Surgical Science
Vol.4 No.1(2013), Article ID:27431,7 pages DOI:10.4236/ss.2013.41018
The Role of Vitamin E in Cerebral Hypoxia: An Ultrastructural Study
1Anatomy Department, King Saud University, Riyadh, KSA
2Anatomy Department, Faculty of Medicine, Al-Azhar University, Cairo, Egypt
3Anatomy Department, Salman Bin Abdulaziz University, Al-Kharj, KSA
4Anatomy Department, Faculty of Medicine, Cairo University, Giza, Egypt
5Pathology Department, King Saud University, Riyadh, KSA
Email: *alihassan3750@yahoo.com, *a.ali@sau.edu.sa
Received October 19, 2012; revised November 30, 2012; accepted December 8, 2012
Keywords: Hypoxia; Vitamin E; Ischemia
ABSTRACT
Hypoxia, due to impaired cerebral blood flow, has hazardous effects on brain structure and function. To minimize as much as possible the neurological consequences from hypoxic-ischemic (HI) brain injury, neuro-protective strategies are urgently required. Vitamin E has been shown to have protective effects against cerebral ischemia, possibly due to its anti-oxidant effects. Thirty white mice, of both sexes, were obtained from the animal house at King Khalid University Hospital, King Saud University. They were divided into three groups; each included 10 animals: Group A was considered as a control one, animals of Group B were subjected to a permanent link to the carotid arteries on both sides and animals of Group C underwent permanent link to carotid arteries on both sides and concomitantly were given Vitamin E as an anti-oxidant. Animals of Group C were injected by Vitamin E (equivalent to 15 mg/day), into the peritoneal cavity as a single dose for a week and after the prescribed period the mice were sacrificed under deep anesthesia and their brains were extracted and prepared for an electron microscopic study of brain tissue. Specimens from animals of Group B showed a large number of neurons that had been deteriorated. Mitochondria were the most affected organelles. There were a large number of dark cells which probably resulted from shrunken nerve cells and exhibited opaque nuclei. The number of affected nerve cells was much lower in brain tissues from animals of the Group C which revealed absence of dark cells. The study did not disclose any similar changes in brain tissues of the control group animals. Our results suggested that treatment with Vitamin E after hypoxia-ischemia led to a neuro-protective effect that appeared in reduction of cell death of neurons. Thus, the present study provides an evidence that Vitamin E protects the brain tissue of the consequences of hypoxia caused by ischemia in the tested experimental animals. It could be recommended in the treatment of cerebrovascular stroke and neurodegenerative diseases.
1. Introduction
Integrity of the brain depends on blood supply of continuous oxygen and glucose to meet energy demand in the tissues. Cerebral hypoxia refers to deprivation of oxygen supply to brain tissue. It can cause reversible confusion and fainting.
When the flow of blood to brain tissue is interrupted, injury ensues from lack of oxygenation and then from subsequent re-oxygenation (ischemia-reperfusion) [1].
Within brain, oxygen free radicals impair capillary endothelium that maintain water and electrolyte homeostasis, alter membrane fluidity characteristics and contribute to synaptic damage. Because brain has a high concentration of polyunsaturated fatty acids, it is very susceptible to injury by lipid peroxidation.
Following injury, astrocytes appear to exercise a neuroprotective effect by expressing antioxidant enzymes. Intrinsic defense mechanisms may not be sufficient, however, to meet the acute demands of oxidant stress induced byischemia/reperfusion [2]. In experimental models, pretreatment with alpha-tocopherol has been shown to attenuate lipid peroxidation during reperfusion.
It would be of interest to examine the progression of neuronal injury that occurred after a brief interruption of blood flow to the brain and define the changes associated with cerebral ischemia and reperfusion, to identify capable therapies that would modify the extent of neuronal damage [3].
Such an investigation seems to be highly important in the clinic because if the flow interruption is not reversed during the first hour the cells within the most severely ischemic region will inevitably die [4]. Because there is a narrow therapeutical window after cerebral ischemia to reverse or interfere with the progression of neuronal damage the search for effective agents capable of reducing the progression of neuronal injury occurring after ischemia further supports the research. According to this purpose, the present study was undertaken to determine the effectiveness of several agents, including antioxidants, in alleviating cell injury that followed the withdrawal of metabolic inhibitors, tomimic neuronal reperfusion after situations of hypoxia, hypoglycemia or ischemia.
Hypoxia is a critical factor in brain ischemia more than any other organ of the body. The combination of hypoxia to ischemia may trigger pathological events. The reduction of cerebral blood flow in conjunction with hypoxia may induce spontaneous thrombus formation which reduces blood perfusion further [5].
According to reference [6] it was found that the vascular adhesion induced by hypoxia and re-oxygenation was increased leading to further impairment of the condition compared with that in normal oxygen concentrations. Antioxidants protect lipids from peroxidation by free radicals through giving up their own electrons to the free radicals. When a free radical gains the electron from an antioxidant, it no longer needs to attack the cell and the chain reaction of oxidation is broken.
