Journal of Cancer Therapy
Vol.2 No.2(2011), Article ID:5479,7 pages DOI:10.4236/jct.2011.22025
Identification of Small and Discriminative Gene Signatures for Chemosensitivity Prediction in Breast Cancer
![]()
Department of Computer Science, Houghton College, Houghton, New York, USA.
Email: wei.hu@houghton.edu
Received January 22nd, 2011; revised April 5th, 2011; accepted April 15th, 2011.
Keywords: Genetic Algorithm, Gene Signature, Breast Cancer, Sparse Logistic Regression, Predictor, Chemosensitivity
ABSTRACT
Various gene signatures of chemosensitivity in breast cancer have been discovered. One previous study employed t-test to find a signature of 31 probe sets (27 genes) from a group of patients who received weekly preoperative chemotherapy. Based on this signature, a 30-probe set diagonal linear discriminant analysis (DLDA-30) classifier of pathologic complete response (pCR) was constructed. In this study, we sought to uncover a signature that is much smaller than the 31 probe sets and yet has enhanced predictive performance. A signature of this nature could inform us what genes are essential in response prediction. Genetic algorithms (GAs) and sparse logistic regression (SLR) were employed to identify two such small signatures. The first had 13 probe sets (10 genes) selected from the 31 probe sets and was used to build a SLR predictor of pCR (SLR-13), and the second had 14 probe sets (14 genes) selected from the genes involved in Notch signaling pathway and was used to develop another SLR predictor of pCR (SLR-Notch-14). The SLR-13 and SLR-Notch-14 had a higher accuracy and a higher positive predictive value than the DLDA-30 with much lower P values, suggesting that our two signatures had their own discriminative power with high statistical significance. The SLR prediction model also suggested the dual role of gene RNUX1 in promoting residual disease (RD) or pCR in breast cancer. Our results demonstrated that the multivariable techniques such as GAs and SLR are effective in finding significant genes in chemosensitivity prediction. They have the advantage of revealing the interacting genes, which might be missed by single variable techniques such as t-test.
1. Introduction
The molecular and pathological characteristics observed in breast cancer patients implies that breast cancer is a heterogeneous disease. Distinct molecular subtypes of breast cancer identified by patterns of gene expression could lead to different clinical outcomes. In current practice, chemotherapy is applied empirically, and does not benefit all patients, illustrating the imperative needs for a more personalized approach in cancer treatment. Therefore, the ability to predict whether an individual patient will benefit from a specific therapy is of great clinical significance. Single clinical or molecular indicators, such as tumor size, tumor grade, histology, hormone receptor or human epidermal growth factor receptor 2 (HER2) expression, does not always give reliable predictions of response to a treatment. With gene expression profiling, it is possible to find multiple genes that can used to build an enhanced predictor of response to chemotherapy in breast cancer [1-7].
In [8], t-test for unequal-variance was applied to find a signature of 31 probe sets (27 genes) with highest differentially expressed values between pRC and residual disease (RD). A 30-probe set diagnal linear discriminant analysis (DLDA-30) predictor of pCR was constructed based on this signature. The value of this predictor is the ability to identify those patients most likely to benefit from a particular treatment, the neoadjuvant chemotherapy, in this case. This predictor is able to recognize not all responsive patients but exclusively those that will benefit the most, as defined by attaining a pCR.
As a single variable technique, the t-test used in [8] analyzes one gene at a time and might miss the interactions between genes. We hypothesized that the signature of 31 probe sets in [8] could be optimized with help of multivariable techniques such as genetic algorithms (GAs), which can explore multiple solutions concurrently to find interacting and informative genes. In this study, our aim was to identify a signature that is a small subset of the 31 probe sets in [8] and use it to develop a predictor of pCR that can achieve better predictions than the DLDA-30. A gene signature of this nature captures the essence of response prediction.
