Paper Menu >>
Journal Menu >>
 apoptosis mechanism and functions of various ABSTRACT apoptosis proteins, it will be helpful to obtain infor- mation about their subcellular location. This is Apoptosis proteins have a central role in thebecause the subcellular location of apoptosis proteins development and homeostasis of an organism.is closely related to their function [5,6]. It has been These proteins are very important for under-known that there are 732 archetypical proteins with standing the mechanism of programmed cell“apoptosis” domains [7], and only 98 of these pro- death, and their function is related to theirteins are known to be the apoptosis protein (for more types. The apoptosis proteins are categorizeddetails, one can visit: http://www.apoptosis-db.org). into the following four types: (1) Cytoplasmic Scientists usually deal with a number of protein protein; (2) Plasma membrane-bound protein; sequences already known belonging to apoptosis pro- (3) Mitochondrial inner and outer proteins; (4) teins. However, it is both time-consuming and costly Other proteins. A novel method, the Hilbert-to determine which specific subcellular location a Huang transform, is applied for predicting the given apoptosis protein belongs to. Confronted with type of a given apoptosis protein with supportsuch a situation, can we develop a fast and effective vector machine. High success rates wereway to predict the subcellular location for a given obtained by the re-substitute test (98/98=100%)apoptosis protein based on its amino acid sequence? and jackknife test (91/98 = 92.9%).Recently, Guo-ping Zhou [7] attempted to identify the subcellular location of apoptosis proteins accord- ing to their sequences by means of the covariant discriminant function, which was established on the basis of the Mahalanobis distance and Chou's invariance theorem [7,8,9].The results were quite 1. INTRODUCTIONpromising, indicating that the subcellular location of Apoptosis, or programmed cell death, is a fundamen-apoptosis proteins are predictable to a considerably tal process controlling normal tissue homeostasis by accurate extent if a good vector representation of pro- regulating a balance between cell proliferation and tein can be established. It is expected that, with a con- death [1]. This process entails the autolytic degrada-tinuous improvement of vector representation meth- tion of cellular components, and is characterized byods by incorporating amino acid properties, and by blebbing of cell membranes, shrinkage of cell vol-using more powerful mathematics methods, some the- umes, and condensation of nuclei [2], and is currentlyory predicting method might eventually become a use- an area of intense investigation. Cell death andful tool in this area because the function of an renewal are responsible for maintaining the proper apoptosis protein is closely related to its subcellular turnover of cells, which ensures a constant controlled location. The present study was initiated in an flux of fresh cells. Programmed cell death and cellattempt to address this problem. proliferation are tightly coupled. When apoptosis Chou and Elrod made an extensive research in pre- malfunctions, a variety of formidable diseases can dicting subcellular location mainly based on the ensue: blocking apoptosis is associated with cancer amino acid composition. Subsequently, in order to and autoimmune disease, whereas unwanted take into account the sequence-order effects and apoptosis can possibly lead to ischemic damage orimproved the prediction quality, Chou has further neurodegenerative disease [3]. Apoptosis is consid-incorporated the quasi-sequence order effect [5] and ered to have a key role in these several devastatingintroduced the concept of “pseudo-amino-acid com- diseases and, in principle, provides many targets forposition”[9]. For example, Chou [10] classified mem- therapeutic intervention [4]. To understand the brane proteins into five different types and proposed Keywords:Hilbert