According to references [7-9] and [10] antioxidants inhibit re-oxygenation injury, and minimize neuronal damage. The human body has an antioxidant defense system there are two lines of antioxidant defense within the cell; the first line is found in the fat-soluble cellular membrane consisting of Vitamin E, beta-carotene and coenzyme Q; of these Vitamin E is considered the most potent chain breaking antioxidant within the membrane of the cell. The second line is found inside the cell; the water soluble antioxidant scavengers include Vitamin C, glutathione peroxidase, superoxide dismutase.
Vitamin E (a-tocopherol) has also shown promise in modifying oxidative stress pathways and improving neurological outcome in many animal studies. In an animal head-injury study, administration of Vitamin E caused a neuro-protective effect by decreasing the rate of lipid peroxidation [11].
There are two families of fat-soluble compounds, the tocopherols and the tocotrienols constitute Vitamin E. Alpha-tocopherol is the most biologically active of these compounds. Natural occurring-tocopherol is found only in the D isomer, while synthetic tocopherol is a racemic mixture of the D and L isomers, with approximately 75% of the biologic activity of the pure D-tocopherol. One milligram of the racemic form is the equivalent of one IU of Vitamin E activity. The primary dietary source of Vitamin E is vegetable oil, specially soybean, corn, safflower and cottonseed oil. It is also found in wheat germ, nuts and green leafy vegetables.
Vitamin E is found naturally in some foods, added to others, and available as a dietary supplement. “Vitamin E” is the collective name for a group of fat-soluble compounds with distinctive antioxidant activities, naturally occurring Vitamin E exists in eight chemical forms (alpha-, beta-, gamma-, and delta-tocopherol and alpha-, beta-, gamma-, and delta-tocotrienol) [12].
According to Leonarduzzi et al., (2010) [13] alphatocopherol is the most potent antioxidant that acts upon cell membranes and has the ability to neutralize compounds which may potentially disrupt membrane stability. Alpha-tocopherol is the only form that is recognized to meet human requirements.
Reference [14] discusses that as Vitamin E is the major lipophilic antioxidant in the brain and this implies its efficacy as an antioxidant against oxidative stress underlies the molecular pathogenesis of neural disorders. This signifies the importance of Vitamin E in neuro-inflammatory conditions.
The present study aims to evaluate the role of Vitamin E as an antioxidant in protection of brain tissue against experimentally induced ischemia.
2. Materials and Methods
Thirty Wistar albino rats of either sex, procured from the colony at the King Khalid University Hospital, King Saud University, were caged and kept on a 12-hour light/dark schedule. They were fed with a standard laboratory diet and tap water and housed in cages (seven rats per cage). All experiments were carried out according to recommendation of King Saud University of Experimental Animals Ethics Committee which is matched with international ethics for handling of experimental animals.
2.1. Experimental Design
All animals were divided equally into three groups. Each group consisted of ten rats. Group B underwent permanent bilateral carotid ligation. Group C underwent permanent bilateral carotid ligation with concomitant administration of Vitamin E as antioxidant, and Group A served as control. Group C animals were injected with Vitamin E (equivalent to 15 mg/day) intra-peritonealy as a single dose for one week. The animals of Groups B & C were anesthetized, the neck was incised in the midline, and the common carotid arteries were ligated with 4-0 silk suture and severed between sutures bilaterally. Total time of the surgery never exceeded 7 minutes. Approximately 4 to 6 hours after surgery, the animals were exposed to one hour period of hypoxia (6.5% O2) by placing them in an airtight container partially submerged in a 37˚C water bath. Seven days later the rats were sacrificed under deep anesthesia and their brains were extracted and cerebral tissues were prepared for electron microscopy.
2.2. Electron Microscopy
The obtained cerebral tissues were diced into proper sized pieces (1mm cubes) and immediately fixed in buffered 2.5% glutaraldehyde (phosphate buffer, pH 7.2) for at least 6 hrs. The tissue specimens were then post fixed in 1% osmium tetroxide (OSO4) and subsequently dehydrated in graded ethanol series. After infiltration with resin mixture (Epon: Araldite mixture), the tissue specimens were embedded. Ultra-thin sections (70 - 80 nm thickness) were prepared and contrasted with uranyl acetate and lead citrate, and finally examined and photographed under transmission electron microscopy (TEM) (JEOL, 1010 TEM).
3. Results
The cerebral hemispheres of animals of each group looked macroscopically normal. The present ultrastructural study of animals of the control group; Group A, showed normal neurons which exhibited normal distribution of nuclear chromatin and intact nuclear membrane. The cytoplasmic organelles were investigated and seen with normal profiles together with intact neuronal nerve fibers (Figures 1 and 2).
