Paper Menu >>
Journal Menu >>
 J. Biomedical Science and Engineering, 2010, 3, 340-350 JBiSE doi:10.4236/jbise.2010.34047 Published Online April 2010 (http://www.SciRP.org/journal/jbise/). Published Online April 2010 in SciRes. http://www.scirp.org/journal/jbise Characterization of the sequence spectrum of DNA based on the appearance frequency of the nucleotide sequences of the genome ——A new method for analysis of genome structur e Masatoshi Nakahara1, Masaharu Takeda2 1Department of Computer and Information Sciences, Sojo University, Ikeda, Japan; 2Department of Materials and Biological Engineering, Tsuruoka National College of Technology , Tsuruoka, Japan. Email: mtakeda@tsuruoka-nct.ac.jp Received 13 January 2010; revised 25 January 2010; accepted 30 January 2010. ABSTRACT The nucleotide (base) sequence of the genome might reflect biological information beyond the coding se- quences. The appearance frequencies of successive base sequences (key sequences) were calculated for entire genomes. Based on the appearance frequency of the key sequences of the genome, any DNA sequences on the genome could be expressed as a sequence spec- trum with the adjoining base sequences, which could be used to study the corresponding biological phe- nomena. In this paper, we used 64 successive three- base sequences (triplets) as the key sequences, and determined and compared the spectra of specific genes to the chromosome, or specific genes to tRNA genes in Saccharomyces cerevisiae, Schizosaccharomyces pombe and Escherichia coli. Based on these analyses, a gene and its corresponding position on the chromosome showed highly similar spectra with the same fold enlargement (approximately 400-fold) in the S. cere- visiae, S. pombe and E. coli genomes. In addition, the homologous structure of genes that encode proteins was also observed with appropriate tRNA gene(s) in the genome. This analytical method might faithfully reflect the encoded biological information, that is, the conservation of the base sequences was to make sense the conservation of the translated amino acids se- quence in the coding region, and might be universally applicable to other genomes, even those that consisted of multiple chromosomes. Keywords: Appearance Frequency of Triplet in Genome Base Sequence; Self-Similarity; Analytical Method of Genome Structure 1. INTRODUCTION It was well known that there were structural hierarchies in the genome, such as the chromosome, nucleosome, ORF (open reading frame) and so on [1]. Among them, much attention have been paid to the ORF, and many research projects were being performed from the view- point of protein function using methods such as pro- teome and transcriptome analyses [2-5]. Many studies of entire genome sequences have been reported [6-11], al- though complete genome base sequences have only been revealed within the last 10 years or so. However, we currently have limited tools to analyze a large-scale molecule such as a whole genome, including pertinent hard-and software. It was very important to investigate the structural features of the entire genome because the four bases could be arranged in a sophisticated fashion in the genome, and in principle the base sequences might be reflected in the conformations of protein, RNA and DNA. In other words, if we could identify a meaningful structure, or an analytical method for analysis of the ge- nome, we could also obtain important information about the functions of protein, RNA and DNA from that struc- ture. The four bases in genomic DNA were arranged sophisticatedly in all organisms and distinguish the cod- ing-and the non-coding region clearly on the genome. By analyzing the appearance frequency of the bases, it was shown that first, the symmetry [8-11], second, the bias [12-15] and third, the fractality [16-19] could b e n e c- essary to generate genome base sequences. We analyzed genome structure based on the appearance frequency of genome base sequences [20]. We have studied many ge- nome sequences down-loaded from databases like NCBI [21], and calculated the appearance frequencies of an optional base sequence (key sequence) in a genome. Subsequently, we determined the sequence spectra of chromosome, gene and DNA from the key sequence of the genome (chromosome), and analyzed both the cod- ing-and non-coding sequences because the key se-  M. Nakahara et al. / J. Biomedical Science and Engineering 3 (2010) 340-350 Copyright © 2010 SciRes. JBiSE 341 quences were used throughout the genome in cells. However, in the coding regions in the DNA, the appear- ance frequencies of the key sequences of an individual gene should vary in the genome because the pro- tein-encoding gene and the adjoining (5’- and 3’-) non-coding base sequences were different. In other words, the appearance frequencies of the base sequences should be different for each gene. Even if the base se- quences of the gene were identical, the adjoining base sequences differ, suggesting that each DNA sequence might have an effect on the expression of the gene and function as an informative DNA molecule [20]. Each gene was transcribed to mRNA, and translated to a protein on the ribosome (polyribosome) according to the DNA sequence of each coding region. In other words, the