 International Journal of Organic Chemistry, 2011, 1, 242-249 doi:10.4236/ijoc.2011.14035 Published Online December 2011 (http://www.SciRP.org/journal/ijoc) Copyright © 2011 SciRes. IJOC A Simple and Efficient Procedure for a 2-Pyridones Synthesis under Solvent-Free Conditions Zahira Kibou1, Nawel Cheikh1,2, Didier Villemin2*, Noureddine Choukchou-Braham1, Bachir Mostefa-Kara1, Mohamed Benabdallah1 1Laboratoire de Catalyse et Synthèse en Chimie Organique, Faculté des Sciences, Université Aboubekr Belkaïd, Tlemcen, Algérie 2ENSICAEN, LCMT, Caen, France E-mail: *didier.villemin@ensicaen.fr Received June 7, 2011; revised July 21, 2011; accepted August 9, 2011 Abstract A new series of 3-cyano-2-pyridones derivatives have been prepared by reaction of enaminonitriles with pri- mary amine under solvent free condition. This procedure have the advantage of high yields and being environ- mentally-friendly. Keywords: 2-Pyridones, Solvent-Free, Enaminonitriles, Green Chemistry 1. Introduction Nowadays, one powerful solution in the Green & Sus- tainable Chemistry movement is the replacement of tra- ditional synthetic methods, which use harmful stoichio- metric reagents that produce vast amounts of wastes, with clean and simple catalytic alternatives with high atom efficiency [1,2]. Solvent-free and dominos reac- tions represent very powerful green chemical technology procedures from both the economical and synthetic point of view and represent a possible instrument to perform a near-ideal synthesis because they enhance the rate of many organic reactions and afford quantitative yields [3-8]. Heteroaromatic rings containing atoms frequently play an important role as the scaffolds of bioactive sub- stances [9]. It is well-known that the pyridone [9] and its derivatives are among the most popular N-heteroaro- matic compounds integrated into the structures of many pharmaceutical compounds and the structural units occur in various molecules exhibiting diverse biological activi- ties [10-12]. This can easily be demonstrated using the following examples (Figure 1) [13]. Pyridone L-697,661 [13] has been recognized as a specific non-nucleoside reverse transcriptase inhibitor of human immunodefi- ciency virus-1 (HIV-1) [13], Milrinone WIN 47203 [9,14], Amrinone WIN 40680 [9,14] and their analogues are well time- honored positive inotropic and vasodilata- tory agents, used in the clinical treatment of heart failure [9,14]. Some others are reported to show antitumor [15], antibacterial activity, evaluated as human rhinovirus (HRV) 3C-protease (3CP) inhibitors [15] and other biological activities. Others, that share the 2-Pyridone and its deriva- tives, illustrate a large class as ligands in coordination chemistry [16,17]. The various research teams around the world were and are still interested in the synthesis of 2-Pyridones (Pyri- din-2(1H)-ones). The various synthetic approaches to 2- pyridones of this type are described. Many literature sources [18-26] describe more general approaches in- volving the condensation of unsaturated ketones with methylene active amides, using cyanoacetamide. A num- ber of Milrinone (Figure 1) analogues have been ob- tained [18-26]. Departing from the previous literature, and as part of our continuing interest in the progress of new synthetic methods in heterocyclic chemistry in our laboratory [27-29], we started the development of a new preparative procedure for this class of heterocyclic scaf- fold compounds. 