It was observed that bilateral carotid ligation induced ultrastructural changes in animals of Group B. The striking lesions were consistently found in the neuronal cell bodies. The cytopathological changes seen in brain tis-
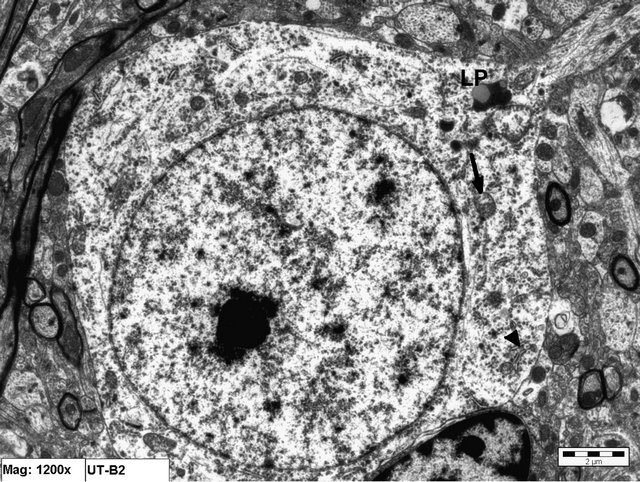
Figure 1. Electron micrograph of a neuronal cell from an animal in Group A (control). Notice the cytoplasmic organelles; mitochondria (arrow), rER (arrow head) that appear normal. Normal nuclear membrane is also seen. LP = lipofuscin pigment (×12,000).
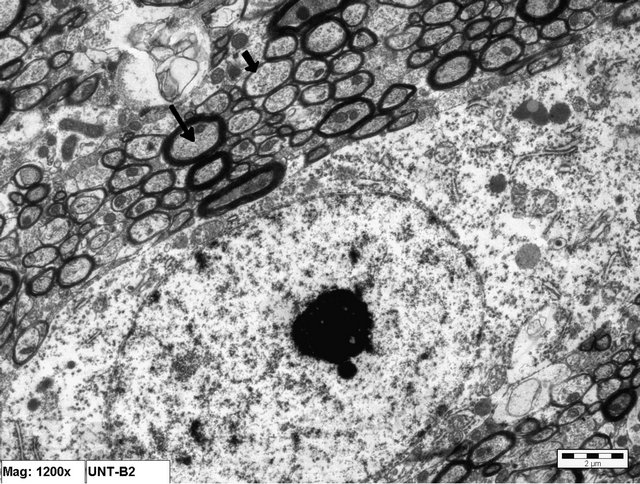
Figure 2. Electron micrograph of a normal neuronal cell from an animal in Group A (control). Notice the surrounding myelinated nerve fibers (arrow) (×12000).
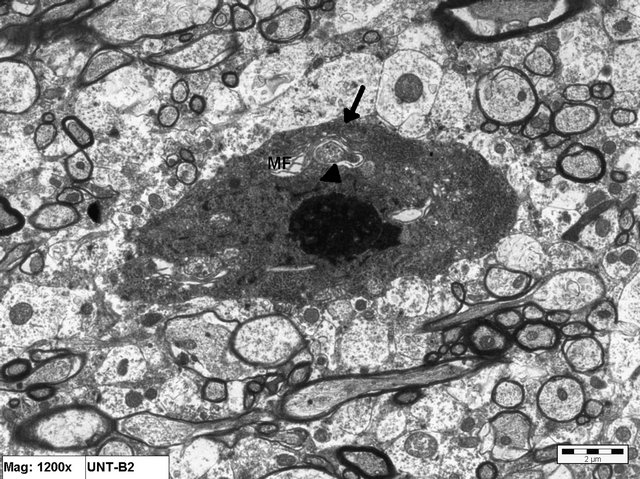
Figure 3. Electron micrograph of brain tissue from an animal in Group B showing a degenerating neuron with a dense body (arrow) > Notice the degenerated mitochondria (arrow head). MF = myelin figure (×12000).
sues of animals of this group consisted primarily of a degenerative reaction affecting the cytoplasmic compartment of some neuronal cell somas, represented by shrunken electrondense ischemic neurons (Figure 3).