biological information of DNA (base sequence) should be transferred to protein via mRNA (base se- quence). That is, the information of the base sequence of DNA was transformed to the amino acid sequence by tRNAs corresponding to the base sequences of the mRNA on the ribosome [22]. However, the coding regions varied in individual ge- nomes and species [23,24]. The non-coding sequences might be necessary to precisely, rapidly, and consistently regulate gene expression [24,25]. In other words, the genome might be a “field” on which the four bases were sophisticatedly arranged into genes that were regulated and expressed to carry out the biological phenomena of life. Therefore, analytical methods to characterize ge- nome structure were needed to understand the encoded biologic al phenom e na . In this study, we developed an analytical method based on the frequencies of the nucleotide (base) se- quences in the whole genome according to the flow of biological information, and focused on the self-similarity in the genomes of S. cerevisiae and S. pombe, where most of the genes had introns, and E. coli, in which most of genes were in operons. 2. MATERIALS AND METHODS 2.1. Sequence Spectrum Method (SSM) The outline of the proposed method was as follows. The base sequence of interest was sectioned by a small num- ber of bases from the top (5’-end). The sectioned base sequence was called the key sequence. In the case of three successive base sequences (d = 3), the appearance frequencies of the 64 triplets (the genetic codon) were shown in Ta b l e 1 (key sequence at d = 3). The key se- quences of the nine successive base sequences (d = 9) was 262,144 sequences (= 49, ref. 20). The appearance frequency of the key sequence was counted in the entire genome, and was plotted at the position of the first base of the key sequence as described in the next pa ragraph of the Materials and Methods. These procedures were car- ried out for the entire base sequence of interest with one base shift (p = 1). The next step was to average the ap- pearance frequencies so that a recognizable pattern of appearance frequency was obtained for the base se- quence. This pattern of the averaged appearance fre- quency was called the “sequence spectrum”. Finally, the homology factor between two sequence spectra was cal- culated to determine the degree of homology. The exact procedure was explained below in a mathematic al way. Let S be an entire set of base sequences, and B = [bi] be a partial set of interest in S. A base element was de- noted by bi (I=1, ... , M), and M was the base sequence size of B. The base element bi become A (adenine), T (thymine), G (guanine) or C (cytosine). The key se- quence ki and the appearance frequency fi were defined for bi as follows. Key sequence ki: base sequence comprised of sequen- tial base elements bi~bi+d-1 (d : base size of the key se- quence) Appearance frequency fi: appearance count of ki in S The key sequence ki was compared with the base se- quence of the entire set S, and the appearance frequency fi was increased by one every time the key sequence ki matches the partial base sequence of the entire set S. This procedure was iterated for all key sequences ki to obtain fi (I = 1, ... , M). Consequently, the appearance frequency vector F = [fi] (I = 1, ... , M) was determined (actually, the appearance frequencies for the last (d-1) base elements of B could not be calculated; however, this was neglected because M >> d-1). Next, the appearance frequency fi was averaged as fol- lows: 1 21 im s ij jim f f m where the parameter m was average width. This aver- aged appearance frequency Fs = [fsi] (I = 1, … , M) was called the “sequence spectrum”. The next step was to calculate the homology factor to determine the degree of homology. The homology factor determines the homologous region of a target base se- quence with respect to a reference base sequence. In order to derive the homology factor, the mutual correla- tion function MF was calculated as 1 1 1 1 1 1 (,)( )*() ()*() ()*() 1 1 Mr kiikk i k Mr ii i Mr kikkikk i Mr i i Mr kik i M FFsr Fstfsrfsrfstfst Fsr Fst Fsrfsr fsrfsr fsr Fstfst fstfstfst fsr fsr Mr fst fst Mr  M. Nakahara et al. / J. Biomedical Science and Engineering 3 (2010) 340-350 Copyright © 2010 SciRes. JBiSE 342 Table 1. Key sequences of the three succssive base sequences*1 in genome*2. Triplet Frequency Triplet Frequency Triplet Frequency Triplet Frequency (a) S. cerevisiae AAA 478,677 AAT 359,378 AAG 263,401 AAC 219,288 ATA 302,770 ATT 358,051 ATG 221,867 ATC 214,197 AGA 246,395 AGT 184,087 AGG 138,976 AGC 139,262 ACA 208,942 ACT 183,292 ACG 106,020 ACC 141,084 TAA 271,996 TAT 301,699 TAG 156,650 TAC 172,399 TTA 271,724 TTT 475,621 TTG 279,349 TTC 286,655 TGA 244,596 TGT 207,422 TGG 179,858 TGC 150,406 TCA 245,024 TCT 244,505 TCG 110,351 TCC 154,145 GAA 288,804 GAT 213,000 GAG 136,067 GAC 118,074 GTA 172,583 GTT 218,208 GTG 128,946 GTC 117,316 GGA 154,364 GGT 139,691 GGG 81,268 GGC 95,122 GCA 150,888 GCT 139,012 GCG 67,875 GCC 95,478 CAA 281,266 CAT 222,808 CAG 152,602 CAC 129,575 CTA 155,668 CTT 261,471 CTG 152,121 CTC 135,857 CGA 110,589 CGT 105,859 CGG 70,348 CGC 68,463 CCA 181,394 CCT 138,308 CCG 71,012 CCC 82,880 ( b ) S. p ombe AAA 569,684 AAT 409,666 AAG 277,238 