Huang transform; Sup- port vector machine; Subcellular location predict Hilbert Huang transform for predicting proteins subcellular location Hilbert Huang transform for predicting proteins subcellular location Feng Shi, Qiu-Jian Chen & Na-na Li School of Science, Huazhong Agricultural University, Wuhan, Hubei, China. Correspondence should be addressed to Feng Shi (shifeng@mail.hzau.edu.cn). J. Biomedical Science and Engineering, 2008, 1, 59-63Scientific Research Publishing JBiSE Published Online May 2008 in SciRes.http://www.srpublishing.org/journal/jbise SciRes Copyright ©2008  a covariant discriminant algorithm to predict theand the lower envelop (linked by local minima) are types of membrane proteins. Recently, Cai et al. [11] zero at every point. applied neural network to this problem. To improve TheEMDprocessisasfollows.According to the prediction quality, Chou [5] proposed a newHilbert-Huang transform(HHT)[14], once the method in which the covariant discriminate algo-extrema of a time seriesx(t) are identified, all the rithmwasaugmentedtoincorporatethequasi-local maxima and minima are connected by two spe- sequence-order effect. This method uses the amino cial lines as the upper and lower envelopes respec- acidcompositionandthesequence-order-couplingtively. Their mean is designated as m, and the differ- 1 numbers (reflecting the sequence order effect) in ence between x(t) andm is x(t)-m =h . If h is not an 1111 order to improve the prediction quality. Feng [12] pro-IMF, h is treated as the data and undergoes the pro- 1 posed a new representation of unified attribute vector,cedure above, thenh-m=h . Repeat this sifting 111 11 that each protein can be represented by a vector, procedure k times until h is an IMF, that ish- which is 20-D vector in Hilbert space with unified1k1(k-1) length. Hence, all of proteins have their representa-m=h, thus the first IMF component is obtained, 1k1k tive points on the surface of the 20-D globe. The rep-i.e. . Then separate IMF from the original time series 1 resentative points of the proteins in the same familyby x(t)-IMF=r. Treatr as the new data and subject 111 or with the higher sequence identity are closer on theit to the same sifting process above. Repeat this pro- surface. The overall predictive accuracy could be cedure on all the subsequent r , i.e. r-IMF =r,r- improved from 3% to 5% for different databases [12] j1222 with this simply modification of the usage of the IMF =r ,,r-IMF=r . 33 n-1nn amino acid composition. Recently, a series of new So the result is: powerful approaches have been developed by Chou and his co-workers [13]. Encouraged by the great suc- cesses of the previous invertigators in the area, here we would like to use a different strategy, the support vector machines, to approach this very important but also very difficult problem in the hope that our 2.2. Hilbert transform approach can play a complementary role to the exist-Having obtained the intrinsic mode function compo- ing methods.nents IMF (denoted as c), one will have no difficulty ii in applying the Hilbert transform to each IMF compo- 2. HILBERT HUANG TRANSFORMnent, The HHT consists of two parts: empirical mode decomposition (EMD) and Hilbert spectral analysis (HSA). This method is potentially viable for nonlin- ear and nonstationary data analysis, especially for time-frequency-energy representations. It has beenin which the PV indicates the principal value of the tested and validated exhaustively, but only empiri-singular integral. With the hilbert transform, the ana- cally. In all the cases studied, the HHT gave results lytic signal is defined as much sharper than those from any of the traditional analysis methods in time-frequency-energy represen- tations.Additionally, the HHT revealed true physicalHere, a(t)is the instantaneous amplitude, and(t) meanings in many of the data examined. Powerful asii it is, the method is entirely empirical. In order to is the phase function, make the method more robust and rigorous, many