In such nerve cells, both the cytoplasm and nucleoplasm were uniformly dense and dark stained, accompanied by an increased number of lysosomes and lipofuscin pigments (Figures 4 and 5). It was, in addition, readily confirmed that the mitochondria were much affected in some of brain specimens of animals of this group; some were appeared elongated, condensed and their membranes assumed a thickened dark compact appearance (Figure 6). In other ultrathin sections, the mitochondria were shown to be swollen and typically displayed a small spherical profile (Figures 6 and 7). An outstanding feature of the edematous neural degenerative reaction was, in addition, obvious in the dissolution of the endoplasmic reticular
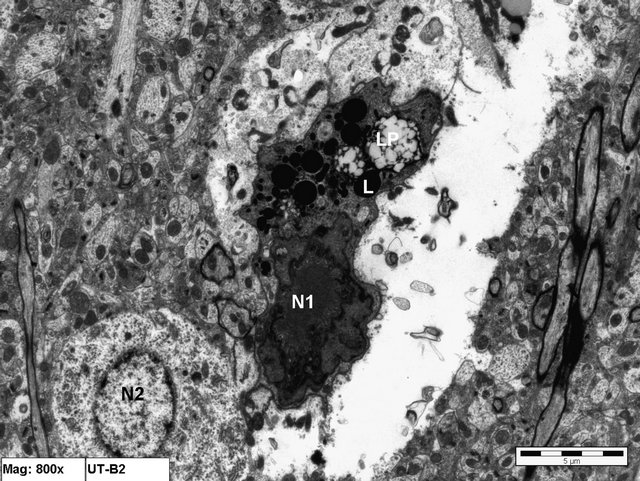
Figure 4. Electron micrograph of brain tissue from an animal in Group B showing a shrunken electron—dense ischemic neuron (N1) and a more normal neuron (N2). Notice the large number of Lysosomes (L) and lipofuscin pigments (LP) in the degenerated neuron (×8000).

Figure 5. A higher magnification of a shrunken electron— dense ischemic neuron in brain tissue from specimens of animals of Group B (arrow). Notice the massive surrounding edematous tissue (*) (×10,000).
system with formation of vacuoles bounded by rough membranes and disaggregated polysomes (Figure 6).
In the specimens of animals of Group C (treated by Vitamin E), the signs of neuronal edematous degeneration were not much pronounced. The mitochondria appeared less swollen, the rER appeared with normal profile and the nucleus presented with less clumping of chromatin (Figures 8 and 9).
Ischemia is defined as a severe reduction or blockage of blood flow and is a pathophysiological event that causes cerebral damage [15]. Although the precise mechanism responsible for ischemic brain damage is still unclear, a number of interconnecting biochemical events appear to be involved, such as energy depletion, cellular acidosis, ion homeostasis breakdown, Ca2+ influx and
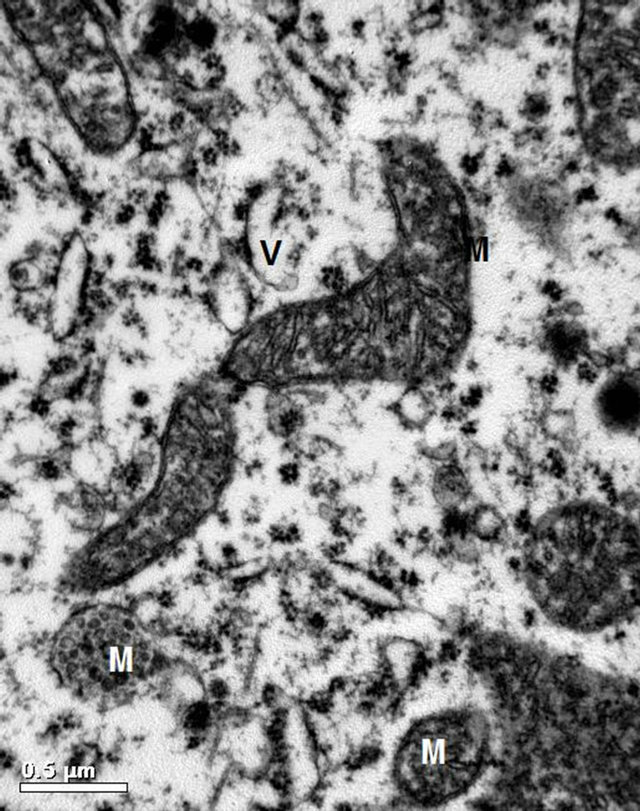
Figure 6. A higher magnification of cytoplasmic structures of a neuronal cell in brain tissue from specimens of animals of Group B showing deteriorated spherical mitochondria (M) and dilated cisternae of rER (V) (×30,000).
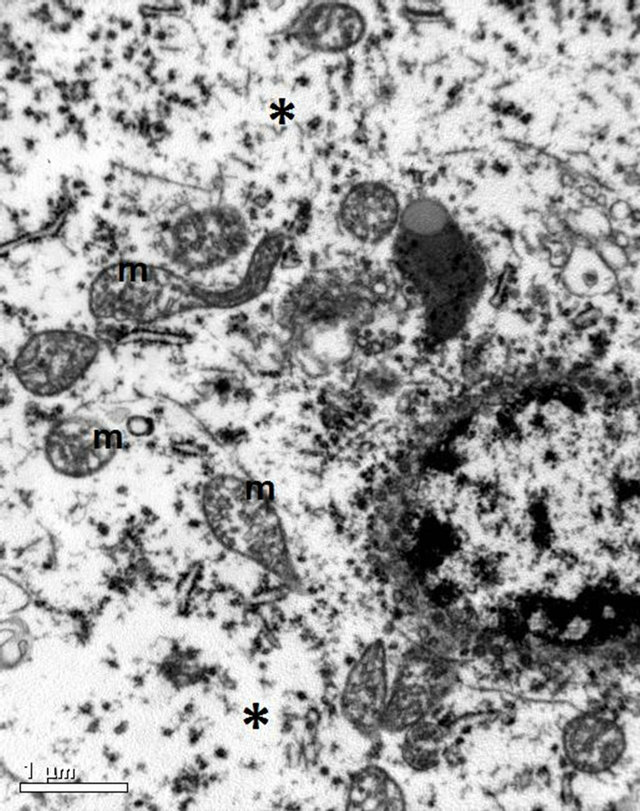
Figuer 7. Electron micrograph of brain tissue from an animal in Group B showing an oedematous cytoplasmic compartment of a degenerated neuron (*). Notice also that the mitochondria (m) are spherical, edematous and swollen. (×25,000).