2. Patients and Methods
2.1. Patient Cohorts and Clinical Information
One breast cancer patient cohort was obtained from a previous publication [8] (n = 133). Needle-biopsy samples were collected from 133 patients with stage I, II, or III breast cancer who received preoperative weekly paclitaxel and a combination of fluorouracil, doxorubicin, and cyclophosphamide (T/FAC). These 133 patients were divided into two subsets, one training set of size 81 and one validation set of size 52. These data contain clinical information including patient age, gender, race, histological classification, stage, nuclear grade, ER (estrogen receptor), PR (progesterone receptor), and HER2 (human epidermal growth factor 2) status, pathologic complete response, and residual disease. These data also contain each patient’s genome-scale gene expression profiles generated using Affymetrix U133A chip (Santa Clara, CA). pCR was defined as no residual invasive cancer in the breast or lymph nodes. pCR is presently accepted as a reasonable early indicator for long-term survival.
2.2. Sparse Logistic Regression
A standard least squared linear regression solves the following problem:
Given data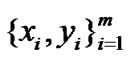 , find
, find 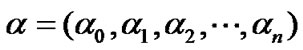 such that
such that
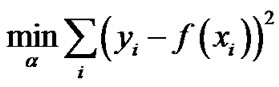 where
where 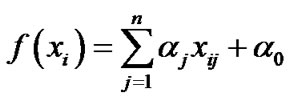
The LASSO regression, studied by Tibshirani R [9], deals with the following problem:
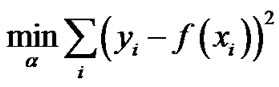 with
with 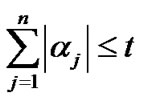 where t controls the
where t controls the 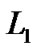 norm of
norm of . This constraint on
. This constraint on 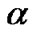 produces a sparse model, i.e., many components of
produces a sparse model, i.e., many components of 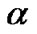 can be zero. Following the idea of LASSO, Shevade at el. studied the following problem for sparse logistic regression, when
can be zero. Following the idea of LASSO, Shevade at el. studied the following problem for sparse logistic regression, when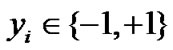
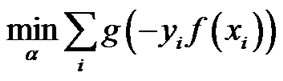 with
with 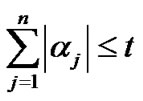 where
where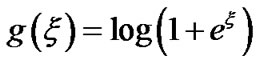 , which is the negative loglikelihood function associated with the probability model Prob
, which is the negative loglikelihood function associated with the probability model Prob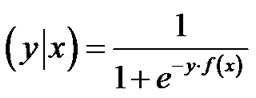 .
.
Cawley GC et al. [10] utilized a novel technique to solve this sparse logistic regression problem efficiently. In our study, we used +1 to label those cases of RD status, and used –1 to label those cases of pCR status.
2.3. Notch Signaling Pathway
Notch genes encode highly conserved cell surface receptors. The Notch signaling pathway consists of Notch receptors, ligands, negative and positive modifiers, and transcription factors. It plays a key role in the normal development of many tissues and cell types, through diverse effects on cell regulation, proliferation, and differentiation. Aberrant Notch signaling has been observed in several human cancers, including acute T-cell lymphoblastic leukemia, cervical cancer, and breast cancer [11,12]. The Oligo GEArray Human Notch Signaling Pathway Microarray [13] was designed for profiling expression of 113 genes (Table 1) involved in Notch signaling.
2.4. Prediction Accuracy Evaluation
In order to evaluate the significance of our predictions, we need to compare them with random guesses. For each dataset, a random-label permutation was conducted while keeping the number of instances in each group fixed. The matches between the permuted labels and the original ones were recorded. The standard P value was the percentage of 1000 random predictions with higher accuracy than the calculated predictions.
2.5. Genetic Algorithms
Genetic algorithms (GAs), a particular class of evolutionary algorithms, are search algorithms that adopt some common processes in genetics such as selection, mutation, and inheritance. The GAs outperform other traditional search algorithms in various applications.