AAC 234,759 ATA 310,191 ATT 409,111 ATG 227,572 ATC 207,984 AGA 225,118 AGT 196,340 AGG 128,892 AGC 158,220 ACA 212,145 ACT 193,959 ACG 110,332 ACC 123,580 TAA 334,648 TAT 310,127 TAG 162,059 TAC 183,503 TTA 334,208 TTT 572,331 TTG 296,280 TTC 292,897 TGA 244,964 TGT 213,557 TGG 156,002 TGC 157,500 TCA 245,161 TCT 227,278 TCG 123,339 TCC 149,364 GAA 291,250 GAT 207,564 GAG 134,381 GAC 108,437 GTA 185,292 GTT 236,486 GTG 113,029 GTC 109,314 GGA 148,699 GGT 123,656 GGG 67,242 GGC 75,049 GCA 157,454 GCT 157,621 GCG 64,622 GCC 75,416 CAA 295,764 CAT 227,501 CAG 134,892 CAC 113,317 CTA 160,646 CTT 277,788 CTG 135,142 CTC 134,949 CGA 122,848 CGT 110,569 CGG 62,511 CGC 64,344 CCA 156,714 CCT 129,667 CCG 61,979 CCC 67,351 ( c ) E. coli AAA 108,901 AAT 82,992 AAG 63,364 AAC 82,578 ATA 63,692 ATT 83,395 ATG 76,229 ATC 86,476 AGA 56,618 AGT 49,774 AGG 50,611 AGC 80,848 ACA 58,633 ACT 49,863 ACG 73,263 ACC 74,899 TAA 68,837 TAT 63,279 TAG 27,241 TAC 52,591 TTA 68,824 TTT 109,825 TTG 76,968 TTC 83,846 TGA 83,483 TGT 58,369 TGG 85,132 TGC 95,221 TCA 84,033 TCT 55,469 TCG 71,733 TCC 56,025 GAA 83,490 GAT 86,547 GAG 42,460 GAC 54,737 GTA 52,670 GTT 82,590 GTG 66,108 GTC 54,225 GGA 56,199 GGT 74,291 GGG 47,470 GGC 92,123 GCA 96,010 GCT 80,285 GCG 114,609 GCC 92,961 CAA 76,607 CAT 76,974 CAG 104,785 CAC 66,752 CTA 26,762 CTT 63,653 CTG 102,900 CTC 42,714 CGA 70,934 CGT 73,159 CGG 86,870 CGC 115,673 CCA 86,442 CCT 50,412 CCG 87,031 CCC 47,764 *1; 5'- to 3'-end correspond to the left to the right letter of each triplet. *2; S. cerevisiae g e nome is composed of 16 c hromosomes pl us mtDNA. S. pombe genome is composed of 3 chromosomes plus mtDNA.  M. Nakahara et al. / J. Biomedical Science and Engineering 3 (2010) 340-350 Copyright © 2010 SciRes. JBiSE 343 where Fsr: sequence spectrum of the reference base sequence with base size Mr Fst: sequence spectrum of the target base sequence with base size Mt (> Mr) The mutual correlation function MF ranges from –1 to 1, and then the homology factor HF was defined as 1 ,*100 % 2 k k MF HFFsr Fst The higher the homology factor, the more homologous the sequence spectra were. The homologous regions of the target base sequence with respect to the reference base sequence were obtained by calculating the homol- ogy factors HFk for all k (k = 0, ... , Mt-Mr), and target- ing the regions with hi g he r ho mology factors . When the target base sequence was very large, ele- ments of the target sequence spectrum were skipped by the size factor p to reduce the size as follows. 1* 1 iip fst fst For instance, when p = 2 123 135 ,, ,, f st fstfstfst fstfst This operation reduced the size to 1/p. The base sequences of the genomes were obtained from the databases listed below. Saccharomyces cerevisiae: http://www.mips.biochem.mpg.de/ Schizosaccharomyces pombe: http://www.sanger.ac.uk / Escherichia coli: http://bmb.med.miami.ed u/Eco gene/ecoWeb / 2.2. Appearance Frequencies of Bases or Base Sequences. In order to analyze the structure of the base sequence, the most appropriate parameter was considered to be the appearance frequency. For three successive bases (trip- lets), the appearance frequency was counted for the en- tire genome by matching the triplet from the start of the base sequence in a genome with one base shift (p = 1) as follows. Ex. T riplet bases: AAT AAT (one base shift) BaseSequence: 5’-ATCGAATCCGTAATTCGGAGTCGAATT-3’ Count of AAT: 1 2 3 3. RESULTS 3.1. Sequence Spectrum Figure 1 showed the sequen ce spectrum of the F1F0-ATPase subunit gene ATP1 [26, YBL099W] in Saccharomyces cerevisiae. In this figure, the vertical parameter of the sequence spectrum fsi was not designated, and it was scaled properly because the shape of the sequence spec- trum only makes sense in this manuscript. The horizontal parameter was the base sequence number i (I=1, ... , M), and it was also omitted in the following figures because it was easily derived from the base sequence size M. Controllable parameters in the sequence spectrum were the base size d of the key sequence, the average width m, and the size factor p (skipped base numbers). The parameter d determines the highest resolution for extracting the structural features of the base sequence. In this report, we used the key sequence as d = 3 (appear- ance frequency table of triplet, Table 1) for numerical experiments of the homologous structure discussed in the following sections. However, as shown in Figure 1, smaller m-values caused a harder zigzag pattern of the sequence spectrum, and eventually it become more difficult to identify the structure of the base sequence (Figure 1(a)). Therefore, large m-values were usually used to obtain the overall features of the structure, and smaller m-values were ap- plied to investigate the structure in detail (Figure 1(b)). The value of m normally ranges from 1/10 to 1/100 of the base sequence size. In this manuscript, m = 2 for a tRNA, m = 60 for a gene, and m = 8,000 for a chromo- some. The size factor p was adjusted to the base se- quence size especially when the homology factor between a small reference and a large target was calculated. The possible appearance frequencies fi of ke y sequences ki were calculated for the entire set S in advance. The ap- pearance frequency table depended on the entire set S, and in general S was the genome of the t arget spec i es . 