2. Results and Discussions In our work, we developed a new method for an easier, simpler and more universal synthesis to prepare this type of heterocycles “2-pyridone”, while trying to respect the criteria of the green chemistry, in which we employed, as a key step, the synthesis of enaminonitrile and in the pre- sence of a catalytic amount of NH4OAc (Scheme 1). From Scheme 1, we found that the synthesis under solvent-free of new nitrogen heterocyclic compounds of “2-pyridone derivatives” can be obtained, by a simple, effective, fast and cleaner method, using the three fol- lowing steps: 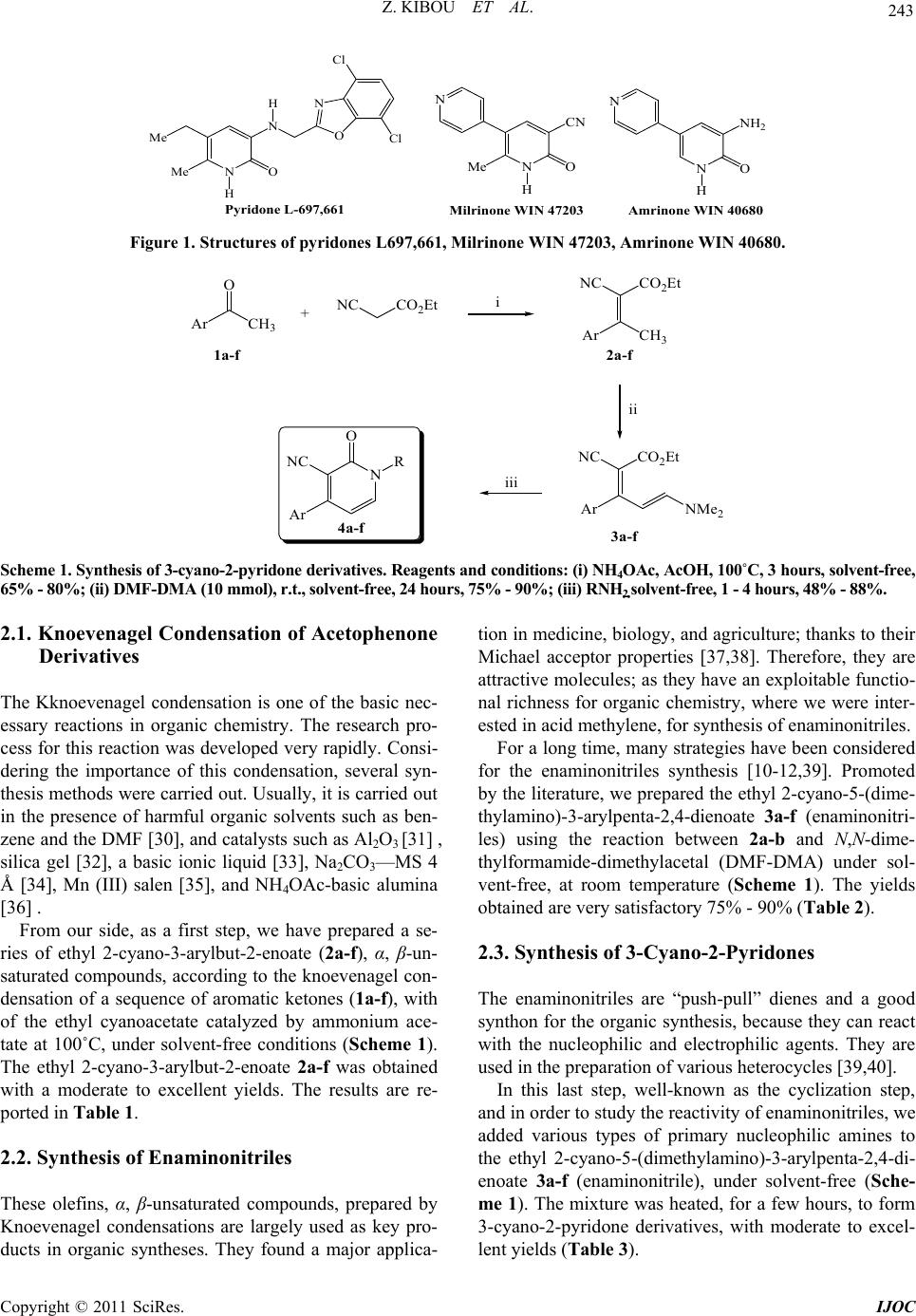 243 Z. KIBOU ET AL. N NH 2 O H N Amrinone WIN 40680 N CN O H Me N Milrinone WIN 47203 N O H N Me Me O N Cl Cl H Pyridone L-697,661 Figure 1. Structures of pyridones L697,661, Milrinone WIN 47203, Amrinone WIN 40680. Ar CH 3 O CO 2 EtNC Ar CH 3 NC CO 2 Et Ar NC CO 2 Et NR O Ar NC +i iii NMe 2 ii 1a-f 2a-f 3a-f 4a-f Scheme 1. Synthesis of 3-cyano-2-pyridone derivatives. Reagents and conditions: (i) NH4OAc, AcOH, 100˚C, 3 hours, solvent-free, 65% - 80%; (ii) DMF-DMA (10 mmol), r.t., solvent-free, 24 hours, 75% - 90%; (iii) RNH2 solvent-free, 1 - 4 hours, 48% - 88%. 2.1. Knoevenagel Condensation of Acetophenone Derivatives The Kknoevenagel condensation is one of the basic nec- essary reactions in organic chemistry. The research pro- cess for this reaction was developed very rapidly. Consi- dering the importance of this condensation, several syn- thesis methods were carried out. Usually, it is carried out in the presence of harmful organic solvents such as ben- zene and the DMF [30], and catalysts such as Al2O3 [31] , silica gel [32], a basic ionic liquid [33], Na2CO3—MS 4 Å [34], Mn (III) salen [35], and NH4OAc-basic alumina [36] . From our side, as a first step, we have prepared a se- ries of ethyl 2-cyano-3-arylbut-2-enoate (2a-f), α, β-un- saturated compounds, according to the knoevenagel con- densation of a sequence of aromatic ketones (1a-f), with of the ethyl cyanoacetate catalyzed by ammonium ace- tate at 100˚C, under solvent-free conditions (Scheme 1). The ethyl 2-cyano-3-arylbut-2-enoate 2a-f was obtained with a moderate to excellent yields. The results are re- ported in Table 1. 2.2. Synthesis of Enaminonitriles These olefins, α, β-unsaturated compounds, prepared by Knoevenagel condensations are largely used as key pro- ducts in organic syntheses. They found a major applica- tion in medicine, biology, and agriculture; thanks to their Michael acceptor properties [37,38]. Therefore, they are attractive molecules; as they have an exploitable functio- nal richness for organic chemistry, where we were inter- ested in acid methylene, for synthesis of enaminonitriles. For a long time, many strategies have been considered for the enaminonitriles synthesis [10-12,39]. Promoted by the literature, we prepared the ethyl 2-cyano-5-(dime- thylamino)-3-arylpenta-2,4-dienoate 3a-f (enaminonitri- les) using the reaction between 2a-b and N,N-dime- thylformamide-dimethylacetal (DMF-DMA) under sol- vent-free, at room temperature (Scheme 1). The yields obtained are very satisfactory 75% - 90% (Table 2). 