Pseudo-code for genetic algorithms is as follows:
• Generate an initial population of individuals
• Evaluate initial population
• Repeat Perform selection Apply genetic operations such as mutation and crossover to generate a new generation of individuals Evaluate individuals in the population
• Until some stopping criteria is satisfied
3. Results
To search for a subset of the 31 probe sets, we represented our solution, referred to as an individual in GAs, as a binary vector of size 31 to indicate the presence (1)

Table 1. Genes involved in notch signaling pathway as described in [13].
or absence (0) of each probe set in the 31 probe sets. Our fitness value was the prediction accuracy of SLR based on the training set. In each generation of GA, we randomly divided the training set (n = 82) into five equal subsets for five-fold cross validation, where four subsets were used as a training set for SLR and one subset as a test set to get the accuracy of SLR on this test set. We ran the GA algorithm with population size 40, individual size 31, and 100 generations. In each generation, the top 50% of the individuals with highest fitness values were selected as parents to produce the next generation. A single point crossover and point mutations were applied to each individual in the population. As a result of GA selection, a collection of individuals with high prediction accuracy were discovered. The following individual has this property with training set accuracy equal to 0.89.

The above binary representation means the following 13 probe sets (10 genes) were selected (Table 2).
This 13 probe set signature highlighted the critical genes for response prediction, and therefore provided clear insight into the molecular mechanisms that regulate the chemosensitivity in breast cancer. Encouraged by the finding of the 13 probe sets from the 31 probe sets in [8], we applied the same selection procedure using GAs to the 113 genes involved in Notch signaling pathway (Table 1), resulting in a signature of 14 probe sets (SLR-Notch-14) (Figure 1). It is worth noting that our search for a smaller and yet more discriminative set of genes from the 31 probe sets was accomplished by two size reductions. First, the size of informative genes was reduced from 31 probe sets to 13 by GAs, and then SLR was able to further reduce this 13 probe sets to 10 probe sets (Figure 1).
We report in Tables 2 and 3 the t-test statistic, P value, and gene names for the 13 probe sets from the 31 probe sets in [8] and the 14 probe sets from the genes involved in Notch signaling pathway. The P values in these two tables showed that the most discriminative genes as a group measured by SLR were not necessarily those with the smallest P values as individual genes. In general, the genes that were highly expressed in the RD cases contributed positively to the prediction of RD and those highly expressed in the pCR cases contributed positively to the prediction of pCR.
In addition to testing the SLR-13 and SLR-Notch14 on the training set (Table 4), we also tested them on the validation set (Tables 5 and 6) for prediction performance and P values. Table 5 suggests that the two predictors, SLR-13 and SLR-Notch-14, complemented each other in their specificity and sensitivity. On the other hand the confusion matrix in Table 6 contains information about actual and predicted classifications obtained through the three predictors. The SLR-Notch-14 had three nonzero P values and they were all less than 0.05,
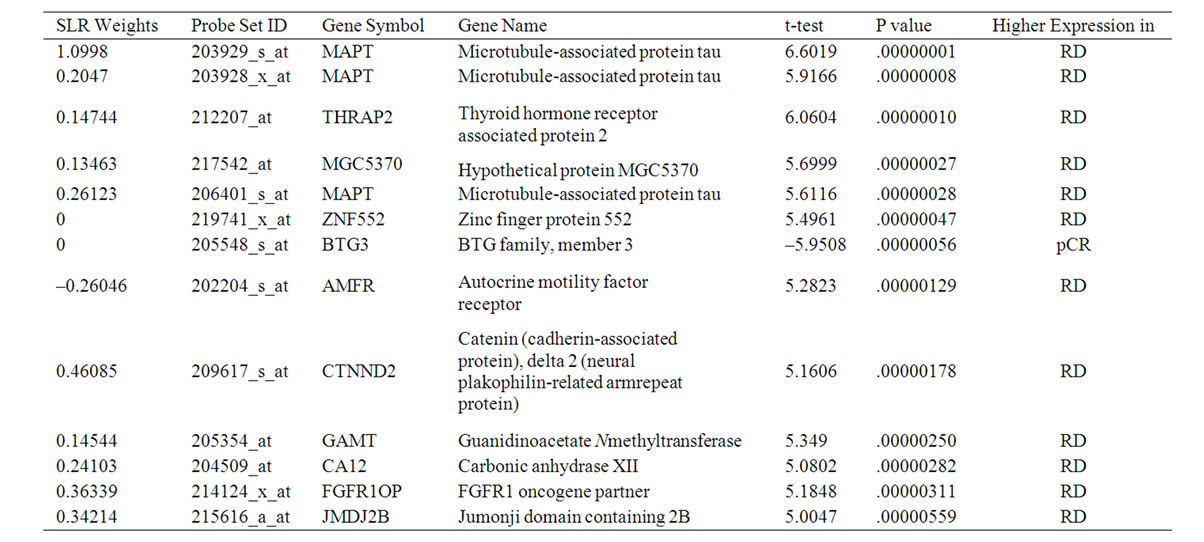
Table 2. 13 informative probe sets selected by GA and SLR from the 31 probe sets in [8].