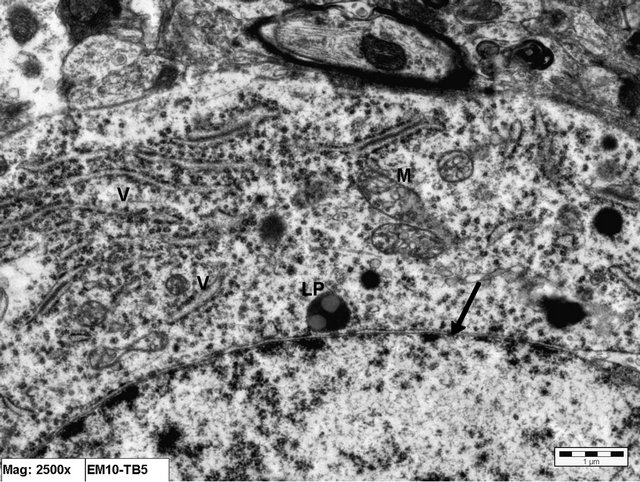
Figure 8. Electron micrograph of brain tissue from an animal in Group C (treated with Vitamin E) showing part of an intact nuclear membrane of a neuron (arrow). The cisternae of rER (V) appear normal. The mitochondria (m) are slightly vacuolated. LP = lipofuscin pigment (×25,000).
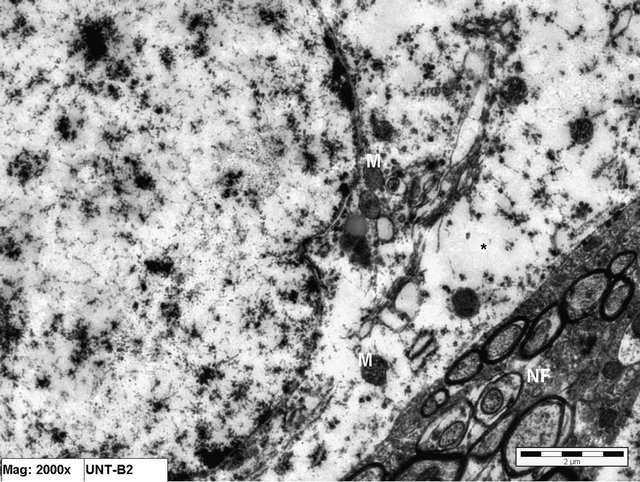
Figure 9. Electron micrograph of brain tissue from an animal in Group C (treated with Vitamin E) showing normal distribution of nuclear chromatin of a neuron together with intact mitochondria (m). Notice the slight cytoplasmic edema (*) and the normal surrounding nerve fibers (NF). (×20,000).
activation of phospholipasesand proteases [15]. Most of these events are accompanied by generation of oxygen free radicals, thus oxidative stress is an important mechanism of brain injury [16,17].
The present study demonstrated ultrastructural changes in brain tissues of animals subjected to permanent bilateral carotid ligation. The dark cells observed in this experimental group of animals were most probably apoptotic neuronal cells. Furthermore, marked neuronal cell degeneration was also noticed in animals of this group. The results of this study are in accordance with Edinger [18] and Tsujimoto and Shimizu (2004) [19] and Faisal et al. (2006) [20] who reported vacuolization and extensive lysis of cytoplasmic contents in the damaged neuronal cells, suggesting induction of an autophagic-lysosomal compartment of programmed cell death. Reference [21] concluded that cerebral ischemia may result in the formation of apoptotic bodies, which supports the obtained results in the present study.
Cessation or severe reduction of blood flow results in almost permanent functional deficits which become rapidly irreversible unless blood flow is promptly restored [22]. Regional oxygen is increased in the beginning of cerebral ischemia, compensating for reduction of cerebral blood flow (CBF), to maintain the cerebral metabolic rate of oxygen close to the normal value [23]. Subsequently, there is a reduction of oxygen and cerebral metabolic rate of oxygen which indicates imminent infarction [24]. These results indicate decreased oxygen metabolism and suggest that hypoxia modulates the fate of ischemic tissues.