2.3. Synthesis of 3-Cyano-2-Pyridones The enaminonitriles are “push-pull” dienes and a good synthon for the organic synthesis, because they can react with the nucleophilic and electrophilic agents. They are used in the preparation of various heterocycles [39,40]. In this last step, well-known as the cyclization step, and in order to study the reactivity of enaminonitriles, we added various types of primary nucleophilic amines to the ethyl 2-cyano-5-(dimethylamino)-3-arylpenta-2,4-di- enoate 3a-f (enaminonitrile), under solvent-free (Sche- me 1). The mixture was heated, for a few hours, to form 3-cyano-2-pyridone derivatives, with moderate to excel- lent yields (Table 3). Copyright © 2011 SciRes. IJOC 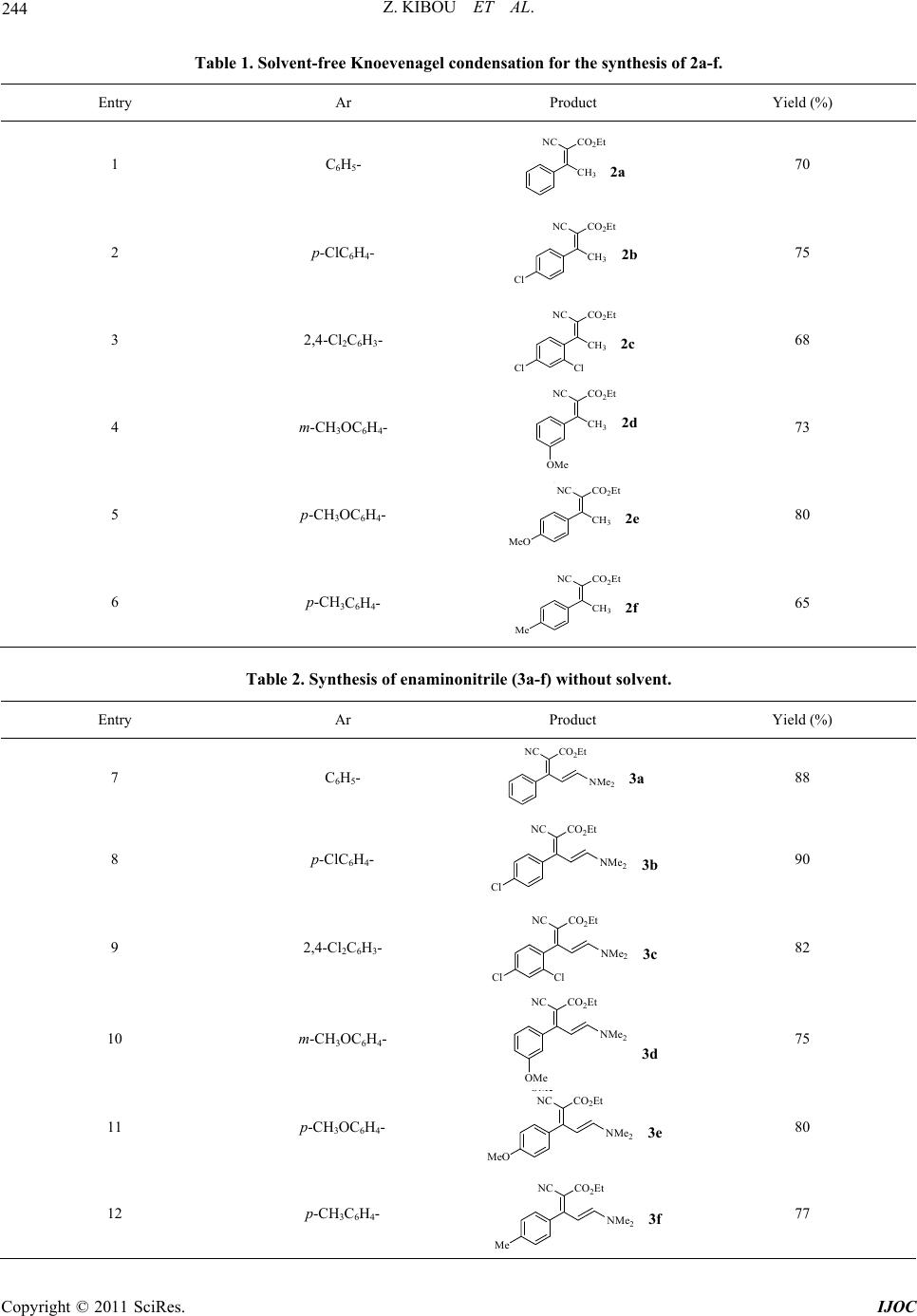 Z. KIBOU ET AL. 244 Table 1. Solvent-free Knoevenagel condensation for the synthesis of 2a-f. Entry Ar Product Yield (%) 1 C6H5- CH 3 NC CO 2 Et 2a 70 2 p-ClC6H4- CH 3 NC CO 2 Et Cl 2 75 3 2,4-Cl2C6H3- CH 3 NC CO 2 Et ClCl 2c 68 4 m-CH3OC6H4- CH 3 NC CO 2 Et 2 OMe 73 5 p-CH3OC6H4- CH3 NC CO2Et MeO 2e 80 6 p-CH3C6H4- CH 3 NC CO 2 Et Me 2f 65 Table 2. Synthesis of enaminonitrile (3a-f) without solvent. Entry Ar Product Yield (%) 7 C6H5- NC CO 2 Et NMe 2 3a 88 8 p-ClC6H4- NC CO 2 Et Cl NMe 2 3b 90 9 2,4-Cl2C6H3- NC CO 2 Et ClCl NMe 2 3c 82 10 m-CH3OC6H4- NC CO 2 Et NMe 2 OMe 3d 75 11 p-CH3OC6H4- NC CO 2 Et MeO NMe 2 3e 80 12 p-CH3C6H4- NC CO 2 Et Me NMe 2 3f 77 Copyright © 2011 SciRes. IJOC  245 Z. KIBOU ET AL. Table 3. Synthesis of the 3-cyano-2-pyridones. Entry Enaminonitrile R Product Yield (%) 13 CH3- NC 4a 1 N O 66 14 CH2=CH-CH2- NC 4a 2 N 70 15 C6H5CH2- NC 4a 3 N O 73 16 3a (CH3)2CH- NC N O 4a 4 48 17 CH3- 76 18 CH2=CH-CH2- 80 19 C6H5CH2- 85 20 3b (CH3)2CH- NC NC NC NC N O N O N O N O Cl Cl Cl Cl 4b 4b 2 4b 3 4b 4 50 21 CH3- 75 22 CH2=CH-CH2- 80 23 C6H5CH2- 84 24 3c (CH3)2CH- NC NC NC NC N O N O N O N O Cl Cl Cl Cl Cl Cl Cl Cl 4c 1 4c 2 4c 3 4c 52 25 CH3- 73 26 CH2=CH-CH2- 79 27 C6H5CH2- 80 28 3d (CH3)2CH- NC NC NC NC 4d 1 4d 2 4d 3 N N O N O N O 4d 4 OMe OMe OMe 53 Copyright © 2011 SciRes. IJOC  Z. KIBOU ET AL. Copyright © 2011 SciRes. IJOC 246 29 CH3- 74 30 CH2=CH-CH2- 81 31 C6H5CH2- 88 32 3e (CH3)2CH- NC NC NC NC N O N O N O N O MeO MeO MeO MeO 4e 1 4e 2 4e 3 4e 4 51 33 CH3- 80 34 CH2=CH-CH2- 83 35 C6H5CH2- 88 36 3f (CH3)2CH- NC NC NC NC N O N O N O N O Me Me Me Me 4f 1 4f 2 4f 3 4f 4 49 3. Conclusions In summary, we have developed a simple, efficient and rapid method for the synthesis of 3-cyano-2-pyridones, following three steps, i.e. the Knoevenagel condensation catalyzed by NH4OAc, the enaminonitriles synthesis, and finally the synthesis of the 3-cyano-2-pyridone under sol- vent-free conditions. This procedure has the advantages of being a mild conditions reaction, using a catalytic qua- ntity of NH4OAc, with moderate to excellent yields, and where we operate with simplicity while respecting the criteria of Green Chemistry. 4. Experimental The melting points were measured using a Bank Kofler HEIZBANK apparatus standard WME 50-260˚C and were uncorrected. IR spectra were obtained with solids with a Fourier transform Perkin Elmer Spectrum One wi- th ATR accessory. Only significant absorptions are listed. The 1H NMR spectra were recorded at 400 MHz, on a Brüker AC 400 spectrometers and 13C NMR spectra were recorded in the same spectrometers at 100.6 MHz. Sam- ples were registered in CDCl3 solutions using TMS as an internal standard. The chemical shifts are expressed in units (ppm) and quoted downfield from TMS. The multi- plicities are reported as: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. General procedure 1: Synthesis of ethyl 2-cyano-3- (aryl) but- 2-enoate 2a-b A mixture of acetophenone or substituted acetophe- none (10 mmol), ethyl cyanoacetate (10 mmol), ammoni- um acetate (10 mmol) and some drops of icy acetic acid were stirred and heated at 100˚C during 3 hours. The re- action mixture was cooled down to room temperature, di- luted with 30 ml of CH2Cl2. The organic layer obtained was washed with (3 × 20 ml) of water, (10 ml) of satura- ted NaCl, dried on MgSO4, filtered then evaporated un- der vacuum. The compounds 2a-f were obtained as col- ourless oil. Ethyl 2-cyano-3-phenylbut-2-enoate 2a. The general procedure 1, using (1.20 g, 10 mmol) of acetophenone, (1.13 g, 10 mmol) of ethyl cyanoacetate, (0.77 g, 10 mmol) of ammonium acetate and some drops of icy ace- tic acid, gave 70% of 2a as burn oil. 1H NMR (400 MHz, CDCl3): 7.53 - 7.14 (m, 5H), 4.20 (q, 2H, JH-H = 7.2 Hz), 2.57 (s, 3H), 1.26 (t, 3H, JH-H = 7.2 Hz). 