3.2. Reverse-Complement Symmetry in the Appearance Frequency Table Table 1 showed the appearance frequencies (3 succes- sive base sequences = triplet, d = 3) of the key sequence for Saccharomyces cerevisiae (a), Schizosaccharomyces pombe (b), and Escherichia coli (c). This table gave some important features about the genome. In the case of S. cerevisiae, first, it was notable that the appearance frequencies of the key sequence and its reverse- complementary key sequence were almost the same. The reverse-complement key sequence was derived from reversing the base order of the original key sequence in S. cerevisiae, exchanging A and T, and exchanging G and C. For example, the appearance frequency of 5’-ATT is 358,051 an d that of 5’-A AT was 359,378. The difference was less than 1%. The largest difference was about 2% for 5’-GGG (81,268) and 5’-CCC (82,880). This fact is valid regardless of the species, such as Escherichia coli (Table 1(b)) or Schizosaccharomyces pombe (Table 1(c)). This reverse-complement symmetry led to the fact that the numbers of A and T were almost equal, and the number s of G and C we r e almost equ a l.  M. Nakahara et al. / J. Biomedical Science and Engineering 3 (2010) 340-350 Copyright © 2010 SciRes. JBiSE 344 (a) ATP1, m = 0 (b) ATP1, m = 60 Figure 1. Sequence spectrum of ATP1. Sequence spectra of AT P1 [26-28] from Saccharomyces cerevisiae with different average widths (a) m = 0, and (b) m = 60. The vertical parame- ter (appearance frequency of the triplet, d = 3) of the sequence spectrum is not designated, and it is scaled properly. The hori- zontal axis is the base sequence of AT P1 (1,638 nt designated as M, ATG = start codon – TAA = stop codon). The skipped base numbers (p) are shown in the figures. The zigzag motif becomes more moderate and the resolution becomes lower as the average width of m becomes larger. Generally it was well known that the numbers of A and T and the numbers of G and C were the same due to the double helix structure of DNA. However, in this case, this coincidence of base numbers occurred in the genome, so it had nothing to do with the double helix structure. Therefore, the coincidence of base numbers occurred when the base sequence size was very large even in a sin- gle strand. Actually this reverse-complement symmetry occurred in each chromosome as well. On the other hand, it did not occur when the base se- quence size was not large enough. For instance, the base sequence size of a single gene was not adequate. The fact that the appearance frequencies of the key sequence and its reverse-complementary key sequence were almost equal implies that there must be a certain amount of symmetry in the genome. Second, the appearance frequency (in parentheses) for each key sequence was not random, but some of the key sequences had very close appearance frequencies even when they did not have a complementary relationship. For example, in the case of S. cerevisiae, the key se- quences 5’-AAC (219,288), 5’-ATC (214,197) and 5’-ACA (208,942) had close appearance frequencies of about 210,000, and those of the key sequences 5’-ACG (106,020), 5’-CGA (110,589) and 5’-GAC (110,874) were about 110,000. These different key sequences with close appearance frequencies might have a similar effect on the sequence spectrum. In other words, sin- gle-stranded DNA with base-symmetry might be able to make many double-helical stems in a molecule, and the peaks of the sequence spectrum, the “up” of the dou- ble-helical stem might have the same effect on the “down” of it. Needless to say, these facts were valid re- gardless of the species. 3.3. Homologous Structure in Genomes (Enlargement-Reduction of the base Sequence) ATP1 (YBL099W) of S. cerevisiae was present on the left arm of chromosome II (37,045-38,679 from the left telomere). Figure 2 showed the spectra of ATP1 (1,638 nt, Figure 1(b)), and (a) chromosome II (813,139 nt), respectively. The red arrowhead indicated the position of ATP1 on chromoso me II [27, 28]. When the spectrum of ATP1 (1,638 nt) was skipped 3 bases and the homology analyzed between chromosome II and the skipped-ATP1, the red-region (20,401 ~ 60,401 = 40,000 nt) of chromo- some II was homologous to the 3 bases-skipped-ATP1 (1,341 ~ 1,638 = 297 nt) (Figure 2(b), HF of the red-region of chromosome II to the purple-region of ATP1 = 95%). When ATP1 was skipped 10, or 16 bases, the ho- mologous area of ATP1 to the red-region of chromosome II was enlarged to 990 nt (Figure 2(c), 648 ~ 1,638), or 1,584 nt (Figure 2(d), 54 ~ 1,638), r espectively. That is, the base sequence of the complete ATP1 gene had self-similarity to the gene-position on chromosome II. Other genes of S. cerevisiae were highly homologous with the gene-position of each chromosome irrespective to the sizes, the order, the direction of transcription and the chromosomes. The fold-enlargement of the gene to each chromosome was calculated as approximately 400-fold (Table 2(a)). The same relationship of the enlargement-reduction of the chromosome-gene was observed in S. pombe (eu- karyotic cells, Tab le 2(b)) and E. coli (prokaryotic cells, Tab l e 2 (c )). In the case of small intron-containing