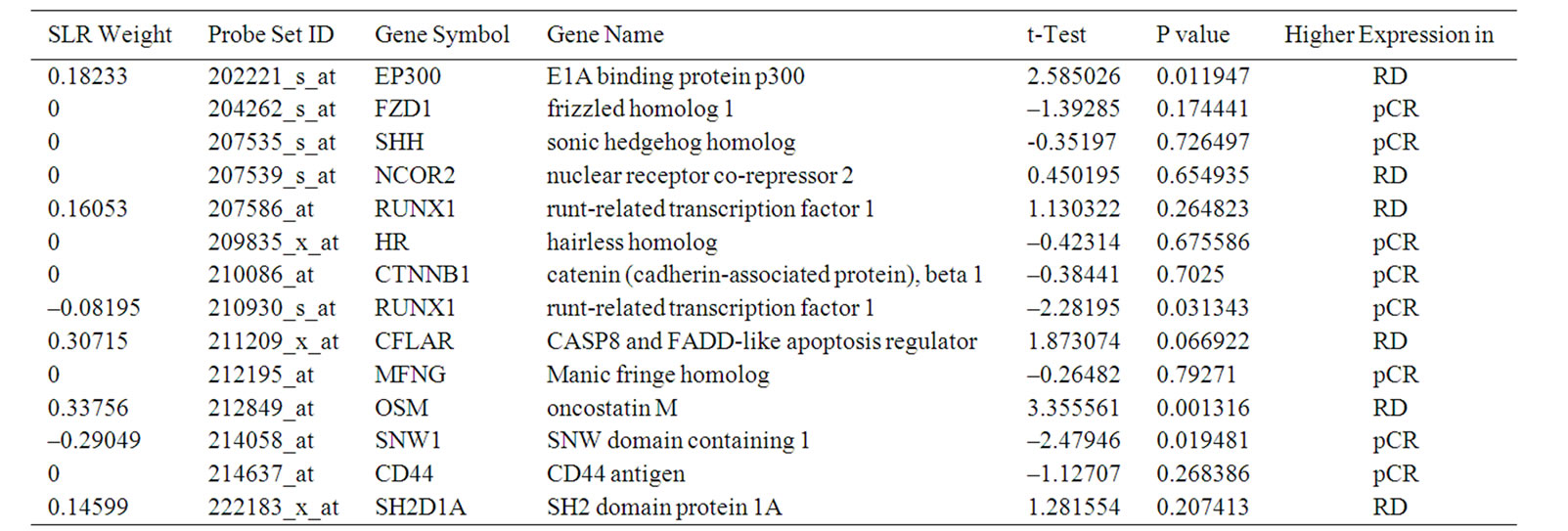
Table 3. 14 informative probe sets selected by GA and SLR from Notch signaling pathway.