13C NMR (100 MHz, CDCl3): 172.21, 162.12, 136.91 - 128.10, 116.01, 104.95, 61.83, 24.50, 13.75. IR (neat, cm–1): 2225, 1747, 1682. Ethyl 3-(4-chlorophenyl)-2-cyanobut-2-enoate 2b. The general procedure 1, using (1.54 g, 10 mmol) of 4-chlo- roacetophenone, (1.13 g, 10 mmol) of ethyl cyanoacetate and (0.77 g, 10 mmol) of ammonium acetate and some drops of icy acetic acid, gave 75% of 2b as burn oil. 1H NMR (400 MHz, CDCl3): 7.51 - 7.18 (m, 4H), 4.20 (q, 2H, JH-H = 7.2 Hz), 2.62 (s, 3H), 1.29 (t, 3H, JH-H = 7.2 Hz). 13C NMR (100 MHz, CDCl3): 174.81, 164.23, 136.75 - 128.60, 115.38, 102.00, 62.10, 24.50, 24.75. IR (neat, cm–1): 2222, 1747, 1682.  247 Z. KIBOU ET AL. General procedure 2: Synthesis of ethyl 2-cyano-5- (dimethylamino)-3-arylpenta-2, 4-dienoate 3a-b A mixture of ethyl 2-cyano-3-arylbut-2-enoate (10 mmol) 2a-b, N,N-dimethylformamide dimethyl acetal (10 mmol) were stirred at room temperature without solvent during 24 hours. The solution takes a colouring increas- ingly dark burn. The purple solid obtained was washed several times with diethyl ether and crystallised in abso- lute ethanol to provide products 3a-b. Ethyl 2-cyano-5-(dimethylamino)-3-phenylpenta-2, 4-dienoate 3a. The general procedure 2, using (2.15 g, 10 mmol) of 2a and (1.19 g, 10 mmol) of N, N-dime- thylformamide dimethyl acetal, gave 85% of compound 3a as yellow solid, mp 142˚C. 1 H NMR (400 MHz, CDCl3): 7.32 - 7.25 (m, 5H), 6.57 (d, 1H, JH-H = 12.8 Hz), 5.98 (d, 1H, JH-H = 12.8 Hz), 4.32 (q, 2H, JH-H = 7.2 Hz), 3.07 (s, 3H), 3.04 (s, 3H), 1,39 (t, 3H, JH-H = 7.2 Hz), 13C NMR (100 MHz, CDCl3): 169.41, 165.56, 156.71, 137.80 - 127.70, 119.88, 99.75, 86.04, 60.07, 45.43, 37.48, 14.37. IR (neat, cm–1): 2191, 1674, 1609, 1508. Ethyl 3-(4-chlorophenyl)-2-cyano-5-(dimethy-lami- no) penta-2,4-dienoate 3b. The general procedure 2, using (2.49 g, 10 mmol) of 2b, and (1.19 g,10 mmol) of N,N-dimethylformamide dimethyl acetal, gave 90% of compound 3b as yellow solid, mp 208˚C. 1H NMR (400 MHz, CDCl3): 7.35 - 7.20 (m, 4H), 6.48 (d, 1H, JH-H = 12.8 Hz), 5,90 (d, 1H, JH-H = 12.8 Hz), 4.26 (q, 2H, JH-H = 7.2 Hz,), 3.01 (s, 3H), 2.99 (s, 3H), 1.33 (t, 3H, JH-H = 7.2 Hz). 13C NMR (100 MHz, CDCl3): 169.88, 158.38, 140.75, 137.20 - 128.75, 115.41, 107, 89.08, 60.01, 47.80, 38.77, 14.28. IR (neat, cm–1 ): 2199, 1680, 1604, 1507. General procedure 3: Synthesis of 2-Pyridones 4ai-bi A mixture of ethyl 2-cyano-5-(dimethylamino)-3-ar- ylpenta-2,4-dienoate 3a-b (2 mmol) and primary amine (2 mmol) were heated for a few hours. After cooling, the solid obtained was washed several times with diethyl ether to give 2-pyridone derivatives 4ai-bi. 1,2-dihydro-1-methyl-2-oxo-4-phenylpyridine-3-car bonitrile 4a1. The general procedure 3, using (0.43 g, 2 mmol) of 3a and (0.06 g, 2 mmol) of methylamine, gave 66% of compound 4a1 as white solid, mp 174˚C. 1 H NMR (400 MHz, CDCl3): 7.55 (d, 1H, JH-H = 6.7 Hz), 7.51 - 7.50 (m, 5H), 6.62 (d, 1H, JH-H = 6.4 Hz), 3.62 (s, 3H). 13C NMR (100 MHz, CDCl3): 159.82, 158.87, 134.46, 129.69 - 127.00, 114.57, 106.04, 101.42, 37.12. IR (neat, cm–1): 2220, 1645, 1597. 