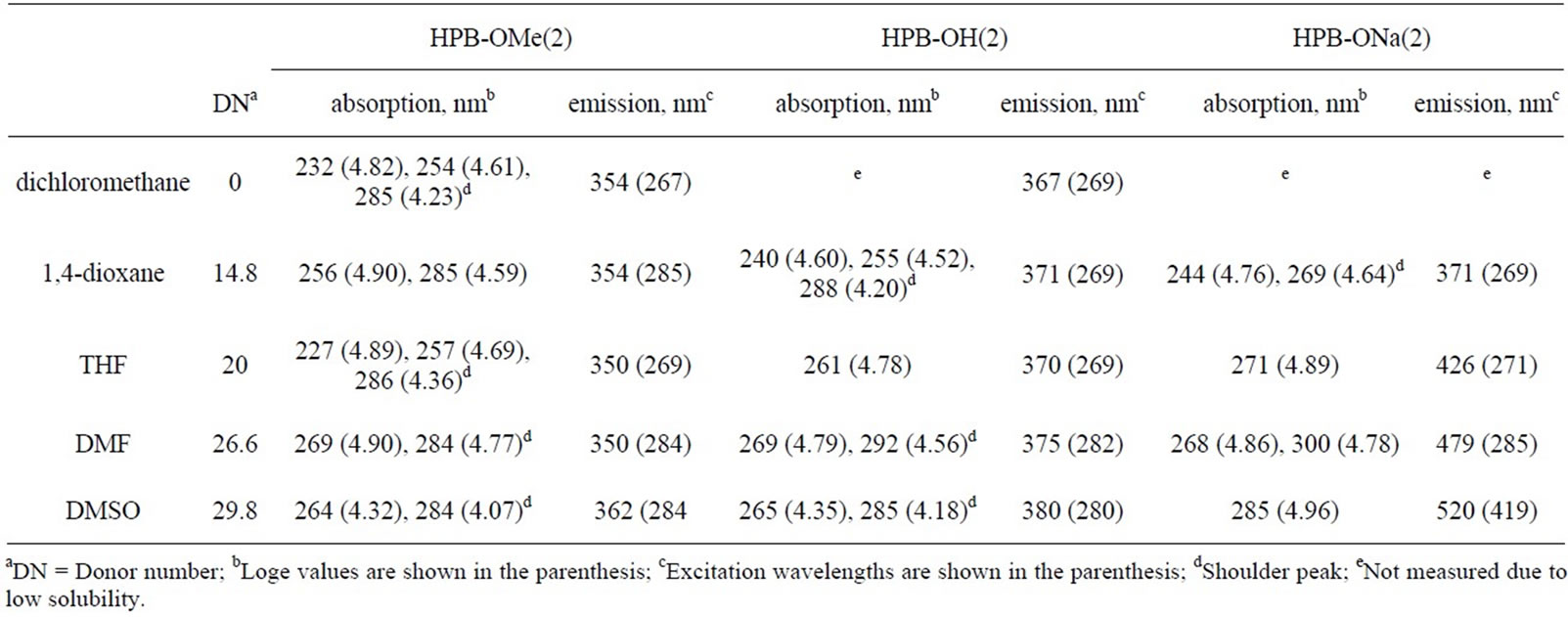
Table 3. Optical data of HPB-OMe(2), HPB-OH(2), and HPB-ONa(2).