out- standing mathematical problems related to the HHT method need to be resolved. In this section, a brief introduction to the methodology of the HHT will be given. Readers interested in the complete details should consult [14]. and the instantaneous frequency is simply 2.1. The empirical mode decomposition method (the sifting process) In this method any time series, including non-linear and non-stationary series, can be decomposed into a With the Hilbert Spectrum defined, we can also finite number of intrinsic mode functions (IMFs) definethemarginal spectrum h(w) as through empirical mode decomposition (EMD) pro- cess.An IMF is a function which must follow two con- ditions: (1) the difference between the numbers of extrema and zero-crossings is of 1 ; and (2) the The marginal spectrum offers a measure of the mean of the upper envelop (linked by local maxima)total amplitude (or energy) contribution from each SciRes JBiSE Copyright ©2008 F. Shi et al./J. Biomedical Science and Engineering 1 (2008) 59-63 60 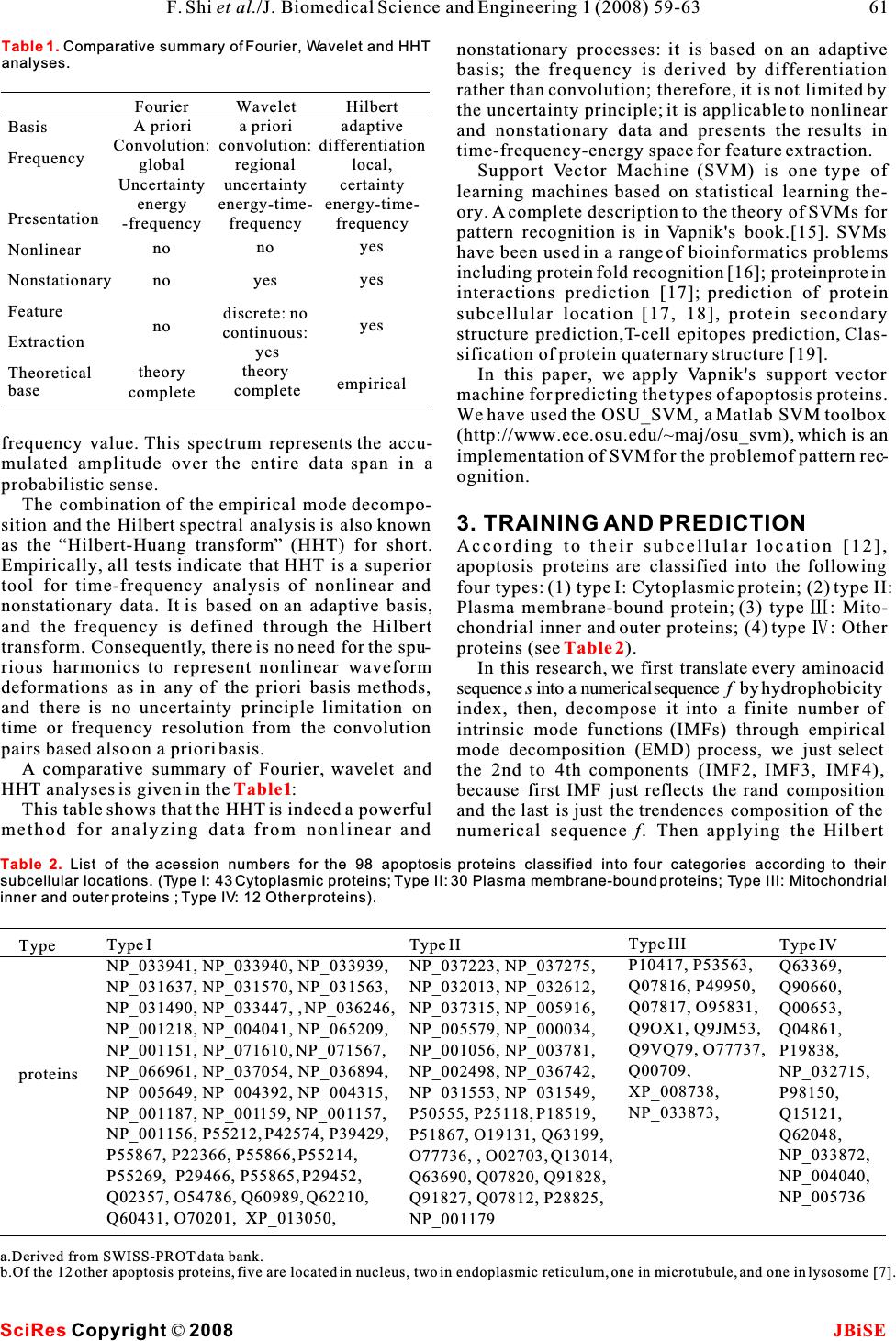 nonstationary processes: it is based on an adaptive basis; the frequency is derived by differentiation rather than convolution; therefore, it is not limited by the uncertainty principle; it is applicable to nonlinear and nonstationary data and presents the results in time-frequency-energy space for feature extraction. Support Vector Machine (SVM) is one type of learning machines based on statistical learning the- ory. A complete description to the theory of SVMs for pattern recognition is in Vapnik's book.[15]. SVMs have been used in a range of bioinformatics problems including protein fold recognition [16]; proteinprotein interactions prediction [17]; prediction of protein subcellular location [17, 18], protein secondary structure prediction,T-cell epitopes prediction, Clas- sification of protein quaternary structure [19]. In this paper, we apply Vapnik's support vector machine for predicting the types of apoptosis proteins. We have used the OSU_SVM, a Matlab SVM toolbox (http://www.ece.osu.edu/~maj/osu_svm), which is an frequency value. This spectrum represents the accu-implementation of SVM for the problem of pattern rec- mulated amplitude over the entire data span in aognition. probabilistic sense. The combination of the empirical mode decompo- sition and the Hilbert spectral analysis is also known 3. TRAINING AND PREDICTION as the“Hilbert-Huang transform” (HHT) for short.According to their subcellular location [12], Empirically, all tests indicate that HHT is a superiorapoptosis proteins are classified into the following tool for time-frequency analysis of nonlinear and four types: (1) type I: Cytoplasmic protein; (2) type II: nonstationary data. It is based on an adaptive basis,Plasma membrane-bound protein; (3) type : Mito- and the frequency is defined through the Hilbertchondrial inner and outer proteins; (4) type : Other transform. Consequently, there is no need for the spu-proteins (see). rious harmonics to represent nonlinear waveformIn this research, we first translate every aminoacid deformations as in any of the priori basis methods, sequences into a numerical sequencef by hydrophobicity and there is no uncertainty principle limitation onindex, then, decompose it into a finite number of time or frequency resolution from the convolution intrinsic mode functions (IMFs) through empirical pairs based also on a priori basis.mode decomposition (EMD) process, we just select A comparative summary of Fourier, wavelet andthe 2nd to 4th components (IMF2, IMF3, IMF4), HHT analyses is given in the:because first IMF just reflects the rand composition This table shows that the HHT is indeed a powerfuland the last is just the trendences composition of the method for analyzing data from nonlinear and numerical sequence f. Then applying the Hilbert Table 2 Table1 SciRes JBiSE Copyright ©2008 61 F. Shi et al./J. Biomedical Science and Engineering 1 (2008) 59-63 Type I