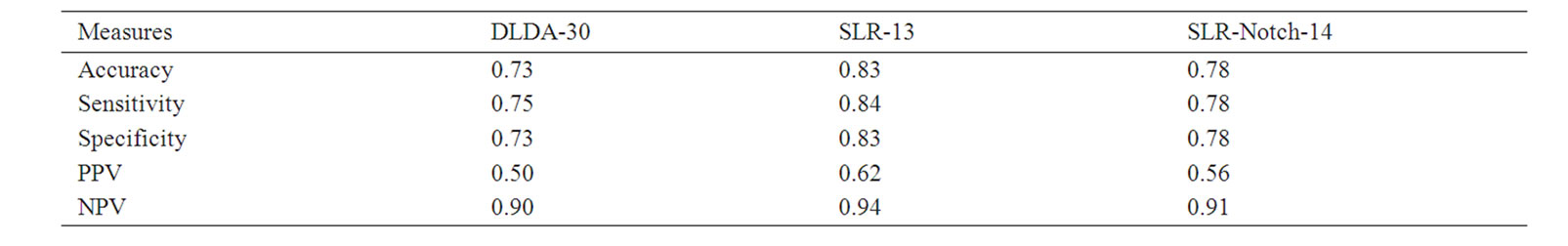
Table 4. Prediction measures of SLR-13 and SLR-Notch-14 on the training set (five-fold cross validation).
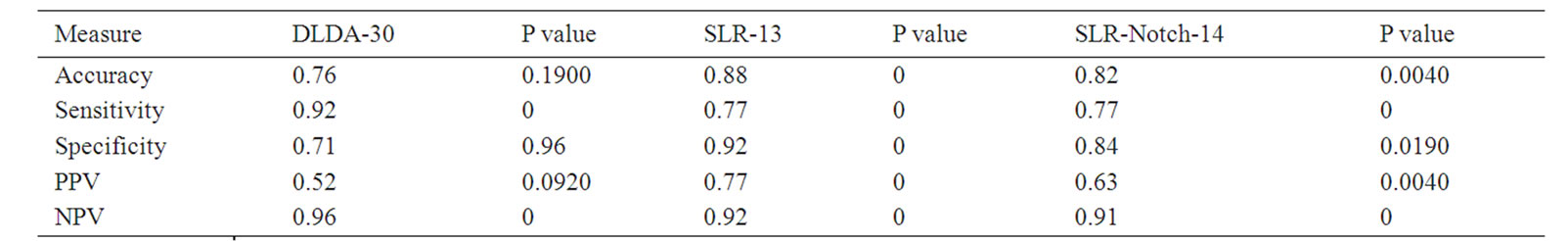
Table 5. Prediction measures of DLDA-30, SLR-13, and SLR-Notch-14 on the validation set.

Table 6. Confusion matrices for DLDA-30, SLR-13, and SLR-Notch-14 on the validation set.
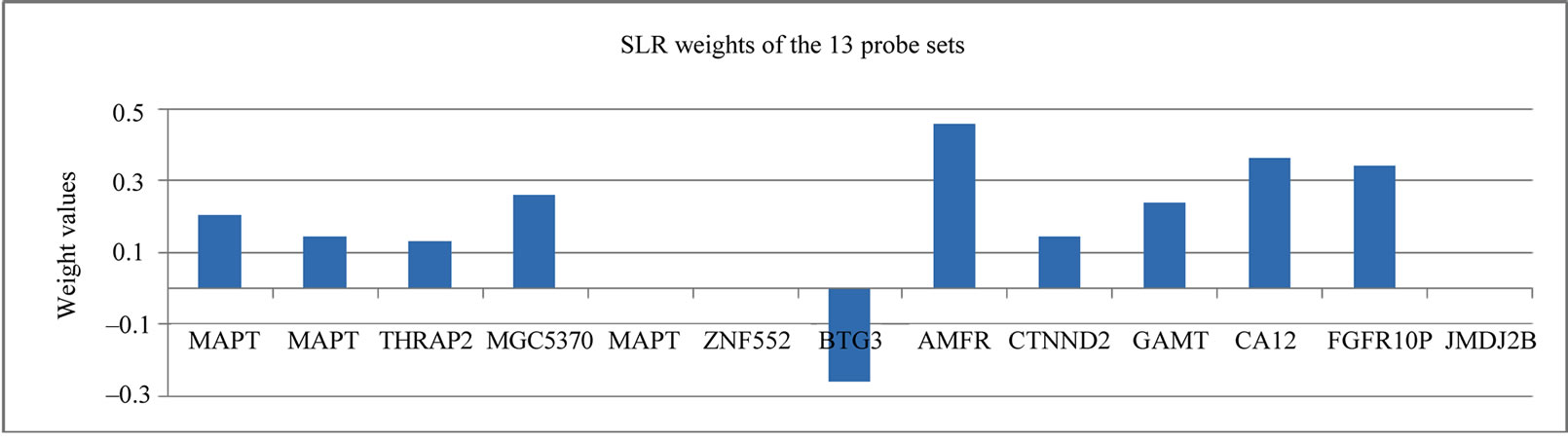
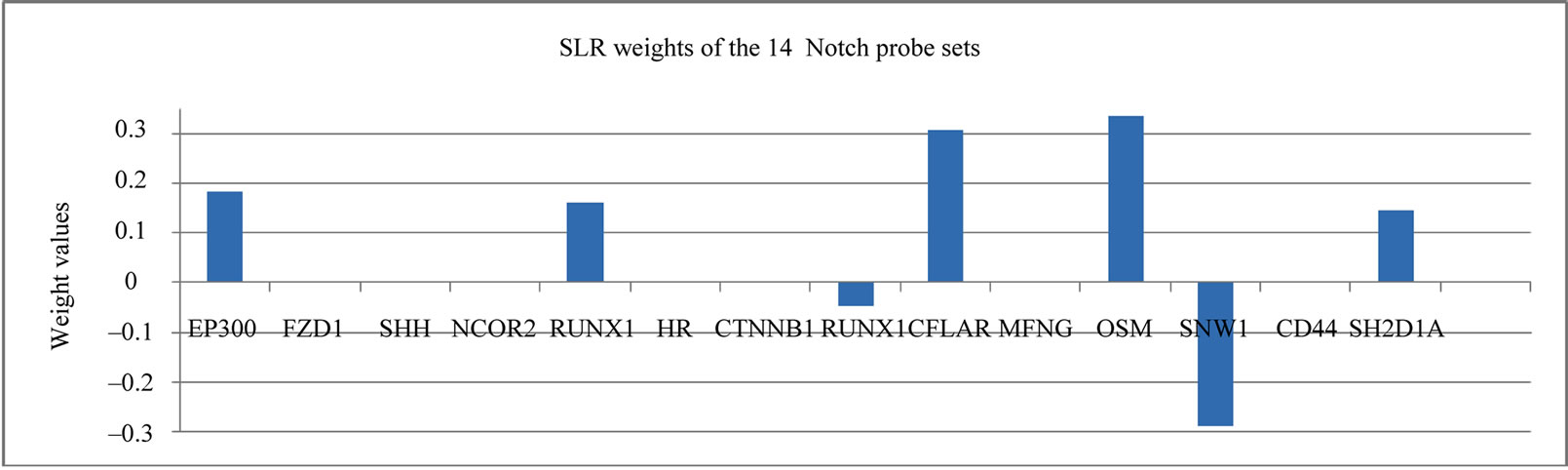
Figure 1. The SLR weights of the 13 probe set and 14 probe set signature.
implying that the prediction accuracies of SLR-13 and SLR-Notch14 were statistically significant. The DLDA-30 had three nonzero P values but they were all larger than 0.05, especially those for accuracy and specificity.
In Table 2, the genes were all highly expressed in the RD cases with one exception, whereas Table 3 has more balanced gene numbers in the RD and pCR cases, which could partially contribute to the distinct predicting power of SLR-13 and SLR-Notch-14 (Tables 5 and 6). In the SLR model, genes with a positive weight contributed positively to the RD prediction and those with a negative weight contributed positively to the prediction of pCR. Obviously, genes of zero weight did not contribute to response prediction. The gene BTG3 [14] was highly expressed in the pCR cases, and so it was weighted positively toward the prediction of pCR in the SLR-13 signature (Figure 1). However, in the SLR-Notch-14 signature one UNIX1 probe set was expressed highly in the RD cases and another UNIX1 probe set was highly expressed in the pCR cases, which were also reflected by their opposite SLR weights (Figure 1). RUNX1 has a dual role in promoting cell cycle progression and differentiation [15] and can function as a transcription activator or as a repressor [16].