genes in S. pombe , and genes in operons in the E. coli genome, the homology condition of the base width was also 100 nt, like that of the S. cerevisiae genome. Therefore, the homology pattern in a wide range of organisms might be dependent on the base sequence sizes for the gene analyzed. In any case, in the S. cerevisiae, S. pombe and E. coli genomes, genes and the base sequence near the chromosomal position of the gene had self-similarity with each other in the same ratio, approximately 400-fold. In some preliminary experiments, we observed the self-  M. Nakahara et al. / J. Biomedical Science and Engineering 3 (2010) 340-350 Copyright © 2010 SciRes. JBiSE 345 (a) Chromosome Ⅱ (b) ATP1, p = 3 HF = 95.0% (c) ATP1, p = 10 HF = 95.9% (d) ATP1, p = 16 HF = 92.4% Figure 2. Homology of chromosome II to ATP1. (a) Sac- charomyces cerevisiae chromosome II (813,139 nt, from the left telomere sequence to the right telomere sequence), m = 8,000, d = 3, p = 400. The ATP1 gene is located 37,001 bases from the left telomere of chromosome II (arrowhead) [26-28]. The red-region is composed of 40,000 nt (the numbers on the abscissa 20,401 – 60,401). The numbers on the abscissa indi- cate the base number from the left telomere according to MIPS. (b) ATP1 gene (1,638 nt, F1F0-ATPase complex subunit) (26), m = 60, d = 3, p = 3. (c) ATP 1 (1,638 nt), m = 60, d = 3, p = 10. (d) ATP1 (1,638 nt), m = 60, d = 3, p = 16. The homologous region (purple) of ATP1 to the red-region was designated the base number of the initiated base “A” (the start codon, ATG) of the coding region of AT P1 as 1 [26, 28]. similarity of a gene to the chromosomal position in H. sapiens (for instance, Hs.5174 and chromosome 22; data not shown). This self-similarity might be universal in all species. 3.4. Homologous Structure in tRNAs (Enlargement-Reduction of the Base Sequence) If a homologous structure was general, it must exist not only in protein-coding genes but also in RNA genes. Ac- tually, the sequence spectrum of each gene was more than 80% similar to the tRNA genes in S. cerevisiae, S. pombe and E. coli ( Table 3). Most amino acids have plural genetic codons. Each genetic codon had plural tRNA genes on several different chromosomes. How were the plural tRNA genes used properly to construct proteins during the transformation of the biological in- formation in organisms? The genetic codons for gluta- mate (Glu) were 5’-GAA and 5’-GAG. In S. cerevisiae, the nuclear-encoded Glu(GAA)-tRNA genes were 14 on various chromosomes, and all of them were composed of 72 identical nucleotides (bases). Three out of these 14 Glu(GAA)-tRNA genes were present on chromosome V (576,869 bp), located at positions 177,098 ~ 177,169, 354,930 ~ 355,001 and 487,397 ~ 487-326, and were designated Glu (GAA-1), Glu (GAA-2) and Glu (GAA-3), respectively [29-31, Figure 3 lower panel]. Figure 3 showed that the sequence spectra of these 3 Glu (GAA)-tRNA genes on chromosome V and ATP1 [26-28] were depicted. The window length of the tRNA gene was 70 nt in the analysis because Glu (GAA)-tRNA genes were composed of 72 nt (bold-black bar in upper panel). In addition, the Glu (GAA)-tRNA spectra analy- sis used DNA sequences (112 bp) adjoined to the 5’-, 3’- 20 nucleotides (green letters) added to these three Glu (GAA)-tRNA genes (72 bp, black letters). As a result, the homology factors (HF) of ATP1 to these three Glu (GAA)-tRNA genes were different; that is, 77.0% for GAA-1, 77.0% for GAA-2 and 88.5% for GAA-3, re- spectively, although these Glu (GAA)-tRNA genes were all composed of 72 identical nucleotides. The sequence spectra of ATP1 (1,638 nt) and the nu- clear-encoded 14 Glu (GAA)-tRNA (72 nt) were fairly homologous. The red area of the Glu (GAA)-tRNA gene was homologous to the homologous area (purple) of the ATP1 gene (1,638 bp), and the bracket in Figure 3 showed the Glu (GAA)-tRNA gene consisting of 72 bp. The homologous area (red) of the Glu (GAA)-tRNA to the ATP 1 gene overlapped with a part of the adjoining sequences of the tRNA-gene (the homologous region of the tRNA gene with the ATP1 gene was also indicated from the red-base to the red base in the lower panel of Figure 3). In other words, the sequence spectrum analy- ses based on the frequencies of the base sequences in the genome indicated that the sequence spectrum of the gene might be influenced by the adjoined DNA sequences. The smaller the base numbers of the DNA sequence, such as for the tRNA-genes, the greater these effects. In the same way, other nuclear-encoded 11 Glu (GAA)- tRNA genes on several different chromosomes were generally homolog ous to the ATP1 gene on chromosome II, which encoded the subunit of the F1F0-ATPase complex [26-28], but their homology factors (HF) varied. The maximum homologous Glu (GAA) tRNA gene was on chromosome IX (HF = 89.2%, position, 370,414-370,485,  M. Nakahara et al. / J. Biomedical Science and Engineering 3 (2010) 340-350 Copyright © 2010 SciRes. JBiSE 346 Table 2. Self-similarity with a gene to the chromosome. Gene nt (*1) Chromosom e (*2)nt (*3) intron # p-value (*4) HF (%) (*5) (a) S. cerevisiae SEO1 1,779 1, left 230,203 0 17 61.2 FLO1 4,611 1, right 0 46 73.3 ATP1 1,638 2, left 813,139 0 16 92.4 SUP45 1,311 2, right 0 13 72.4 PRD1 2,136 3, left 315,350 