NP_033941, NP_033940, NP_033939, NP_031637, NP_031570, NP_031563, NP_031490, NP_033447, , NP_036246, NP_001218, NP_004041, NP_065209, NP_001151, NP_071610, NP_071567, NP_066961, NP_037054, NP_036894, NP_005649, NP_004392, NP_004315, NP_001187, NP_001159, NP_001157, NP_001156, P55212, P42574, P39429, P55867, P22366, P55866, P55214, P55269, P29466, P55865, P29452, Q02357, O54786, Q60989, Q62210, Q60431, O70201, XP_013050, Type proteins Type II NP_037223, NP_037275, NP_032013, NP_032612, NP_037315, NP_005916, NP_005579, NP_000034, NP_001056, NP_003781, NP_002498, NP_036742, NP_031553, NP_031549, P50555, P25118, P18519, P51867, O19131, Q63199, O77736, , O02703, Q13014, Q63690, Q07820, Q91828, Q91827, Q07812, P28825, NP_001179 Type III P10417, P53563, Q07816, P49950, Q07817, O95831, Q9OX1, Q9JM53, Q9VQ79, O77737, Q00709, XP_008738, NP_033873, Type IV Q63369, Q90660, Q00653, Q04861, P19838, NP_032715, P98150, Q15121, Q62048, NP_033872, NP_004040, NP_005736 a.Derived from SWISS-PROT data bank. b.Of the 12 other apoptosis proteins, five are located in nucleus, two in endoplasmic reticulum, one in microtubule, and one in lysosome [7]. Table 2.List of the acession numbers for the 98 apoptosis proteins classified into four categories according to their subcellular locations. (Type I: 43 Cytoplasmic proteins; Type II: 30 Plasma membrane-bound proteins; Type III: Mitochondrial inner and outer proteins ; Type IV: 12 Other proteins). Basis Frequency Presentation Nonlinear Nonstationary Feature Extraction Theoretical base Wavelet a priori convolution: regional uncertainty energy-time- frequency no yes discrete: no continuous: yes theory complete Fourier A priori Convolution: global Uncertainty energy -frequency no no no theory complete Hilbert adaptive differentiation local, certainty energy-time- frequency yes yes yes empirical Table 1. Comparative summary of Fourier, Wavelet and HHT analyses.  transform to each IMF component, we get the instan-When the re-substitution test was performed for the taneous amplitudea(t),thengettheenergy valuecurrent study, the type of each apoptosis protein in a idata set was in turn identified using the rule parame- e= , (t=2, 3, 4). Next, get its energy ratio iters derived from the same data set, the so-called training data set. As shown in, the overall suc- .Last every protein was represented as a cess rate thus obtained for the 98 apoptosis proteins point or a vector in a 23-D space. The first 20 compo-in was 100%, indicating an excellent self- nents of its vector were supposed to be the occur-consistency. rence frequencies of the 20 amino acids in the protein However, during the process of the re-substitution concerned, the last three components were its energytest, the rule parameters derived from the training ratio times a weight, there, we set the weight is 0.2. data set include the information of the query protein The computations were carried out on a PC. Alsolater plugged back in the test. This will certainly for the SVM, the width of the Gaussian RBFs is underestimate the error and enhance the success rate selected as that which minimized an estimate of the because the same proteins are used to derive the rule VC-dimension.After being trained, the hyper-plane parameters and to test themselves. Nevertheless, the output by the SVM was obtained. The SVM method is re-substitution test is absolutely necessary because it applied to two-class problems. In this paper, for the reflects the self-consistency of a prediction method, four-class problems, we have used a simple andespecially for its algorithm part. A prediction algo- effective method:“one-against-others” method [16]rithm certainly cannot be deemed as a good one if its to transfer it into two-class problems. We first test the self-consistency is poor. In other words, the re- selfconsistency and leave-one-out cross-validationsubstitution test is necessary but not sufficient for (jackknife test) of the method, followed by testingevaluating a prediction method. As a complement, a the