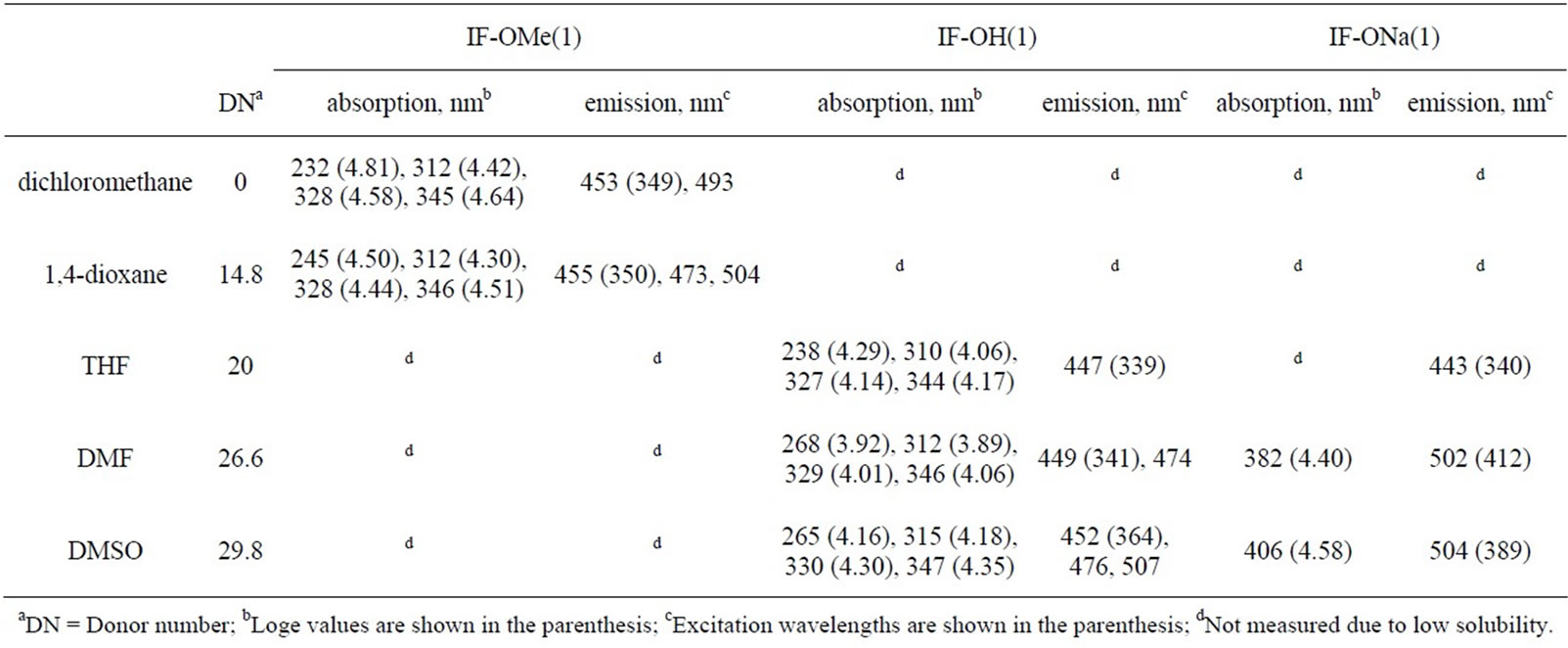
Table 4. Optical data of IF-OMe, IF-OH, and IF-ONa.
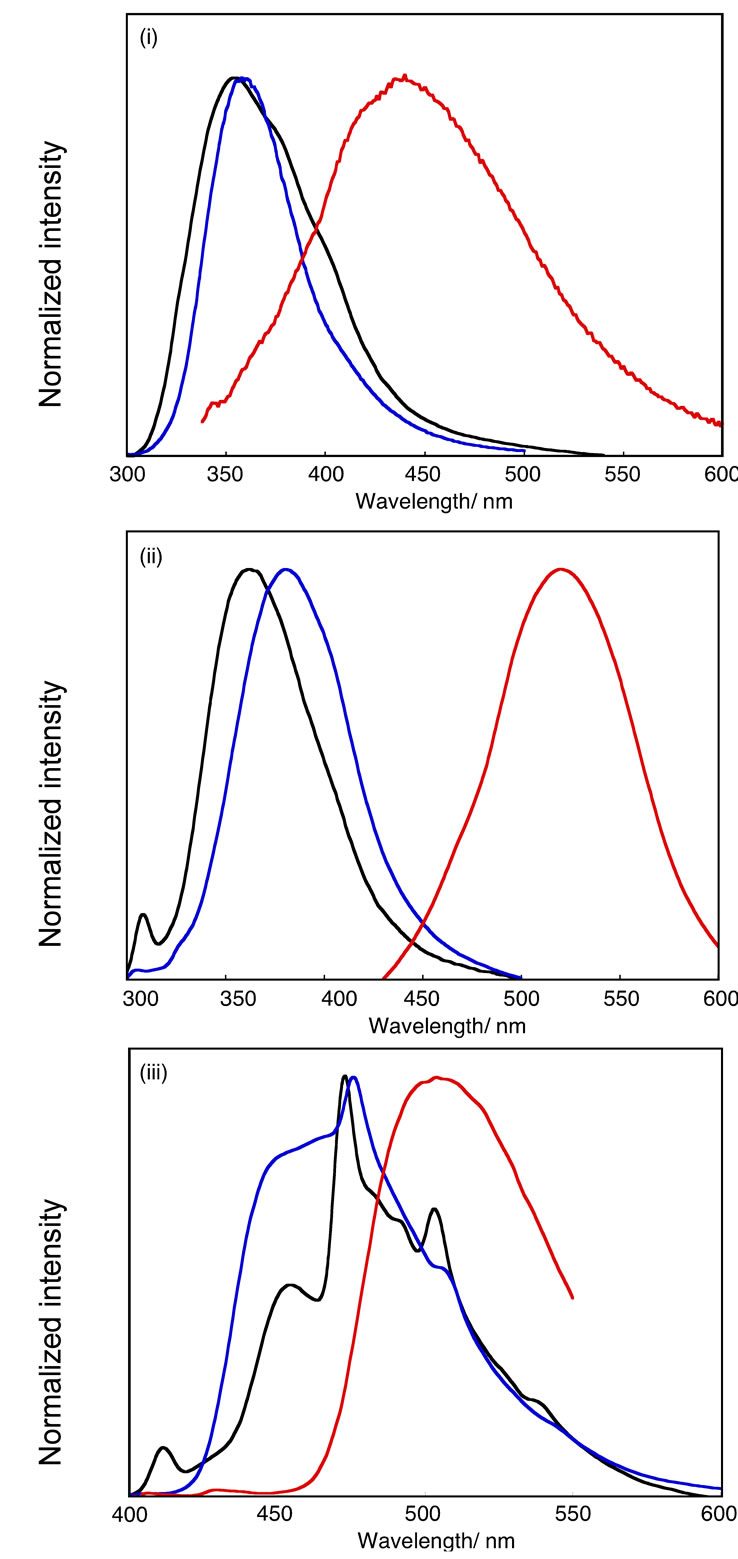
Figure 4. PL spectra of HPB-OMe(1) (i; black curve), HPB-OMe(2) (ii; black curve), HPB-OH(1) (i; blue curve), HPB-OH(2) (ii; blue curve), IF-OH (iii; blue curve), and their deprotonated species (i, ii, iii; red curve) in DMSO and IF-OMe (iii; black curve) in 1,4-dioxane.
The treatment of the DMSO solutions of HBC-OH, HPB-OH(1), HPB-OH(2), and IF-OH(1) with NaH causes a bathochromic shift in absorption bands by approximately 20-60 nm. The formation of HBC-ONa, HPB-ONa(1), HPB-ONa(2), and IF-ONa(1) was mainly responsible for the shift of λmax towards a longer wavelength. To prove that these observations were due to the deprotonation of the OH groups after treatment with NaH, we confirmed that there was no change in the absorption spectra of HBC-OMe, HPB-OMe(1), HPB-OMe(2), and IF-OMe(1) upon the addition of NaH. The absorption peaks at 238 nm, 262 nm, and 288 nm in the UV-Vis spectrum of HPB-OH(1) correspond to the π-π* transition of the m-, o-, and p-terphenyl moieties in this compound, respectively. These peaks shifted to longer wavelengths after treatment with NaH, by 5 nm, 5 nm, and 20 nm, respectively. The degree of bathochromic shift in the peak that corresponds to the electric transition along the p-terphenyl moiety was the largest of the three described. This result suggests that ICT from the ONa group to the inner benzene rings was preferred through the p-terphenyl moiety. No significant bathochromic shift was observed in HPB-ONa(2). This is because of the bond twisting between the 2,6-dimethyl -4-methoxyphenyl and central benzene rings. The UV-Vis spectrum of IF-ONa was somewhat ambiguous because it was partly soluble in organic solvents. However, bathochromic shifts were observed in the case of IF-ONa.