0 21 77.7 PHO87 2,769 3, right 0 27 75.2 ATP16 480 4, left 1,531,929 0 4 93 RAD9 3,927 4, right 0 39 74.2 PAU2 360 5, left 576,870 0 3 85.2 GLC7 1,461 5, right 0 14 73.6 EMP47 1,335 6, left 270,148 0 13 81.4 PHO4 939 6, right 0 9 82.8 POX1 2,244 7, left 1,090,936 0 22 80.9 TFC4 3,075 7, right 0 30 69.9 GUT1(STE20) 2,127 8, left 562,638 0 21 61.6 IRE1(NDT80) 3,345 8, right 0 33 80 HOP1 1,815 9, left 439,885 0 17 64.5 MRS1(PAN1) 1,089 9, right 0 10 91.7 CYR1 6,078 10, left 745,440 0 61 79.7 ATP2 1,533 10, right 0 15 75.1 SDH1 1,920 11, left 666,445 0 17 71.1 CCP1((NUP133) 1,083 11, right 0 10 76.2 HSP104 2,724 12, left 1,078,173 0 27 68.4 MAS1 1,386 12, right 0 13 81.2 CYB2(CAT2) 1,773 13, left 924,430 0 17 88.4 HXT2(AAC1) 1,623 13, right 0 16 70.1 RAS2 966 14, left 784,328 0 9 86.1 POP2 1,299 14, right 0 13 75.4 ADH1 1,044 15, left 948,061 0 10 85.2 ADE2 1,713 15, right 0 17 78.8 TBF1(PHO85) 1,686 16, left 948,061 0 16 67.7 PZF1 1,287 16, right 0 12 91.2 (b) S. pombe ATP2 1,578 1 (968,783) 5,579,133 0 15 78.6 RPL37 337 1 (1,275,535) 1 3 81.3 RPL37(exon) 270 2 77.5 CDC24 1,823 1 (2,863,965) 6 18 81.3 CDC24(exon) 1,506 15 77.5 ATP1 2,049 1 (5,256,781) 2 20 75 ATP1(exon) 1,611 16 76.1 MEU6 2,083 2 (454,230) 4,539,804 2 20 82.3 MEU6(exon) 1,956 19 82.3 CDC2 1,189 2 (1,500,340) 4 11 76.4 CDC2(exon) 894 8 78.8 ATP16 483 2 (3,046,873) 0 4 90.4 SPO4 1,672 2 (3,827,178) 2 16 85.1 SPO4(exon) 1,290 12 74.6 RAF1 1,917 3 (100,255) 2,455,984 0 19 73 HIF2 1,875 3 (194,552) 3 18 71.9 HIF2(exon) 1,695 16 N.D.(*6) SRK1 3,932 3 (1,302,900) 1 39 64 SPK1(exon) 1,743 17 69  M. Nakahara et al. / J. Biomedical Science and Engineering 3 (2010) 340-350 Copyright © 2010 SciRes. JBiSE 347 Gene nt (*1) Chromosom e (*2)nt (*3) intron # p-value (*4) HF (%) (*5) GAF1 2,568 3 (1,666,310) 0 25 76.2 TIF6 1,104 3 (2,223,154) 2 11 86.4 TIF6(exon) 735 7 76 ATP5 838 3 (2,268,884) 2 8 74.8 ATP5(exon) 651 6 93 (c) E. coli (K12) araA 1,503 66,835 4,639,221 0 15 74.5 lacZ 3,075 362,455 0 30 66.3 galE 1,017 790,262 0 10 87.7 trpD 1,596 1,317,813 0 15 77.5 cybB 531 1,488,926 0 5 87.8 galF 894 2,111,458 0 8 88 argA 1,332 2,947,264 0 13 68.9 secY 1,332 3,440,788 0 13 82.8 atpA 1,741 3,916,339 0 17 73.9 purA 1,299 4,402,710 0 12 83 *1, Base numbers of the gene without intron. *2, Gene position on the chromosome (from the left to the right = S. pombe). *3, Size (base numbers) of chromosome or genome. *4, Skipped base numbers of the g ene (max.p-value). *5, Entire gene in the max.p- value-chromosome HF (%) in the homologous region. *6, not determined. Table 3. Self-similarity with a protein to tRNA gene. Gene size (nt) (*1) chromosome (*2)tRNA (*3) size (nt, *4) chromosome (*5) p (*6) (S. cerevisiae) ATP1 1,638 2 Glu(GAA) 72 12 16 RAS2 936 14 Lys(AAG) 72 6 9 ADH1 1,047 15 Arg(AGG) 72 10 10 TFC4 3,075 7 Ser(TCG) 103 3 30 PAU2 360 5 Ser(AGC) 101 6 3 CYR1 6,078 10 Ser(AGC) 101 6 60 (S. pombe) ATP1 1,611 1 Tyr(TAC) 84 2 16 YPT3 645 1 Arg(AGA) 73 2 6 CDC2 894 2 Ser(TCT) 82 1 8 SPO4 1,290 2 Thr(ACT) 72 3 12 GAF1 2,568 3 Ser(AGC) 95 2 25 TIF6 735 3 Arg(AGA) 73 3 7 (E. coli) galE 1,017 K12 genome Ser(TCC) 88 K12 genome 10 atpA 1,735 Ser(AGC) 93 17 cybB 531 Ser(TCC) 88 5 lacZ 3,075 Arg(CGT) 77 30 *1; base numbers of gene without intron *2; Chromosome presented the gene *3; Homologous tRNA gene. *4; Size of tRNA gene. *5; Chromosome presented the tRNA gene. *6; Skipped base numbers of the gene . 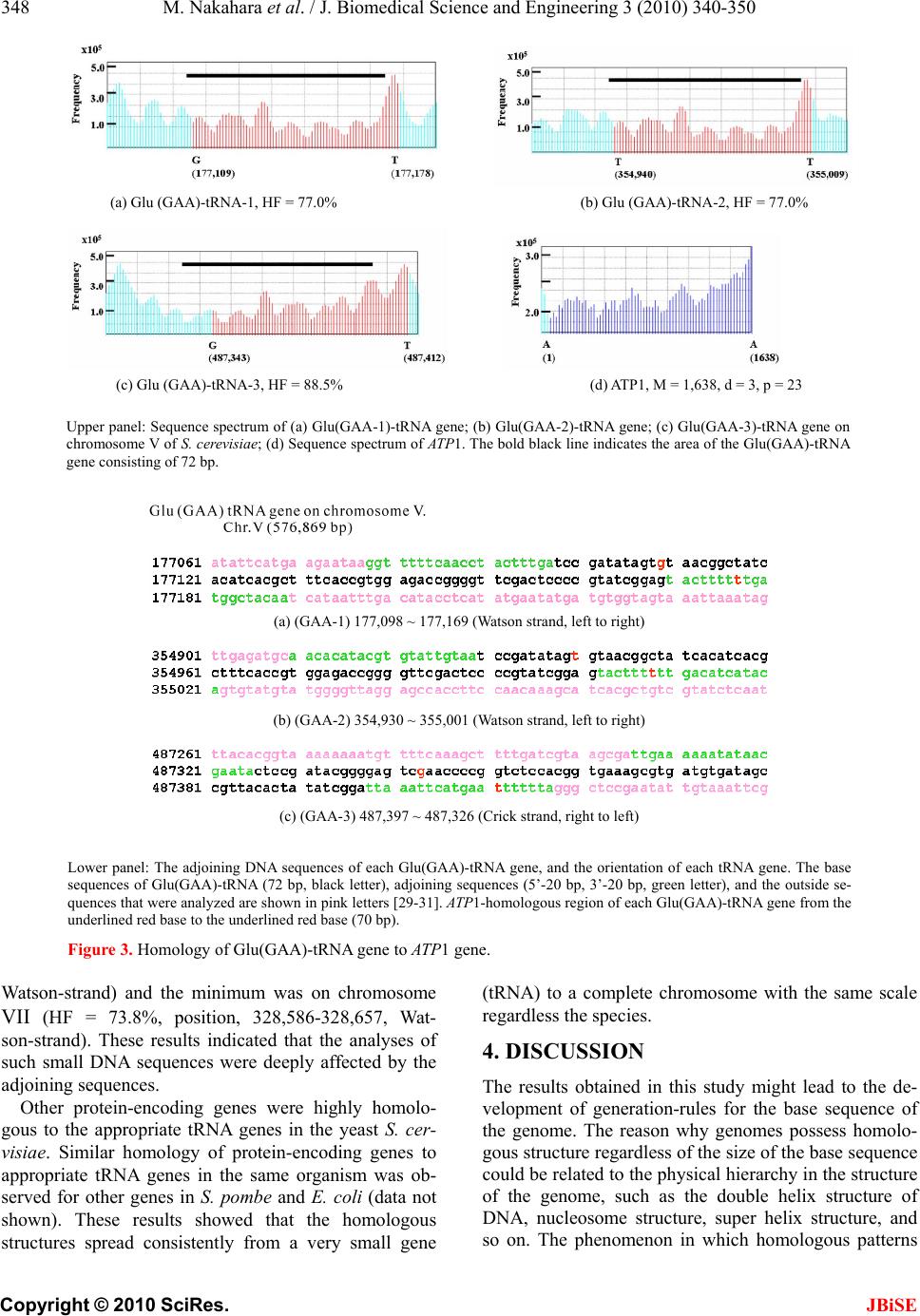 M. Nakahara et al. / J. Biomedical Science and Engineering 3 (2010) 340-350 Copyright © 2010 SciRes. JBiSE 348 (a) Glu (GAA)-tRNA-1, HF = 77.0% (b) Glu (GAA)-tRNA-2, HF = 77.0% (c) Glu (GAA)-tRNA-3, HF = 88.5% (d) ATP1, M = 1,638, d = 3, p = 23 Upper panel: Sequence spectrum of (a) Glu(GAA-1)-tRNA gene; (b) Glu(GAA-2)-tRNA gene; (c) Glu(GAA-3)-tRNA gene on chromosome V of S. cerevisiae; (d) Sequence spectrum of ATP1. The bold black line indicates the area of the Glu(GAA)-tRNA gene consisting of 72 bp. (a) (GAA-1) 177,098 ~ 177,169 (Watson strand, left to right) (b) (GAA-2) 354,9 30 ~ 355,001 (Watson strand, left t o right) (c) (GAA-3) 4 87,397 ~ 487,326 (Crick strand, right to left) Lower panel: The adjoining DNA sequences of each Glu(GAA)-tRNA gene, and the orientation of each tRNA gene. The base sequences of Glu(GAA)-tRNA (72 bp, black letter), adjoining sequences (5’-20 bp, 3’-20 bp, green letter), and the outside se- quences that were analyzed are shown in pink letters [29-31]. ATP1-homologous region of each Glu(GAA)-tRNA gene from the underlined red base to the underlined red base (70 bp). Figure 3. Homology of Glu(GAA)-tRNA gene to ATP1 gene. Watson-strand) and the minimum was on chromosome VII (HF = 73.8%, position, 328,586-328,657, Wat- son-strand). These results indicated that the analyses of such small DNA sequences were deeply affected by the adjoining sequences. Other protein-encoding genes were highly homolo- gous to the appropriate tRNA genes in the yeast S. cer- visiae. Similar homology of protein-encoding genes to appropriate tRNA genes in the same organism was ob- served for other genes in S. pombe and E. coli (data not shown). These results showed that the homologous structures spread consistently from a very small gene (tRNA) to a complete chromosome with the same scale regardless the species. 