method by prediction of an independent dataset. cross-validation test for an independent testing data As a result, the rates of self-consistency, cross-set is needed because it can reflect the effectiveness validation of prediction were quite high.of a prediction method in practical application. This In addition to the prediction algorithm, we also is important especially for checking the validity of a need to construct a training data set to complete thetraining data set-whether it contains sufficient infor- establishment of a statistical prediction method. Tomation to reflect all the important features concerned realize this, based on the SWISS-PROT data bank, 98 so as to field a high success rate in application. apoptosis proteins (the date were taken from Zhou [7]) were classified into the following four subcellular locations: (1) cytoplasmic, (2) plasma membrane-4.2. Jackknife test bound, (3) mitochondrial, and (4) other ().As is well known, the independent data set test, sub- sampling test, and jackknife test are the three meth- 4RESULTS AND DISCUSSIONods often used for cross-validation in statistical pre- By means of the SVM algorithm described in the lastdiction. Among these three, however, the jackknife section, a statistical prediction was performed for the test is deemed as the most effective and objective one 98 apoptosis proteins listed in. The predic-for a comprehensive discussion about this). During tion was conducted by two different approaches, the jackknifing, each protein in the data set is in turn sin- re-substitution test and the jackknife test. The resultsgled out as a tested protein and all the rule parameters aregiven in.are calculated based on the remaining proteins. In other words, the subcellular location of each apoptosis protein is identified by the rule parameters 4.1. Re-substitution testderived using all the other apoptosis proteins except The so-called re-substitution test is an examination the one that is being identified. During the process of for the self-consistency of a prediction method[7]. yi Las ever ti Table 3 Table 1 Table 1 Table 2 Table 3 Test method Success Rate Re-substitute Jack-knife covariant SVM HHT covariant SVM HHT Type 43/43=100% 42/43=97.70% 43/43=100% 42/43=97.7% 39/43=91.4% 41/43=95.3% Type 30/30=100% 30/30=100% 30/30=100% 22/30=73.3% 28/30=93.3% 29/30=96.7% Type 9/13=60.2% 13/13=100% 13/13=100% 4/13=30.8% 12/13=92.5% 12/13=96.7% Type 7/12=58.3% 12/12=100% 12/12=100% 3/12=25.0% 9/12=75.0% 9/12=75.7 Overall 89/98=90.8% 97/98=99.0% 98/98=100% 71/98=72.5% 88/98=89.8% 91/98=92.9% SciRes JBiSE Copyright ©2008 62F. Shi et al./J. Biomedical Science and Engineering 1 (2008) 59-63 Table 3.Tested results for the 98 apoptosis prtoeins in Table 2 by both Re-substitution test and Jackknife test.All use Gauss RBF kernel function, while the value C =15, and the gama= 80.  [8]Chou, K C. A. novel approach to predicting protein structural jackknifing, both the training data set and testingclasses in a (20-1)-D amino acid composition space. data set are actually open, and a protein will in turn Proteins:Structure, Function and Genetics 1995, 21: 319-344. move from one to the other.As expected, the success [9]Chou, K. C. Prediction of protein cellular attributes using prediction rates by jackknife test were decreased in pseudo-amino-acid-composition. Proteins :Structure, Function, and Genetics 2001, 43:246-255 (Erratum:ibid., 2001, vol. 44, comparison with those by the re-substitution test. 60). Such a decrement is particularly more remarkable for[10]Chou, J. J., Li, H., Salvesen G.S., Yuan, J. & G. Wagner. Solu- small subsets. This is because the cluster-tolerant tion structure of BID, an intracellular amplifier of apoptotic capacity for small subsets is usually low. And hence signaling. Cell 1999, 96:615-624. [11]Cai,Y. D., Liu, X. J. & Chou, K. C. Artificial neural network the information loss resulting from jackknifing will model for predicting membrane protein types. J. Biomol. have a greater