1-allyl-1,2-dihydro-2-oxo-4-phenylpyridine-3-carbo nitrile 4a2. The general procedure 3, using (0.43 g, 2 mmol) 3a and (0.11g, 2 mmol) of allylamine, gave 76% of compound 4a2 as white solid, mp 99˚C - 100˚C. 1 H NMR (400 MHz, CDCl3): 7.61 (d, 1H), 7.53 - 7.49 (m, 5H), 6.36 (d, 1H, JH-H = 6.7 Hz), 6.02 - 5.92 (m, 1H), 5.37 - 5.30 (m, 2H), 4.64-4.62 (m, 2H). 13C NMR (100 MHz, CDCl3): 160.2, 159.78, 140.41, 135.46, 131.29- 128.03, 120.29, 115.55, 107.26, 102.76, 51.26. IR (neat, cm–1): 2216, 1634, 1591. 1-benzyl-1,2-dihydro-2-oxo-4-phénylpyridine-3-car bonitrile 4a3. The general procedure 3, using (0.43 g, 2 mmol) of 3a and (0.21 g, 2 mmol) of benzylamine, gave 73% of compound 4a3 as white solid, mp 134˚C. 1 H NMR (400 MHz, CDCl3): 7.60 (d, 1H, JH-H = 7.2 Hz), 7.58 - 7.38 (m, 25H), 6.31 (d, 1H, JH-H = 7.2 Hz), 5.19 (s, 2H). 13C NMR (100 MHz, CDCl3): 160.55, 159.66, 140.44, 135.43, 134.96 - 128.71, 128.03, 115.98, 107.36, 52.69. IR (neat, cm–1): 2220, 1645, 1597. 1,2-dihydro- 1-isop ropy l-2-ox o-4 -pheny lpyridine-3- carbonitrile 4a4. The general procedure 3, using (0.43 g, 2 mmol) of 3a and of (0.11 g, 2 mmol) isopropylamine gave 48% of compound 4a4 as withe solid, mp 144˚C. 1H NMR (400 MHz, CDCl3): 7.57 (d, 1H, JH-H = 6.8 Hz), 7.49 - 7.50 (m, 5H), 6.38 (d,1H, JH-H = 6.8 Hz), 5.24 - 5.31 (m, 1H), 1.42 (d, 6H, JH-H = 7.2 Hz). 13C NMR (100 MHz, CDCl3): 160.274, 158.80, 136.85, 135.56 - 127.57, 115.86, 107.01, 102.34, 47.68, 21.78. IR (neat, cm–1): 2219, 1640, 1592, 1517. 4-(4-chlorophenyl)-1-methyl-2-oxo-1,2-dihydropyri dine-3-carbonitrile 4b1. The general procedure 3, using (0.60 g , 2 mmol) of 3b and (0.06 g, 2 mmol) of methy- lamine, gave 76% of compound 4b1 as white solid, mp 184˚C. 1H NMR (400 MHz, CDCl3): 7.55 (d, 1H, JH-H = 6.7 Hz), 7.54 - 7.47 (m, 4H), 6.30 (d,1H, JH-H = 6.4 Hz), 3.64 (s, 3H). 13C NMR (100 MHz, CDCl3): 162.12, 157.66, 135.56, 127.33 - 129.88, 115.08, 107.80, 103.34, 34.20. IR (neat, cm–1): 2221, 1644, 1599. 1-allyl-4-(4-chlorophenyl) -2-oxo -1,2- dihydro pyridi ne-3-carbonitrile 4b2. The general procedure 3, using (0.60 g, 2 mmol) of 3b and (0.11 g, 2 mmol) of allylamine, gave 80% of compound 4b2 as white solid, mp 172˚C. 1H NMR (400 MHz, CDCl3): 7.62 (d, 1H), 7.56 - 7.41 (m, 4H), 6.44 (d, 1H, JH-H = 6.7 Hz), 5.93 - 5.58 (m, 1H), 5.34 - 5.28 (m, 2H), 4.76 - 4.68 (m, 2H). 13C NMR (100 MHz, CDCl3): 162.11, 160.22, 143.11, 136.80, 134.2 - 126.50, 122.18, 115.9, 107.80, 103.11, 48.02. IR (neat, cm–1): 2216, 1637, 1595. 4-(4-chlorophenyl)-1-ethyl-2-oxo-1,2-dihydropyridi ne-3-carbonitrile 4b3. The general procedure 3, using (0.60 g, 2 mmol) of 3b and (0.21 g, 2 mmol) of ben- zylamine gave 85% of compound 4b3 as white solid, mp 214˚C. 1H NMR (400 MHz, CDCl3): 7.62 (d, 1H), 7.49- 7.30 (m, 4H), 7.29 - 7.19 (m, 5H), 6.20 (d, 1H), 5.12 (s, 2H). 13C NMR (100 MHz, CDCl3): 167.98, 156.50, 136.67, 135.10, 130.29 - 128.27, 119.84, 100.70, 45.63. IR (neat, cm–1): 2219, 1656, 1597. 4-(4-chlorophenyl)-1-isopropyl-2-oxo-1,2-dihydrop yridine-3-carbonitrile 4b4. The general procedure 3, Copyright © 2011 SciRes. IJOC  Z. KIBOU ET AL. 248 using (0.60g , 2 mmol) of 3b and (0.11 g, 2 mmol) of isopropylamine, gave 50% of compound 4b4 as white solid, mp 142˚C. 1H NMR (400 MHz, CDCl3): 7.52 (d, 1H, JH-H = 6.8 Hz), 7.50 - 7.40 (m, 4H), 6.27 (d, 1H, JH-H = 6.8 Hz), 5.24 - 5.17 (m, 1H), 1.29 (d, 6H, JH-H = 7.2 Hz). 13C NMR (100 MHz, CDCl3): 163.32, 159.18, 134.88, 136.42 - 127.80, 115.9, 106.08, 101.54, 48.78, 21.26. IR (neat, cm–1): 2221, 1648, 1599, 1512. 