A potential consequence of ischemia-hypoxia is autophagy (self-eating), which involves the formation of autophagosomes and autophagolysosomes in degrading cellular constituents for energy production in response to nutrient deprivation [25]. Although autophagy is generally a cell survival mechanism, massive autophagy is associated with cell death [26]. Reference [20] postulate that the combination of ischemia and hypoxia accelerates an energy crisis and precipitates autophagy. References [27] and [28] described that reperfusion of the brain following hypoxia-ischemia leads to the production of oxygen free radicals which attack polyunsaturated fatty acids, leading to membrane lipid peroxidation. The susceptibility of brain to hypoxia depends on the levels of antioxidant enzymes and polyunsaturated fatty acids. Reference [29] suggested that increased concentrations of “oxygen radicals” in the ischemic tissue initiate destructive chain reactions. He also postulated that Vitamin E was thought to be scavenger of active oxygen radicals.
Cerebral ischemia followed by oxygen reperfusion induced apoptosis in hippocampal neurons in stroke-prone spontaneously hypertensive rats (SHRSP) but not in Wistar Kyoto rats [30]. Oxygen radicals were involved in reoxygenation injury after hypoxia in hippocampal slices. Vitamin E inhibited the reoxygenation injury in cultured cortical neurons. In addition, the temporal cortices in Alzheimer’s disease have increased sensitivity to oxygen radicals, and Vitamin E slowed the progression of the disease [30].
Unlike the fine structural changes observed in brain tissues of specimens of animals of Group B (subjected to bilateral carotid ligation), the specimens of animals of Group C (treated with Vitamin E) showed intact nuclear membranes of neurons, normal distribution of neuronal nuclear chromatin, more or less intact mitochondria and non-dilated rough endoplasmic reticulum. According to Tagami et al., 1999 [30] Vitamin E reacts with the radicals and prevents neuronal apoptosis caused by cerebral ischemia and reperfusion. Therefore, Vitamin E seems to be an important agent in lowering radical damage to hippocampal neurons as well as in cortical neurons [8].
The normal cells apoptosis in the present study may be explained by the suggestion of Franz et al. (2003) [31] that the oxidative stress built up upon re-oxygenation may subsequently trigger apoptosis and the presence of antioxidant therefore protect such cells from re-oxygenation induced apoptosis.
Cerebral ischemia followed by overproduction of oxygen-free radicals is implicated in re-oxygenation injury after hypoxia. Antioxidants including Vitamin E react with the radicals and inhibit re-oxygenation injury; thereby preventing apoptosis and necrosis and appear to be the most important agent in limiting free oxygen radical damage in cortical neurons [30]. Vitamin E stops the production of reactive oxygen species (ROS) formed when fat undergoes oxidation. It is under investigation whether, by limiting free-radical production and possibly through other mechanisms, Vitamin E might help to prevent or delay the chronic diseases associated with free radicals [32]. Reference [33] emphasized the relation between cerebral ischemia and delayed neuronal death, and the beneficial antioxidant effects of Vitamin E on attenuating the deleterious consequences of oxidative stress in ischemia.
The present results are in accordance with the conclusion of Ernest et al. (1998) [28], that Vitamin E (α-tocopherol) is the most important lipid-soluble anti-oxidant, that protect the brain from free radical damage.
One potential pathological event in cerebral ischemia is hypoxia-induced fibrin deposition that results from altered anti-coagulant properties of endothelial cells [34, 35] Thus, The induced microvascular thrombosis may also prevent cerebral reperfusion after the release of the large artery occlusion [5]. Vitamin E makes endothelial cells lining the interior surface of blood vessels better able to resist blood-cell components adhering to this surface. Vitamin E also increases the expression of two enzymes that suppress arachidonic acid metabolism, thereby increasing the release of prostacyclin from the endothelium, which, in turn, dilates blood vessels and inhibits platelet aggregation [36]. Therefore, antioxidants appear to be the most important agents in lowering oxygen-free radical damage in cortical neurons.
Conclusively, the present experimental study provides an ultrastructural evidence that Vitamin E protect the brain tissue against the consequences of ischemia-hypoxia which was experimentally induced in the present laboratory animals.
REFERENCES
- R. A. Floyd, “Role of Oxygen Free Radicals in Carcinogenesis and Brain Ischemia,” FASEB Journal, Vol. 4, No. 9, 1990, pp. 2587-2597.