4. DISCUSSION The results obtained in this study might lead to the de- velopment of generation-rules for the base sequence of the genome. The reason why genomes possess homolo- gous structure regardless of the size of the base sequence could be related to the physical hierarchy in the structure of the genome, such as the double helix structure of DNA, nucleosome structure, super helix structure, and so on. The phenomenon in which homologous patterns  M. Nakahara et al. / J. Biomedical Science and Engineering 3 (2010) 340-350 Copyright © 2010 SciRes. JBiSE 349 appear in various size levels is known as “self-similarity” or “fractal”. Therefore, the structure of the genome could be essentially related to the fractal. During the 1990s, many papers reported that the ge- nome bases should follow the fractal-rule [15-18 etc], and Genome Projects for many species had revealed genomic base sequences in the last 10 years. Therefore, analyses of the concrete biological phenomena based on the structures of genomes should be in progress. In this paper, the analyses of the sequence spectrum, m = 2 for a tRNA, m = 60 for a protein, and m = 8,000 for a ch romo s ome w er e us ed. In the c as e of th e se quen ce spectrum of protein, m = 10 (average of 20 nt) or m = 60 (average of 120 nt) was easier to use for the analysis of the sequence spectrum when the m-value corresponded to 6 ~ 7, or 40 amino acid residues, respectively [32]. In the case of the chromosome, m was adjusted to 8,000 (average of 16,000 nt = 80 nucleosomes) or 10,000 (average of 20,000 nt = 100 nucleosomes). In any case, the smaller the adjusted m-value is, the higher the resolution of the sequence spectrum. These results suggested that “m” might be reflected in the higher order structure of a molecule, a gene for tRNA, or protein or chromosome, but the detailed biological meaning of the m-value is in progress [33, 34]. In addition, as described previously, each genetic codon had multiple tRNA genes on several different chromosomes. How were the multiple tRNA genes used properly to construct proteins during the transformation of biological information in organisms? In biological processes, the base sequence of DNA was transcribed to mRNA, and then the base sequence of mRNA was transferred to the amino acid sequence by tRNAs. In such cases, the higher homologous structure (HF) of tRNA genes might be one of the distinctions of an ap- propriated protein. In other words, the base sequence of DNA was reflected in the amino acid sequence through the base sequence of RNA. Therefore, the above method might be applicable to the interactive-sites of DNA, RNA, and protein. In such analyses, the selection of the d- and p-values might be important to obtain the highest resolution of the sequence spectru m correspond ing to the structural features of the target DNAs o r pr ot ei ns. Genomic DNA might be enlarged and reduced be- cause the base sequence of the genomic DNA had frac- tality; therefore, it had similarity to related sites an d was able to prefer a gene over the chromosome. The coding- and non-coding r egions of a genome were different with respect to bases as described. As a result, biases of the four bases occurred on genomic DNA [20]. The analyses based on the appearance frequency of the base sequences in a genome should be universally applicable to everything that was expressed by base se- quences, not only in Saccharomyces cerevisiae, but also Homo sapiens, Escherichia coli and all genomes; there- fore, this method might be applied as a first screen to characterize interaction-sites i n b iological p henomena. 5. CONCLUSIONS The results obtained in this study were summarized as follows. 1) Homologous structure exists in the appear- ance frequency of short base sequences such as triplets over an entire chromosome in the genome, and the 5’- and 3’-adjoining base sequences of the DNA were deeply affected by the homology factor when the target DNA was small in size or located at the boundary, 2) homologous structure was universally observed in a va- riety of species, 3) the homology of the sequence spec- trum of a gene was observed in the appropriate tRNA genes, and the analysis (SSM) of the DNA base se- quences might be reflected in that of protein; in other words, 4) the SSM might be reflected as a vehicle of biological information, and a suitable prediction method to identify interacting regions DNA, RNA or protein by the appropriate conditions of “m”, “d” and “p”, in each gene, or genomic DNA, 5) SSM was faithfully reflected the biological information, therefore, the conservation of the bases sequences of genomic DNA were also con- served the translated amino acids sequence, the protein sequence, in the coding region, 6) SSM could deal con- sistently with molecules that consists of base sequences. 