5. References [1] D. Blasco-Jiménez, A. J. Lopez-Peinado, R. M. Martin- Aranda, M. Ziolek and I. Sobczak, “Sonocatalysis in Solvent-Free Conditions: An Efficient Eco-Friendly Me- thodology to Prepare N-Alkyl Imidazoles Using Amino- Grafted NbMCM-41,” Catalysis Today, Vol. 142, No. 3-4, 2009, pp. 283-287. doi:10.1016/j.cattod.2008.11.028 [2] D. Kumar, V. B. Reddy, B. G. Mishra, R. K. Ranna, M. N. Nadagouda and R. S. Varma, “Nanosized Magnesium Oxide as Catalyst for the Rapid and Green Synthesis of Substituted 2-Amino-2-Chromenes,” Tetrahedron, Vol. 63, No. 15, 2007, pp. 3093-3097. doi:10.1016/j.tet.2007.02.019 [3] L. F. Teitze, “Domino Reactions in Organic Synthesis,” Chemical Reviews, Vol. 96, No. 1, 1996, pp. 115-136. doi:10.1021/cr950027e [4] R. Maggi, R. Ballini and G. Sartori, “Basic Alumina Catalysed Synthesis of Substituted 2-Amino-2-chromenes via Three-Component Reaction,” Tetrahedron Letters, Vol. 45, No. 11, 2004, pp.2297-2299. doi:10.1016/j.tetlet.2004.01.115 [5] J. A. Wang, X. Bokhimi, O. Novaro, T. Lopez, F. Tzom- pantzi, R. Gomez, J. Navarrete, M. E. Llanos and M. Lo- pez-Salinas, “Effects of Structural Defects and Acid-Ba- sic Properties on the Activity and Selectivity of Isopro- panol Decomposition on Nanocrystallite Sol-Gel Alu- mina Catalyst,” Journal of Molecular Catalysis A: Ch- emical, Vol. 137, No. 1-3, 1999, pp. 239-256. doi:10.1016/S1381-1169(98)00077-6 [6] S. Paul, P. Nanda, R. Gupta and A. Loupy, “Ac2O-Py/ Basic Alumina as a Versatile Reagent for Acetylations in Solvent-Free Conditions under Microwave Irradiation,” Tetrahedron Letters, Vol. 43, No. 23, 2002, pp. 4261- 4265. doi:10.1016/S0040-4039(02)00732-3 [7] C. M. Figueiredo, “Preparation of Aluminas from a Basic Aluminium Carbonate,” Catalysis Today, Vol. 5, 1989, pp. 433-442. doi:10.1016/0920-5861(89)80007-0 [8] S. Carre, B. Tapin, N. S. Gnep, R. Revel and P. Magnoux, “Model Reactions as Probe of the Acid-Base Properties of Aluminas: Nature and Strength of Active sites. Corre- lation with Physicochemical Characterization,” Applied Catalysis A: General, Vol. 372, No. 1, 2010, pp. 26-33. doi:10.1016/j.apcata.2009.10.005 [9] M. T. Cocco, C. Congiu and V. Onnis, “Synthesis and Antitumor Activity of 2-Hydroxy-2-pyridones Deriva- tives,” European Journal of Medicinal Chemistry, Vol. 35, No. 5, 2000, pp. 545-552. doi:10.1016/S0223-5234(00)00149-5 [10] F. Manna, F. Chimenti, A. Bolasco, A. Filippelli and E. Lampa, “Antiinflammatory, Analgesic and Antipyfuztic 4,6-Disubstituted 3-Cyanopyridine-2-ones and 3-Cyano- 2-aminopyridines,” Pharmacological Research, Vol. 26, Suppl. 1, 1992, pp. 267-277. doi:10.1016/1043-6618(92)91243-A [11] R. L. Parreira, O. Abrahão and S. E. Galembeck, “Con- formational Preferences of Non-Nucleoside HIV-1 Re- verse Transcriptase Inhibitors,” Tetrahedron, Vol. 57, No. 16, 2001, pp. 3243-3253. doi:10.1016/S0040-4020(01)00193-4 [12] X. Fan, D. Feng, Y. Qu, X. Zhang, J. Wang, P. M. Loiseau, G. Andrei, R. Snoeck, E. De Clercq, “Practical and Efficient Synthesis of Pyrano[3,2-c]pyridone, Pyrano [4,3-b]pyran and Their Hybrids with Nucleoside as Po- tential Antiviraland Antileishmanial Agents,” Bioorganic & Medicinal Chemistry Letters, Vol. 20, No. 3, 2010, pp. 809-813. doi:10.1016/j.bmcl.2009.12.102 [13] E. L. Presti, R. Boggia, A. Feltrin, G. Menozzi, P. Dorigo and L. Mosti, “3-Acetyl-5-acylpyridin-2(1H)-ones and 3- Acetyl-7,8-dihydro-2,5(1H,6H)-quinolinediones: Synthe- sis, Cardiotonic Activity and Computational Studies,” II Farmaco, Vol. 54, No. 7, 1999, pp. 465-447. doi:10.1016/S0014-827X(99)00053-1 [14] W. K. Anderson, D. C. Dean and T. Endo, “Synthesis, Chemistry, and Antineoplastic Activity of α-Halopyridi- nium Salts: Potential Pyridone Prodrugs of Acylated Vi- nylogous Carbinolamine Tumor Inhibitors,” Journal of Medicinal Chemistry, Vol. 33, No. 6, 1990, pp. 1667- 1675. doi:10.1021/jm00168a021 [15] P. S. Dragovich, T. J Prins, Z. Ru, E. L. Brown, F. C. Maldonado, S. A. Fuhrman, L. S. Zalman, L. Iuniland, C. A. Lee and S. T. Worland, “Structure-Based design, Synthesis, and Biological Evaluation of Irreversible Hu- man Rhinovirus 3C Protease Inhibitors. 