The bathochromic shift attributable to deprotonation is dependent on the DN of the solvent used. As summarized in Tables 2-4, absorptions for HPB-OH(1), HPB-OH(2), and their deprotonated species shifted to longer wavelengths as the DN of the solvent was increased. In contrast to the small bathochromic shift for HPB-OH(1) and HPB-OH(2), with an increase in the DN of the solvent, the λmax values of HPB-ONa(1) and HPB-ONa(2) were larger than those of HPB-OH(1) and HPB-OH(2). For example, the λmax value of HPB-ONa(2) varies from 244 nm in 1,4-dioxane (DN = 14.8) to 285 nm in DMSO (DN = 29.8), through to a value of 271 nm in THF (DN = 20.0). Similarly, that of HPB-ONa(1) varies from 267 nm in THF (DN = 20.0) to 304 nm in DMSO (DN = 29.8). The large Δλ value can be attributed to the fact that solvents with a high DN effectively solvate with Na+ to stabilize the deprotonated species in the solutions. Similar solvatochromic behavior was observed in the cases of OPP(n)-OH (n = 4 and 5) and OPP(n)-ONa (n = 4 and 5), as reported earlier [29]. The fact that the Δλ values for HPB-ONa(2) were smaller than those for HPB-ONa(1) corresponds to the reduced length of π-conjugation in HPB-ONa(2) that arises from the bond twisting between 2,6-dimethyl-4-methoxyphenyl and the central benzene rings.
3.4. Photoluminescence
The compounds obtained in this study and their deprotonated species are photoluminescent in solution. The photoluminescence (PL) data are summarized in Tables 2-4. Figure 4 shows the PL spectra of HPB-OMe(1), HPB-OMe(2), HPB-OH(1), HPB-OH(2), IF-OMe, IF-OH, and their deprotonated species in organic solvents. The PL peak positions for HPB-OH(1), HPB-OH(2), and
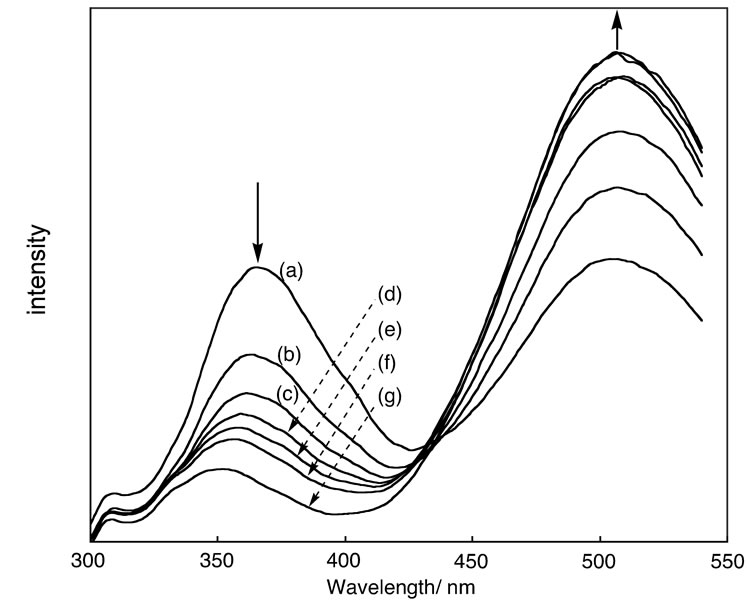
Figure 5. Changes of PL spectra of methanol solution of HPB(1)-OH (5 × 10−5 M) in the presence of various amounts of NaOH. (a): 0 M; (b): 3 × 10−2 M; (c): 4 × 10−2 M; (d): 5 × 10−2 M; (e): 6 × 10−2 M; (f): 7 × 10−2 M; (g): 8 × 10−2 M.