impact on the small subsets than theStruct. Dyn. 2001, 18:607-610. large ones. Nevertheless, as shown in, the[12]Feng, Z. P. Prediction of the subcellular location of overall jackknife rate for the data set of the 98prokaryotic proteins based on a new representation of the amino acid composition. Biopolymers 2001, 58:491-499. apoptosis proteins could still reach 93%. It is [13]Cai, Y. D. & Chou, C. Nearest neighbour algorithm for predict- expected that the success rate for identifying theing protein subcellular location by combing functional subcellular location of apoptosis proteins can be fur-domain composition and pseudo-amino acid composition. ther enhanced by improving the training data of smallBiochem Biophys Res Comm. 2003, 305:407-411. [14]Huang, N. E., Shen, Z., Long, S. R., Wu, M. L., H.H. Shih, subsets by adding into them more new proteins thatZheng, Q., N.C. Yen, C.C. Tung & Liu, H. H. The empirical have been found belonging to the subcellular locationmode decomposition and Hilbert spectrum for nonlinear and defined by these subsets.nonstationary time series analysis. Proc. Roy. Soc. London A 1998, 454:903-995. [15]Vapnik V. Statistical Learning Theory. WileyInterscience 5 CONCLUSIONS1998. The above results, together with those obtained by[16]Ding, C. H. & I. Dubchak. Multiclass protein fold recognition using support vector machines and neural networks. the covariant discriminant prediction algorithm [7],Bioinformatics 2001, 17:349-358. have indicated that the types of apoptosis proteins are [17]Cai, Y. D., Liu, X. J., Xu, X. B.& Chou, K. C. Support vector predictable with a considerable accuracy. It is antici-machines for prediction of protein subcellular location by pated that the HHT, and the SVM, if effectively com-incorporating quasisequenceorder effect. J. Cell. Biochem. 2002, 84:343-348. plemented with each other, will become a powerful[18]Hua, S. J. & Sun, Z. R. Support vector machine approach for tool for predicting the types of apoptosis proteins.protein subcellular localization prediction. Bioinformatics The current study has further demonstrated that the2001, 17:721-728. datasets originally constructed by Zhou[7] will be [19]Hua, S. J. & Sun, Z. R. A novel method of protein secondary structure prediction with high segment overlap measure: sup- very useful for the area of apoptosis study. It isport vector machine approach. J. Mol. Biol. 2001, 308:397- expected that the prediction quality can be further 407. improved if the current HHT can be properly com- bined with pseudoamino acid composition[9] and function domine composition and with other amino acid properties. ACKNOWLEDGMENTS The authors thank Dr. Guo-Ping Zhou for providing the amino acid sequences of the apoptosis proteins and some helpful discussions. The work was partly supported by Huazhong Agricultural Univer- sity, P. R. China REFERENCE [1]Zhou, P., Chou, J. J., Olea RS, Yuan, J. & G. Wagner. Solution structure of Apaf-1 CARD and its interaction with caspase-9 CARD: a structural basis for specific adaptor/caspase interac- tion. Proc Natl Acad Sci USA 1999, 96:11265-11270. [2]Kerr J.F., Wyllie A. H. & A. R. Currie. Apoptosis: a basic bio- logical phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972, 26:239-257. [3]Schulz J. B., Weller M. & M. A. Moskowitz. Caspases as treat- ment targets in stroke and neurodegenerative diseases. Ann Neurol 1999, 45:421-429. [4]Barinaga M. Stroke-damaged neurons may commit cellular sui- cide. Science 1998, 281:1302-1303. [5]Chou, K. C. A new branch of proteomics: prediction of protein cellular attributes. Gene Cloning and Expression Technologies 2002, 4:57-70, [6]Huang, J. & Shi, F. Support vector machines for prodicting apoptosis proteins types. Acta bioinformatics 2005, 53:39-47. [7]Zhou, G. P. & Doctor. K. Subcelluar location of Apoptosis pro- teins. Proteins:Structure, Function, and Genetic 2003, 50:44-48. Table 2 SciRes JBiSE Copyright ©2008 63 F. Shi et al./J. Biomedical Science and Engineering 1 (2008) 59-63 |