- E. Shohami, E. Beit-Yannai, M. Horowitz and R. Kohen, “Oxidative Stress in Closed-Head Injury: Brain Antioxidant Capacity as an Indicator of Functional Outcome,” Journal of Cerebral Blood Flow and Metabolism, Vol. 17, 1997, pp. 1007-1019. doi:10.1097/00004647-199710000-00002
- A. R. Green, A. H. Hainsworth and D. M. Jackson, “GABA Potentiation: A logical Pharmacological Approach for The Treatment of Acute Ischemic Stroke,” Neuropharmacology, Vol. 39, No. 9, 2000, pp. 1483-1494. doi:10.1016/S0028-3908(99)00233-6
- N. R. Sims and E. Zaidan, “Biochemical Changes Associated with Selective Neuronal Death Following ShortTerm Cerebral Ischaemia,” International Journal of Biochemistry & Cell Biology, Vol. 27, No. 6, 1995, pp. 531- 550. doi:10.1016/1357-2725(95)00026-L
- S. H. Rezkalla and R. A. Kloner, “No-Reflow Phenomenon,” Circulation, Vol. 105, 2002, pp. 656-662. doi:10.1161/hc0502.102867
- K. Yamagata, T. Kitazawa, M. Shinoda, C. Tagawa, M. Chino and H. Matsufuji, “Stroke Status Evoked Adhesion,” Stroke Research and Treatment, Vol. 2010, 2010, pp. 11-17.
- J. C. Dekkers, L. J. P. van Doornen and H. C. G. Kemper, “The Role of Antioxidant Vitamins and Enzymes in the Prevention of Exercise-Induced Muscle Damage,” Sports Medicine, Vol. 21, No. 3, 1996, pp. 213-238.
- M. Tagami, K. Yamagata, K. Ikeda, Y. Nara, H. Fujino, A. Kubota, F. Numano and Y. Yamori, “Vitamin E Prevents Apoptosis in Cortical Neurons during Hypoxia and Oxygen Reperfusion,” Nature Publishing, New York, 1998, pp. 1415-1429.
- M. Kaczmarski, J. Wojicicki, L. Samochowiee, T. Dutkiewicz and Z. Sych, “The Influence of Exogenous Antioxidants and Physical Exercise on Some Parameters Associated with Production and Removal of Free Radicals,” Pharmazie, Vol. 54, No. 4, 1999, pp. 303-306.
- S. D. Bartolome, A. J. Casillan, J. G. Wood, S. Q. Simpson and A. R. O’Brien-Ladner, “Activated Protein C Infusion Mimics Antioxidant Effects on Hypoxia-Induced Microvascular Injury,” Chest, Vol. 128, No. 4, 2005, p. 377S.
- S. Inci, O. E. Ozcan and K. Kilincx, “Time-Level Relationship for Lipid Peroxidation and the Protective Effect of Alpha-Tocopherol in Experimental Mild and Severe Brain Injury,” Neurosurgery, Vol. 43, No. 2, 1998, pp. 330-335. doi:10.1097/00006123-199808000-00095
- M. G. Traber, “Vitamin E,” In: M. E. Shils, M. Shike, A. C. Ross, B. Caballero and R. Cousins, Eds., Modern Nutrition in Health and Disease, 10th Edition, Lippincott Williams & Wilkins, Baltimore, 2006, pp. 396-411.
- G. Leonarduzzi, B. Sottero and G. Poli, “Targeting Tissue Oxidative Damage by Means of Cell Signaling Modulators: The Antioxidant Concept Revisited,” Pharmacology & Therapeutics, Vol. 128, No. 2, 2010, pp. 336-374. doi:10.1016/j.pharmthera.2010.08.003
- K. Jomova, D. Vondrakova, M. Lawson and M. Valko, “Metals Oxidative Stress and Neurodegenerative Disorders,” Molecular and Cellular Biochemistry, Vol. 345, No. 1-2, 2010, pp. 91-104.
- P. Lipton, “Ischemic Cell Death in Brain Neurons,” Physiological Reviews, Vol. 79, No. 4, 1999, pp. 1-33.
- D. S. Warner, H. Sheng and I. Batinie-Haberle, “Oxidants, Antioxidants and the Ischemic Brain,” Journal of Experimental Biology, Vol. 207, 2004, pp. 3221-3231. doi:10.1242/jeb.01022
- M. M. El Kossi and M. M. Zakhary, “Oxidative Stress in the Context of Acute Cerebrovascular Stroke,” Stroke, Vol. 31, No. 8, 2000, pp. 1889-1892.
- A. L. Edinger and C. B. Thompson, “Death by Design: Apoptosis, Necrosis and Autophagy,” Current Opinion in Cell Biology, Vol. 16, No. 6, 2004, pp. 663-669. doi:10.1016/j.ceb.2004.09.011
- Y. Tsujimoto and S. Shimizu, “Another Way to Die: Autophagic Programmed,” US Department of Agriculture, Agricultural Research Service, USDA National Nutrient Database for Standard Reference, 2004.