6. ACKNOWLEDGEMENTS The authors wish to thank to Dr. Hiroshi Shibata at Sojo University for his comments about the fractal analysis in this research. REFERENCES [1] Singer, M. and Berg, P. (1991) Genes & genomes – A changing perspective-. University Science Books. [2] Garrel, J.I. (1997) The yeast proteome handbook. Third edition, Beverly, Proteome Inc. [3] Velculescu, V.E., Zhang, L., Zhou, W., Vogelstein, J., Basral, M.A., Bassett, D.E.Jr., Hieter, P., Vogelstein, B. and Kinzler, K.W. (1997) Characterization of the yeast transcriptome. Cell, 88, 243-51. [4] Wan, X.F., VerBerkmoes, N.C., McCue, L.A., Stanek, D., Connlly, H., et al. (2004) Transcriptomic and proteomic characterization of the fur modulon in the metal- reduc- ing bacterium Shewanella oneidensis. The Journal of Bacteriology, 186, 8385-8400. [5] Sakharkar, K.R., Sakharkar, M.K., Culiat, C.T., Chow, V. T. and Pervaiz, S. (2006) Functional and evolutionary analyses on expressed intronless genes in the mouse ge- nome. FEBS Letters, 580, 1472-1478. [6] Karkas, J.D., Rudner, R. and Chargaff, E. (1968) Separa- tion of B. subtilis DNA into complementary strands. II. Template functions and composition as determined by transcription by RNA polymerase. Proceedings of the National Academy of Sciences of the United States of America, 60, 915-920. [7] Bell, S. J., Fordyke, D. R. (1999) Accounting unit of in DNA. Journal of Theoretical Biology, 197, 51-61.  M. Nakahara et al. / J. Biomedical Science and Engineering 3 (2010) 340-350 Copyright © 2010 SciRes. JBiSE 350 [8] Abe, T., Kanaya, S., Kinouchi, M., Kudo, Y., Mori, H. et al. (1999) Gene classification method based on batch- learning SOM. Genome Informatics Seris, 10, 314-315. [9] Baisnee, P.-F., Hampson, S. and Baldi, P. (2002) Why are complement ary DNA strands symmetric? Bioinformatics, 18, 1021-1033. [10] Chen, L. and Zhao, H. (2005) Negative correlation be- tween compositional symmetries and local recombination rates. Bioinformatics, 21, 3951-3958. [11] Albrecht-Buehler, G. (2006) Asymptotically increasing compliance of genomes with Chargaff’s second parity rules through inversions and inverted transpositions. Proceedings of the National Academy of Sciences of the United States of America, 103, 17828-17833. [12] Wilson, J. T., Wilson, L. B., Reddy, V. B., Cavallesco, C., Ghosh, P. K., et al. (1980) Nucleotide sequence of the coding portion of human alpha globin messenger RNA. Journal of Biological Chemistry, 255, 2807-2815. [13] Wada, A., Suyama, A. and Hanai, R. (1991) Phenome- nological theory of GC/AT pressure on DNA base com- position. Journal of Molecular Evolution, 32, 374-378. [14] Nakamura, Y., Itoh, T. and Martin, W. (2007) Rate and polarity of gene and fission in Oryza sativa and Arabi- dopsis thaliana. Molecular Biology and Evolution, 24, 110-121. [15] Paila, U., Kondam, R. and Ranjan, A. (2008) Genome bias influences amino acid choice: analysis of amino acid substitution and re-compilation matrices exclusive to an AT-biased genome. Nucleic Aci ds Research. [16] Voss, R.F. (1992) Evolution of long-range fractal correla- tion and 1/f noise in DNA base sequences. Physical Re- view Letters. 68, 3805-3809. [17] Bains, W. (1993) Local self-similarity of sequence in mammalian nuclear DNA is modulated by a 180 bp pe- riodicity. Journal of Theoretical Biology, 161, 13-143. [18] Weinberger, E.D. and Stadler, P.F. (1993) Why some fitness landscape are fractal. Journal of Theoretical Biol- ogy, 163, 255-275. [19] Lu, X., Sun, Z., Chen, H. and Li, Y. (1998) Characteriz- ing self-similarity in bacteria DNA sequences. Physical Review E—Statistical, 58, 3578-3584. [20] Takeda, M. and Nakahara, M. (2009) Structural Features of the Nucleotide Sequences of Genomes. Journal of Computer A ided Chemis try, 10, 38-52. [21] NCBI Genome Data Base (2009) http://www.ncbi.nlm. nih.gov/sites/entrez?db=genome. [22] Crick, F.H. (1968) The origin of genetic code. Journal of Molecular Biology, 38, 367-379. [23] International Human Genome Sequencing Consortium. (2001) Initial sequencing and analysis of the human ge- nome. Nature, 409, 860-921. [24] Mattick, J.S. (2004) RNA regulation: A new genetics? Nature Reviews Genetics, 5, 316-323. [25] Lynch, M. (2007) The frailty of adaptive hypothesis for the origins of organismal complexity. Proceedings of the National Academy of Sciences of the United States of America, 104, 8597-8604. [26] Takeda, M., Chen, W.-H., Saltzgaber, J. and Douglas, M.G. (1986) Nuclear genes encoding the yeast mito- chondrial ATPase complex-analysis of ATP1 coding the F1-ATPase subunit and its assembly-. Journal of Bio- logical Chemistry, 261, 15126-15 133. [27] Takeda, M., Okushiba, T., Hayashida, T. and Gunge, N. (1994) ATP1 and ATP2, F1F0-ATPase and subunit genes of Saccharomyces cerevisiae, are respectively lo- cated on chromosome II and X. Yeast, 10, 1531-1534. [28] Mewes, H. W., Albermann, K., Bähr, M., Frishmann, D., Gleissner, A., et al. (1997) Overview of the yeast genome. Nature, 387 (supp), 7-65. [29] Dietrich, F. S., Mulligan, J., Hennessy, K., Yelton, M. A., Allen, E., et al. (1997) The nucleotide sequence of Sac- charomyces cerevisiae chromosome V. Nature, 387 (supp), 78-81. [30] Saccharomyce Genome Database. (2009) (http://www. yeastgenome.org/). [31] Transfer RNA data base. (2009) (http://gtrnadb.ucsc.edu/). [32] Matthews, B.W. (1993) Structural and genetic analysis of protein stability. Annual Review of Biochemistry, 62, 139-160. [33] Kornberg, R.D. (1974) Chromatin structure: a repeating unit of histones and DNA. Science, 184, 868-871. [34] van Holde, K. and Zlatonova, J. (1995) Chromatin higher order structure: Chasing a mirage? Journal of Biological Chemistry, 270, 8373-8376. |