6. Struc-ture- Activity Studies of Orally Bioavailable, 2-Pyridone- Con- taining Peptidomimetics,” Journal of Medicinal Chemis- try, Vol. 45, No. 8, 2002, pp. 1607-1623. doi:10.1021/jm010469k [16] B. Kozlevcar, M. Radisek, Z. Jaglicic, F. Merzel, L. Glazar, A. Golobic and P. Segedin, “Strong Antiferro- magnetism in the Dinuclear 2-Pyridone Complex with N-C-O Bridges: A Paddle-Wheel Analogue of the Dinu- clear Tetracarboxylates,” Polyhedron, Vol. 26, 2007, pp. 5414-5419. doi:10.1016/j.poly.2007.08.019 [17] T. Mochida, M. Ueda, C. Aoki and H. Mori, “Structures and Properties of Trans-Dichloro{Tetrakis (5-Chloro-2 (1H)-pyridone-O)}M(II) [M=Mn, Fe, Co, Ni, Cu]; For- mation of Quasi-Macrocyclic Metal Complexes through Hydrogen Bonding,” Inorganica Chimica Acta, Vol. 335, No. 27, 2002, pp. 151-155. doi:10.1016/S0020-1693(02)00817-4 [18] K. R. Gibson, L. Hitzel, R. J. Mortishire-Smith, U. Gerhard, R.A. Jelley, A. J. Reeve, M. Rowley, A. Nadin and A. P. Owens, “Synthesis and Conformational Dy- namics of Tricyclic Pyridones Containing a Fused Seven- Copyright © 2011 SciRes. IJOC  249 Z. KIBOU ET AL. Membered Ring,” Journal of Organic Chemistry, Vol. 67, No. 26, 2002, pp. 9354-9360. doi:10.1021/jo026411a [19] I. Collins, C. Moyes, et al., “3-Heteroaryl-2-pyridones: Benzodiazepine Site Ligands with Functional Delectivity for Alpha 2/Alpha 3-Subtypes of Human GABA (A) Re- ceptor-Ion Channels,” Journal of Medicinal Chemistry, Vol. 45, No. 9, 2002, pp. 1887-1990. doi:10.1021/jm0110789 [20] F. A. Abu-Shanab, A. D. Redhouse, J. R. Thompson and B. J. Wakefield, “Synthesis of 2,3,5,6-Tetrasubtituted Pyridines from Enamines Derived from N, N-Dimethyl- formamide Dimethyl Acetal,” Synthesis, Vol. 5, 1995, pp. 557-560. doi:10.1055/s-1995-3954 [21] W. D. Jones, R. A. Schnettler and E. W. Huber, “A Convenient Synthesis of 5-Acyl-6-substituted 3-cyano-2 (1H)-pyridinones,” Journal of Heterocyclic Chemistry, Vol. 27, No. 3, 1990, pp. 511-518. doi:10.1002/jhet.5570270307 [22] H. Fukatsu, Y. Kato, S. Murase and S. Nakagawa, “Synthesis and Cardiotonic Activity of 5-(2-Substituted thiazol-4-yl)-2-pyridones and Thiazolo[4,5-f] quinolino- nes,” Heterocycles , Vol. 29, No. 8, 1989, pp. 1517-1528. doi:10.3987/COM-89-4984 [23] I. Sircar, B. L. Duell, J. A. Bristol, R. E. Weishaar and D. B. Evans, “Cardiotonic Agents. 5. 1,2-Dihydro-5-[4- (1Himidazol-1-yl)phenyl]-6-methyl-2-oxo-3-pyridinecar- bonitriles and Related Compounds. Synthesis and Ino- tropic Activity,” Journal of Medicinal Chemistry, Vol. 30. No. 6, 1987, pp. 1023-1029. doi:10.1021/jm00389a011 [24] D. W. Robertson, E. E. Beedle, J. K. Swartzendruber, N. D. Jones, T. K. Elzey, R. F. Kauffman, H. Wilson and J. S. Hayes, “Bipyridine Cardiotonics: The Three-Dimen- sional Structures of Amrinone and Milrinone,” Journal of Medicinal Chemistry, Vol. 29, No. 5, 1986, pp. 635-640. doi:10.1021/jm00155a009 [25] J. J. Chen and I. J. Wang, “Synthesis and Colour Assess- ment of Some 3-Cyano-4-pyrenyl-6-substituted-2-pyri- done Derivatives,” Dyes and Pigments, Vol. 29, No. 4, 1995, pp. 305-313. doi:10.1016/0143-7208(95)00055-0 [26] A. H. Abadi, T. M. Ibrahim, K. M. Abouzid, J. Lehmann, H. N. Tinsley, B. D. Gary