- F. Adhami, G. H. Liao, Y. M. Morozov, A. Schloemer, V. J. Schmithorst, J. N. Lorenz, R. S. Dunn, C. V. Vorhees, M. Wills-Karp, J. L. Degen, R. J. Davis, N. Mizushima, P. Rakic, B. J. Dardzinski, S. K. Holland, F. R. Sharp and C.-Y. Kuan, “Cerebral Ischemia-Hypoxia Induces Intravascular Coagulation and Autophagy,” American Journal of Pathology, Vol. 169, No. 2, 2006, pp. 566-583. doi:10.2353/ajpath.2006.051066
- M. R. Pulera, L. M. Adams, H. T. Liu and D. G. Santos, National Institutes of Health Office of Dietary Supplements Bethesda, Maryland, 1998. http://ods.od.nih.gov/
- T. Kirino, “Delayed Neuronal Death in the Gerbil Hippocampus Following Ischemia,” Brain Research, Vol. 239, No. 1, 1982, pp. 57-69. doi:10.1016/0006-8993(82)90833-2
- J. C. Baron, “Perfusion Thresholds in Human Cerebral Ischemia: Historical Perspective and Therapeutic Implications,” Cerebrovascular Diseases, Vol. 11, Suppl. 1, 2001, pp. S2-S8. doi:10.1159/000049119
- J. Sobesky, O. Zaro Weber, F. G. Lehnhardt, V. Hesselmann, M. Neveling, A. Jacobs and W. D. Heiss, “Does the Mismatch Match the Penumbra? Magnetic Resonance Imaging and Positron Emission Tomography in Early Ischemic Stroke,” Stroke, Vol. 36, 2005, pp. 980-985. doi:10.1161/01.STR.0000160751.79241.a3
- B. Levine and D. J. Klionsky, “Development by SelfDigestion: Molecular Mechanisms and Biological Functions of Autophagy,” Developmental Cell, Vol. 6, No. 4, 2004, pp. 463-477. doi:10.1016/S1534-5807(04)00099-1
- P. G. Clarke, “Developmental Cell Death: Morphological Diversity and Multiple Mechanisms,” Anatomy and Embryology, Vol. 181, No. 3, 1990, pp. 195-213. doi:10.1007/BF00174615
- M. Sano, C. Ernesto, R. G. Thomas, M. R. Klauber, K. Schafer and M. Grundman, “A Controlled Trial of Selegiline, Alpha-Tocopherol, or Both as Treatment for Alzehimers Disease,” New England Journal of Medicine, Vol. 336, 1997, pp. 1216-1222. doi:10.1056/NEJM199704243361704
- E. M. Grahama, O. P. Mishrab and M. Delivoria-Papadopoulosb, “Anti-Oxidants and Oxidative Stress Injuries to the Brain in the Perinatal Period,” Seminars in Neonatology, Vol. 3, No. 2, 1998, pp. 75-85.
- M. E. Raichle, “The Pathophysiology of Brain Ischemia,” Annals of Neurology, Vol. 13, No. 1, 1983, pp. 2-10. doi:10.1002/ana.410130103
- M. Tagami, K. Ikeda, K. Yamagata, Y. Nara, H. Fujino, A. Kubota, F. Numano and Y. Yamori, “Vitamin E Prevents Apoptosis in Hippocampal Neurons Caused by Cerebral Ischemia and Reperfusion in Stroke-Prone Spontaneously Hypertensive Rats,” Laboratory Investigation, Vol. 79, No. 5, 1999, pp. 609-615.
- Y. F. Franz, W. P. Tsang and T. T. Kwok, “Antioxidant Suppression of Apoptosis Induction by Re-Oxygenation after Chronic Hypoxia in Human Hepatocellular HepG2 Cells,” Proceedings of the American Association for Cancer Research, Vol. 47, 2003.
- M. G. Traber, “Vitamin E Regulatory Mechanisms,” Annual Review of Nutrition, Vol. 27, 2007, pp. 347-362. doi:10.1146/annurev.nutr.27.061406.093819
- K. Ikeda, H. Negishib and Y. Yamoric, “Antioxidant Nutrients and Hypoxia/Ischemia Brain Injury in Rodents,” Toxicology, Vol. 189, No. 1-2, 2003, pp. 55-61.
- S. F. Yan, N. Mackman, W. Kisiel, D. M. Stern and D. J. Pinsky, “Hypoxia/Hypoxemia-Induced Activation of the Pro-Coagulant Pathways and the Pathogenesis of Ischemia-Associated Thrombosis,” Arteriosclerosis, Thrombosis, and Vascular Biology, Vol. 19, 1999, pp. 2029-2035. doi:10.1161/01.ATV.19.9.2029
- V. S. Ten and D. J. Pinsky, “Endothelial Response to Hypoxia: Physiologic Adaptation and Pathologic Dysfunction,” Current Opinion in Critical Care, Vol. 8, No. 3, 2002, pp. 242-250. doi:10.1097/00075198-200206000-00008
- R. J. Glynn, P. M. Ridker, S. Z. Goldhaber, R. Y. Zee and J. E. Buring, “Effects of Random Allocation to Vitamin E Supplementation on The Occurrence of Venous Thrombo-Embolism: Report from the Women’s Health Study,” Circulation, Vol. 116, 2007, pp. 1497-1503. doi:10.1161/CIRCULATIONAHA.107.716407
NOTES
*Corresponding author.