IF-OH shifted to longer wavelengths after deprotonation with NaH. This shift is comparable to the bathochromic shift observed in the UV-Vis spectra of these compounds. The emission peak positions for HPB-OH(1), HPBOH(2), IF-OH, and their deprotonated species depended on the DN of the solvent. As summarized in Tables 2-4, by varying solvents such as CH2Cl2 and 1,4-dioxane, which have small DN values, with those such as DMF and DMSO, which have large DN values, it is observed that the emission peak positions for HPB-OH(1) and HPB-OH(2) shift by only 3 - 13 nm. However, a significantly large shift in the emission peaks for HPB-ONa(1), HPB-ONa(2), and IF-ONa(1) occurred as the DN of the solvent was increased. These observations are consistent with the result that, with an increase in the DN of the solvent, λmax of HPB-ONa(1), HPB-ONa(2), and IFONa(1) in solution shifts to a longer wavelength than that of HPB-OH(1), HPB-OH(2), and IF-OH(1). The remarkable solvatochromic shift of the PL peak position of HPB-ONa(1) and HPB-ONa(2) may be due to the shift in charge from the phenolate group to the adjacent rings. In addition to the effect of charge shift, a large amount of stabilization energy produced by the solvation of HPBONa(1) and HPB-ONa(2) may contribute to the solvatochromic red shift as the DN of the solvent is increased. There was no change in the PL spectra of HPB-OMe(1), HPB-OMe(2), and IF-OMe(1) upon the addition of NaH, which suggests that the solvatochromism in HPBONa(1), HPB-ONa(2), and IF-ONa(1) can be attributed to the deprotonation of the OH group after treatment with NaH.
Figure 5 shows the PL spectra of the methanol solutions of HPB(1)-OH containing different amounts of NaOH. It is observed that the peak at 371 nm decreases and a new emission peak at 475 nm appears as the concentration of the base is increased. This result confirms the fact that the emission peaks at 371 nm and 475 nm originate from HPB(1)-OH and HPB(1)-ONa, respectively.
The quantum yields of the PLs of the DMF solutions of HPB-ONa(1) and HPB-ONa(2) were 2%, 7%, respectively, while those of HPB-OH(1) and HPB-OH(2) were 24% and 8%, respectively. The fact that the quantum yields of the PLs of HPB-ONa(1) and HPB-ONa(2) are lower than those of HPB-OH(1) and HPB-OH(2) is attributed to the intramolecular charge shift (ICT) in HPB-ONa(1) and HPB-ONa(2). It has been reported that the ICT in π-conjugated molecules reduces their PL emission efficiencies [8].
4. Conslusion
HPBs with hydroxyl groups (HPB-OH species) and their derivatives with an indeno[1,2-b]fluorene structure were obtained by using reactions with transition metal complexes. The treatment of these compounds with a base produced corresponding deprotonated species, whose absorption and PL peak positions in solution shifted towards longer wavelengths with an increase in the DN of the solvent. The optical properties of the HPB-OH compounds were significantly affected by bond twisting between the hydroxyphenyl group and the central benzene ring. The introduction of a planar indeno[1,2-b]fluorene structure to the HPB-OH enhanced its solvatochromic behavior. The results obtained in this study will be useful in providing information for the development of new solvatochromic materials.
5. Acknowledgements
This work was performed under the Cooperative Research Program of “Network Joint Research Center for Materials and Devices” (No. 2011193).