and G. A. Piazza, “Design, Synthesis and Biological Evaluation of Novel Pyridine Derivatives as Anticancer Agents and Phosphodiesterase 3 Inhibitors,” Bioorganic & Medicinal Chemistry, Vol. 17, No. 16, 2009, pp. 5974-5982. doi:10.1016/j.bmc.2009.06.063 [27] D. Villemin, B. Mostefa-Kara, N. Bar, N. Choukchou- Braham, N. Cheikh, A. Benmeddah, H. Hazimeh and C. Ziani-Cherif, “Base Promoted Reaction in Ionic Liquid Solvent: Synthesis of Butenolides,” Letters in Organic Chemitry, Vol. 3, 2006, pp. 558-559. doi:10.2174/157017806778341807 [28] D. Villemin, N. Cheikh, B.Mostefa-Kara, N. Bar, N. Choukchou-Braham and M. A. Didi, “Solvent-Free Re- action on KF-Alumina under Microwave: Serendipitous One-Pot Domino Synthesis of New Isobenzofuran-1(3H) -ones from Alpha-Hydroxyketones,” Tetrahedron Letters, Vol. 47, No. 31, 2006, pp. 5519-5521. doi:10.1016/j.tetlet.2006.05.137 [29] N. Cheikh, N. Bar, et al., “Efficient Synthesis of New Butenolides by Subsequent Reactions: Application for the Synthesis of Original Iminolactones, Bis-Iminolactones and Bis-Lactones,” Tetrahedron, Vol. 67, No. 8, 2011, pp. 1540-1557. doi:10.1016/j.tet.2010.12.062 [30] Q. Shi, J. Chen, Q. Zhunag and X. Wang, “The Conden- sation of Aromatic Aldehydes with Acidic Methylene Compounds in Water,” Chinese Chemical Letters, Vol. 14, 2003, pp. 1242-1245. [31] T. B. FranColse and F. Andre, “Knoevenagel Condensa- tion Catalysed by Aluminium Oxide,” Tetrahedron Let- ters, Vol. 23, No. 47, 1982, pp. 4927-4928. doi:10.1016/S0040-4039(00)85749-4 [32] P. de la Cruz, E. Diez-Barra, A. Loupy and F. Langa, “Silica Gel Catalysed Knoevenagel Condensation in Dry Media under Microwave Irradiation,” Tetrahedron Let- ters, Vol. 37, 1996, pp. 1113-1116. doi:10.1016/0040-4039(95)02318-6 [33] G.-H. Gao, L. Lu, et al., “Basic Ionic Liquid: A Reusable Catalyst for Knoevenagel Condensation in Aqueous Me- dia,” Chemical Research in Chinese Universities, Vol. 23, No. 2, 2007, pp. 169-172. doi:10.1016/S1005-9040(07)60035-X [34] B. Siebenhaar, B. Casagrande, M. Studer and H. U. Blaser, “An Easy-to-Use Heterogeneous Catalyst for the Knoevenagel Condensation,” Canadian Journal of Ch- emistry, Vol. 79, No. 5-6, 2001, pp. 566-569. doi:10.1139/v01-072 [35] M. L. Kantam and B. Bharathi, “Mn(III) Salen Catalyst for Knoevenagel Condensation a Novel Heterogeneous System,” Catalysis Letters, Vol. 55, No. 3-4, 1998, pp. 235-237. doi:10.1023/A:1019051416463 [36] S. Balalaie and N. Nemati, “Ammonium Acetate-Basic Alumina Catalyzed Knoevenagel Condensation under Microwave Irradiation under Solvent-Free Conditions,” Synthetic Communications, Vol. 30, No. 5, 2000, pp. 869- 875. doi:10.1080/00397910008087099 [37] A. McCluskey, P. J. Robinson and T. Hill, “Green Che- mistry Approaches to the Knoevenagel Condensation: Comparison of Ethanol, Water and Solvent Free (Dry Grind) Approaches,” Tetrahedron Letters, Vol. 43, No. 17, 2002, pp. 3117-3120. doi:10.1016/S0040-4039(02)00480-X [38] F. Freeman, “Properties and Reactions of Ylidenema- lononitriles,” Chemical Reviews, Vol. 80, No. 4, 1980, pp. 329-350. doi:10.1021/cr60326a004 [39] A. W. Erian, S. M. Sherif, A. Alassar and Y. M. Elkholy, “β-Enaminonitriles in Heterocyclic Synthesis: A Novel Synthesis and Transformations of α-Substituted-β-enam- inonitriles,” Tetrahedron, Vol. 50, No. 6, 1994, pp. 1877- 1884. doi:10.1016/S0040-4020(01)80859-0 [40] A. W. Erian, “The Chemistry of β-Enamino Nitriles as Versatile Reagents in Heterocyclic Synthesis,” Chemical Reviews, Vol. 93, No. 6, 1993, pp. 1991-2005. doi:10.1021/cr00022a002 Copyright © 2011 SciRes. IJOC
|