4. Discussion
Predicting the treatment response for patients with breast cancer is a great challenge in clinics and is critical in personalized medicine. Various such genes signatures of different predicting power have been identified. The focus of the current study was to find a gene signature that is small but has improved predictions. Our strategy was to apply genetic algorithm, an efficient search algorithm, to refine the genes in a known signature so that only the most relevant genes were remained after this procedure. The findings of this report demonstrated the validity of this approach. We believed that genes in a small signature reveal the essence of association between gene expressions and clinical outcome.
5. Conclusions
Recent studies suggest that gene expression profile correlates with responses to neoadjuvant chemotherapy, with tumors displaying the ER-positive gene signatures being less likely to respond than other types of breast cancer [17]. In this study, we intended to discover a much smaller gene signature than the 31 probe sets in [8] that predicts whether a breast cancer patient will benefit from a preoperative treatment containing paclitaxel followed by fluorouracil, doxorubicin, and cyclophosphamide by achieving a pCR. With the ability to account for multiple gene interactions, two multivariable techniques, genetic algorithms and sparse logistic regression, were employed to identify a 13 probe sets from the 31 probe sets in [8] and a 14 probe set signatures from the genes involved Notch signaling pathway (Table 1). The response predictions of our two signatures had much lower P values than the 31 probe sets in [8], revealing the improved statistical significance of their predictions. The SLR prediction model also verified the dual role of gene RNUX1 in promoting RD or pCR in breast cancer.
6. Acknowledgements
We thank Houghton College for its financial support.
REFERENCES
- M. Chanrion, V. Negre, H. Fontaine, N. Salvetat, F. Bibeau, G. Mac Grogan, L. Mauriac, D. Katsaros, F. Molina, C. Theillet and J. M. Darbon, “A Gene Expression Signature That Can Predict the Recurrence of Tamoxifen-Treated Primary Breast Cancer,” Clinical Cancer Research, Vol. 14, No. 6, 2008, pp. 1744-1752. doi:10.1158/1078-0432.CCR-07-1833
- S. P. Linke, T. M. Bremer, C. D. Herold, G. Sauter and C. Diamond, “A Multimarker Model to Predict Outcome in Tamoxifen-Treated Breast Cancer Patients,” Clinical Cancer Research, Vol. 12, No. 4, 2006, pp. 1175-1183. doi:10.1158/1078-0432.CCR-05-1562
- P. E. Lønning, S. Knappskog, V. Staalesen, R. Chrisanthar and J. R. Lillehaug, “Breast Cancer Prognostication and Prediction in the Postgenomic Era,” Annals of Oncology, Vol. 18, No. 8, 2007, pp. 1293-1306. doi:10.1093/annonc/mdm013
- M. A. Folgueira, D. M. Carraro, H. Brentani, D. F. Patrão, E.M. Barbosa, M. M. Netto, J. R. Caldeira, M. L. Katayama, F. A. Soares, C. T. Oliveira, L. F. Reis, J. H. Kaiano, L. P. Camargo, R. Z. Vêncio, I. M. Snitcovsky, F. B. Makdissi, P. J. e Silva, J. C. Góes and M. M. Brentani, “Gene Expression Profile Associated with Response to Doxorubicin-Based Therapy in Breast Cancer,” Clinical Cancer Research, Vol. 11, No. 20, 2005, pp. 7434-7443. doi:10.1158/1078-0432.CCR-04-0548
- H. K. Dressman, C. Hans, A. Bild, J. A. Olson, E. Rosen, P. K. Marcom, V. B. Liotcheva, E. L. Jones, Z. Vujaskovic, J. Marks, M. W. Dewhirst, M. West, J. R. Nevins and K. Blackwell, “Gene Expression Profiles of Multiple Breast Cancer Phenotypes and Response to Neoadjuvant Chemotherapy,” Clinical Cancer Research, Vol. 12, No. 3, 2006, pp. 819-826. doi:10.1158/1078-0432.CCR-05-1447
- O. Thuerigen, A. Schneeweiss, G. Toedt, P. Warnat, M. Hahn, H. Kramer, B. Brors, C. Rudlowski, A. Benner, F. Schuetz, B. Tews, R. Eils, H.-P. Sinn, C. Sohn and P. Lichter, “Gene Expression Signature Predicting Pathologic Complete Response with Gemcitabine, Epirubicin, and Docetaxel in Primary Breast Cancer,” Journal of Clinical Oncology, Vol. 24, No. 12, 2006, pp. 1839-1845. doi:10.1200/JCO.2005.04.7019
- S. Rathnagiriswaran, Y. W. Wan, J. Abraham, V. Castranova, Y. Qian and N. L. Guo, “A Population-Based Gene Signature is Predictive of Breast Cancer Survival and Chemoresponse,” International Journal of Oncology, Vol. 36, No. 3, 2010, pp. 607-616.