REFERENCES
- M. Era, T. Tsutsui and S. Saito, “Polarized Electroluminescence from Oriented p-Wexiphenyl Vacuum—deposited Film,” Applied Physics Letters, Vol. 67, No. 17, 1995, pp. 2436-2438. doi:10.1063/1.114599
- J. M. Kauffman, P. T. Litak, J. A. Novinski, C. J. Kelly, A. Ghiorghis and Y. Qin, “Electronic Absorption and Emission Spectral Data and Fluorescence Quantum Yields of Bridged p-Oligophenylenes, Bito Deciphenyls, and Related Furans and Carbazoles,” Journal Of Fluorescence, Vol. 5, No. 3, 1995, pp. 295-305. doi:10.1007/BF00723901
- J. M. Tour, “Conjugated Macromolecules of Precise Length and Cssonstitution. Organic Synthesis for the Construction of Nanoarchitectures,” Chemical Reviews, Vol. 96, No. 1, 1996, pp. 537-554. doi:10.1021/cr9500287
- F. Meghdadi, G. Leising, W. Fisher and F. Stelzer, “Multicolour Electroluminescence Diodes Using Oligophenylene and Oligophenylenevinylene Multilayers,” Synthetic Metals, Vol. 76, No. 1-3, 1996, p. 113. doi:10.1016/0379-6779(95)03430-R
- D. J. Grundlach, Y. Y. Lin, T. N. Jackson and D. G. Schlom, “Oligophenyl-Based Organic Thin Film Transistors,” Applied Physics Letters, Vol. 71, No. 26, 1997, pp. 3853-3855. doi:10.1063/1.120524
- Y. Z. Wang, R. G. Sun, F. Meghdadi, G. Leising and A. J. Epstein, “Multicolor Multilayer Light-Emitting Devices Based on Pyridine-containing Conjugated Polymers and Para-Sexiphenyl Oligomer,” Applied Physics Letters, Vol. 74, No. 24, 1999, pp. 3613-3615. doi:10.1063/1.123198
- R. E. Martin and F. Diederich, “Linear Monodisperse π-Conjugated Oligomers: Model Compounds for Polymers and More,” Angewandte Chemie International Edition, Vol. 38, No. 10, 1999, pp. 1350-1377. doi:10.1002/(SICI)1521-3773(19990517)38:10
- V. Sidorov, T. Douglas, S. M. Dzekunov, D. Abdallah, B. Ghebremariam, P. D. Roepe and S. Matile, “Self-Assembled Single-Chain Oligo(p-Phenylene) Amphiphiles: Reversed Micelles, Vesicles and Gels,” Chemical Communications, No. 15, 1999, pp. 1429-1430. doi:10.1039/A903041G
- B. Baumeister and S. Matile, “Programmed Assembly of Expanded Rigid-Rod β-Barrels by Supramolecular Preorganization,” Chemical Communications, No. 11, 2000, pp. 913-914. doi:10.1039/B002113J
- B. Baumeister, N. Sakai, S. Matile, “Giant Artificial Ion Channels Formed by Self-Assembled, Cationic RigidRod β-Barrels,” Angewandte Chemie International Edi- - tion, Vo. 39, No. 11, 2000, pp. 1955-1958. doi:10.1002/1521-3773(20000602)39:11
- R. L. Carroll and C. B. Gorman, “The Genesis of Molecular Electronics,” Angewandte Chemie International Edition, Vol. 41, No. 23, 2002, 4378-4400. doi:10.1002/1521-3773(20021202)41:23
- O. Deeg, P. Kirsch, D. Pauluth and P. Bäuerle, “Combinatorial Parallel Synthesis and Automated Screening of a Novel Class of Liquid Crystalline Materials,” Chemical Communications, No. 23, 2002, pp. 2762-2763. doi:10.1039/B207630F
- Z. N. Yu, H. L. Tu, X. H. Wan, Z. F. Chen and Q. F. Zhou, “Synthesis and Properties of Liquid Crystalline 4,4''-Dialkoxy-2''-Methyl-P-Terphenyls,” Molecular Crystals and Liquid Crystals, Vol. 391, No. 1, 2002, pp. 41-55. doi:10.1080/15421400390193567
- Z.-H. Li, M.-S. Wong, Y. Tao and M. D’Iorio, “Synthesis and Functional Properties of Strongly Luminescent Diphenylamino End-Capped Oligophenylenes,” Journal of Organic Chemistry, Vol. 69, No. 3, 2004, pp. 921-927. doi:10.1021/jo035147y
- K.-H. Ahn, G. Y. Ryu, S.-W. Youn and D.-M. Shin, “The Conjugation Effects on the Luminescence Properties of Oligophenylenes for the OLED,” Materials Science and Engineering: C, Vol. 24, No. 1-2, 2004, pp. 163-165. doi: 10.1016/j.msec.2003.09.010