- K. R. Hess, K. Anderson, W. F. Symmans, et al., “Pharmacogenomic Predictor of Sensitivity to Preoperative Chemotherapy with Paclitaxel and Fluorouracil, Doxorubicin, and Cyclophosphamide in Breast Cancer,” Journal of Clinical Oncology, Vol. 24, No. 26, 2006, pp. 4236-4244.
- R. Tibshirani, “Regression Shrinkage and Selection via the Lasso,” Journal of the Royal Statistical Society B, Vol. 58, No. 1, 1996, pp. 267-288.
- G. C. Cawley and L. C. Talbot, “Gene Selection in Cancer Classification Using Sparse Logistic Regression with Bayesian Regularization,” Bioinformatics, Vol. 22, No. 19, 2006, pp. 2348-2355. doi:10.1093/bioinformatics/btl386
- S. Stylianou, R. B. Clarke, et al., “Activation of Notch Signaling in Human Breast Cancer,” Cancer Research, Vol. 66, No. 3, 2006, pp. 1517-1525. doi:10.1158/0008-5472.CAN-05-3054
- K. Brennan and A. M. C. Brown, “Is There a Role for Notch Signalling in Human Breast Cancer?” Breast Cancer Research, Vol. 5, No. 2, 2003, pp. 69-75. doi:10.1186/bcr559
- http://www.sabiosciences.com/rt_pcr_product/HTML/PAHS-059A.html
- Y. H. Ou, P.-H. Chung, et al., “The Candidate Tumor Suppressor BTG3 Is a Transcriptional Target of P53 That Inhibits E2F1,” The EMBO Journal, Vol. 26, No. 17, 2007, pp. 3968-3980. doi:10.1038/sj.emboj.7601825
- J. Starkova, J. Madzo, G. Cario, T. Kalina, A. Ford, M. Zaliova, O. Hrusak and J. Trka, “The Identification of (ETV6)/RUNX1-Regulated Genes in Lymphopoiesis Using Histone Deacetylase Inhibitors In ETV6/RUNX1- Positive Lymphoid Leukemic Cells,” Clinical Cancer Research, Vol. 13, No. 6, 2007, pp. 1726-1735. doi:10.1158/1078-0432.CCR-06-2569
- F. M. Mikhail, K. K. Sinha, Y. Saunthararajah and G. Nucifora, “Normal and Transforming Functions of RUNX1: A Perspective,” Journal of Cell Physiology, Vol. 207, No. 3, 2006, pp. 582-593. doi:10.1002/jcp.20538
- N. S. Goldstein, D. Decker, D. Severson, et al., “Molecular Classification System Identifies Invasive Breast Carcinoma Patients Who Are Most Likely and Those Who Are Least Likely to Achieve a Complete Pathologic Response after Neoadjuvant Chemotherapy,” Cancel, Vol. 110, No. 8, 2007, pp. 1687-1696. doi:10.1002/cncr.22981