- A. P. H. J. Schenning and E. W. Meijer, “Supramolecular Electronics; Nanowires from Self-Assembled π-Conjugated Systems,” Chemical Communications, No. 26, 2005, pp. 3245-3258. doi: 10.1039/B501804H
- H. Yin, G.-I. Lee, K. A. Sedey, O. Kutzki, H. S. Park, B. P. Orner, J. T. Ernst, H.-G. Wang, S. M. Sebti and A. D. Hamilton, “Terphenyl-Based Bak BH3 α-Helical Proteomimetics as Low-Molecular-Weight Antagonists of Bclx,” Journal of American Chemical Society, Vol. 127, No. 29, 2005, pp. 10191-10196. doi:10.1021/ja050122x
- N. H. Sultana, S. M. Kelly, B. Mansoor and N. O’Neill, “Polycatenar Oligophenylene Liquid Crystals,” Liquid Crystals, Vol. 34, No. 11, 2007, pp. 1307-1316. doi:10.1080/02678290701682357
- I. Yamaguchi, K. Goto and M. Sato, “Synthesis of Oligophenylenes Containing Hydroxyl Group and Their Solvatochromic Behavior,” Tetrahedron, Vol. 65, No. 18, 2009, pp. 3645-3654. doi:10.1016/j.tet.2009.02.073
- I. Yamaguchi, K. Seo and Y. Kawashima, “Synthesis of Dihydroxyoligophenylenes Containing π-Deficient or π- Excess Hetero-Aromatic Rings and Their Solvatochromic Behavior,” Tetrahedron, Vol. 34, No. 7, 2010, pp. 6725- 6732. doi:10.1016/j.tet.2010.06.087
- A. J. Berresheim, M. Müller and K. Müllen, “Polyphenylene Nanostructures,” Chemical Reviews, Vol. 99, No. 7, 1999, pp. 17471786. doi:10.1021/cr970073+
- V. Garg, “Design and Synthesis of Hexaphenylbenzene Based Artificial Photosynthetic Antenna-Reaction Centers,” Umi Dissertation Publishing, Proquest, 2011.
- K. E. Maly, E. Gagnon, T. Maris and J. D. Wuest, “Engineering Hydrogen-Bonded Molecular Crystals Built from Derivatives of Hexaphenylbenzene and Related Compounds,” Journal of American Chemical Society, Vol. 129, No. 14, 2007, pp. 4306-4322. doi:10.1021/ja067571x
- K. Kobayashi, T. Shirasaka, A. Sato, E. Horn and N. Furukawa, “Self-Assembly of a Radially Functionalized Hexagonal Molecule: Hexakis(4-hydroxyl-phenyl)benzene,” Angewandte Chemie International Edition, Vol. 38, No. 23, 1999, pp. 3483-3486. doi:10.1002/(SICI)1521-3773(19991203)38:23
- Y. Xia, Z. He, J. Tong, B. Li, C. Wang, Y. Cao, H. Wu, H. Y. Woo and D. Fan, “Synthesis and Photovoltaic Properties of Alternating Conjugated Polymers Derived from Indeno[1,2-b]fluorene and Bithiophene or Thieno[3,2- b]thiophene-Cored Benzothiadiazole,” Macromolecular Chemistry and Physics, Vol. 212, No. 11, 2011, pp. 1193- 1201. doi:10.1002/macp.201000759
- E. Jeong, S. H. Kim, I. H. Jung, Y. Xia, K. Lee, H. Suh, H.-K. Shim and H. Y. Woo, “Synthesis and Characterization of Indeno[1,2-b]fluorene-based White Light-Emitting Copolymer,” Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 47, No. 14, 2009, pp. 3467-3479. doi:10.1002/pola.23422
- S. H. Wadumethrige and R. Rathore, “A Facile Synthesis of Elusive Alkoxy-Substituted Hexa-peri-hexabenzocoronene,” Organic Letters, Vol. 10, No. 22, 2008, pp. 5139-5142. doi: 10.1021/ol8020429
- N. I. Nijegorodov, W. S. Downey and M. B. Danailov, “Systematic Investigation of Absorption, Fluorescence and Laser Properties of Some pand m-Oligophenylenes,” Spectrochimica Acta A, Vol. 56, No. 4, 2000, pp. 783-795. doi:10.1016/S1386-1425(99)00167-5
- X. Zhang, A. S. Shetty and S. A. Jenekhe, “Electroluminescence and Photophysical Properties of Polyquinolines,” Macromolecules, Vol. 32, No. 22, 1999, pp. 7422- 7429. doi:10.1021/ma990960+
NOTES
*Corresponding author.