Journal of Cancer Therapy
Vol.3 No.6(2012), Article ID:25419,9 pages DOI:10.4236/jct.2012.36143
Immunohistochemical Expression of Ki-67, PCNA, pRb, p16, p53, Bcl-2 and Bax in Esophageal Adenocarcinoma and Barrett’s Associated Dysplasia
![]()
1Department of Gastroenterology, University Hospital “Alexandrovska”, Sofia, Bulgaria; 2Department of Clinical Pathology, “Pirogov” University Hospital, Sofia, Bulgaria; 3First Surgical Clinic, “Pirogov” University Hospital, Sofia, Bulgaria.
Email: dr_andrey_kotzev@abv.bg
Received September 29th, 2012; revised October 31st, 2012; accepted November 11th, 2012
Keywords: Esophageal Adenocarcinoma; Biomarkers; Cell Cycle; Proliferation; Apoptosis
ABSTRACT
Background: Esophageal adenocarcinoma (EAC) has an extremely poor prognosis. There is a need to characterize the molecular alterations in the carcinogenesis of EAC in order to improve the diagnosis and treatment. Materials and Methods: We used 7 markers to explore the changes in the cell cycle, proliferation and apoptosis in patients with EAC and Barrett’s esophagus (BE)-associated dysplasia. The protein expression of Ki-67, PCNA, pRb, p16, p53, Bcl-2 and Bax was evaluated by immunohistochemistry in archival tissue samples, collected from 15 patients with EAC and 5 patients with BE-associated dysplasia. We analyzed also lymph-node, omentum and liver metastases from the primary esophageal tumors. Results: Ki-67, PCNA, pRb, p16, p53, Bcl-2 and Bax expression was observed in 100%, 87%, 60%, 40%, 100%, 7% and 93% of tumor samples, and in 100%, 80%, 0%, 80%, 80%, 20% and 100% of dysplasia samples, respectively. Significant difference in the expression of the markers between EAC and BE-associated dysplasia was detected for pRb (p = 0.006). Ki-67 expression was associated with clinicopathological parameter T (p = 0.012; V = 0.585). Ninefold higher risk to develop EAC was established for the patient with strong p53 expression, than the lacking p53 patient. Patients with strong p53 expression survived 6.8 months longer than the patients with weak p53 expression and 8.6 months longer than the patients with moderate p53 expression. No correlation was found between the expression of the other markers and prognosis. Conclusion: The results suggest that Ki-67, PCNA, pRb, p16, p53 and Bax participate in the pathogenesis of EAC, whereas Bcl-2 does not play essential role in EAC and BE-associated dysplasia. The balance between cell proliferation and apoptosis is lost in EAC and BE-associated dysplasia. Abnormal p53 protein expression has predictive and prognostic value in EAC. Larger prospective studies are needed to confirm these findings.
1. Introduction
Esophageal adenocarcinoma (EAC) is an aggressive cancer with very low survival rate [1]. Early invasion and distant metastases are typical of EAC. The incidence of EAC has been increasing vastly in the last three decades in the USA and in some European countries [2]. The main risk factors for EAC are gastroesophageal reflux disease (GERD), Barrett’s esophagus (BE), obesity, hiatal hernia and male sex [2-5]. Tobacco smoking is a risk factor of developing EAC, while higher alcohol consumption is not associated with increased risk of EAC [6,7]. The role of the diet in the pathogenesis of EAC is unclear and the data are contradictory [8,9].
BE is a known premalignant lesion of EAC and is characterized by a replacement of esophageal stratified squamous epithelium with specialized intestinal epithelium. BE is caused by chronic gastroesophageal reflux and affects 5% - 15% of GERD patients. The patients with BE have considerable increased risk of developing EAC, but the risk of progression to EAC is 0.5% - 1% per year [10,11]. BE could be considered as a successful adaptation to severe chronic gastroesophageal reflux, but at the high cost of elevated cell turnover, which could predispose to malignant transformation. The cell of origin for BE is still not defined, but according to the most popular theory the transdifferentiation of injured esophageal stem cells (de novo metaplasia) initiates the conversion of squamous to columnar epithelium [12]. Neoplastic progression of BE develops consecutively through low-grade dysplasia (LGD) and high grade dysplasia (HGD) and, eventually to cancer. Inflammation is indispensable for occurrence and evolution of BE. The histological alterations in the Barrett’s metaplasia-dysplasia-adenocarcinoma (MDA) sequence are caused by multiple molecular changes. Aberrant expression of oncogenes, tumor suppressor genes and related proteins, is observed in the carcinogenesis of EAC. All these changes impede the sophisticated control of cell cycle, proliferation and apoptosis. The sequence of molecular events is still not strictly delineated, but promoter hypermethylation, loss of heterozygosity (LOH) or mutations of p16 are the earliest events in malignant progression of BE [13-15]. LOH of p53 is also considered as an early molecular event in Barrett’s MDA sequence, which is followed by aneuploidy [16,17]. Upregulation of different growth factors, COX-2, iNOS, telomerase and matrix metaloproteinases, and downregulation of E-cadherin and inhibitors of matrix metaloproteinases are regularly involved in the pathogenesis of EAC [18].
Molecular biomarkers are promising tool for the elucidation of Barrett’s MDA sequence. The number of investigations dealing with molecular biomarkers in EAC pathogenesis is increasing constantly, although none of these markers is used now in daily clinical practice. The accumulating data could help to reveal the biology of EAC, as well to improve the diagnosis and treatment of EAC. Some of the relevant markers could be used as targets in molecular therapy of EAC. Interesting data demonstrated that clonal genetic diversity in BE predicts progression to EAC [19]. It is possible that diverse molecular pathways are engaged in a nonlinear model of EAC carcinogenesis. Therefore, we chose a broad panel of molecular markers and our aim was to analyze the immunohistochemical expression of proliferative markers Ki-67 and PCNA, cell cycle markers pRb and p16, and apoptotic markers p53, Bcl-2 and Bax in EAC and BE-associated dysplasia.
2. Materials and Methods
2.1. Patients and Tissue Samples
In this retrospective study, we used formalin-fixed, embedded in paraffin archival blocks from 15 consecutive patients with EAC and 5 patients with BE-associated dysplasia for histopathological and immunohistochemical analysis in the Department of Clinical Pathology, “Pirogov” University Hospital. The study protocol was approved by the local Ethics Committee. Medical records of the studied patients were reviewed. Tissue samples were obtained from endoscopic biopsies and surgical resections made in the period 1998-2004. All samples were evaluated independently by two experienced histopathologists. All of the included tumors were primary adenocarcinomas with defined esophageal origin, while gastric tumors invading the esophagus were excluded. In dysplasia samples, the inclusion criteria were the availability of dysplasia in the distal esophagus and proven intestinal type of metaplasia adjacent to dysplastic tissue. Mean period of follow up for the patients was 34 months. Four patients with HGD developed EAC for a mean period of 22 months (range 19 - 25), while no progression was detected in the case with LGD. Mean survival for the patients with EAC was 37 months (range 11 - 72). Esophagectomy was performed in 10 patients with EAC, while the others received palliative treatment. None of the patients was treated with neoadjuvant chemotherapy or chemoradiotherapy. TNM system was used for tumor classification. Clinicopathological data of the patients are summarized in Table 1.
2.2. Immunohistochemistry
Protein expression was studied by immunohistochemistry

Table 1. Patient characteristics.
(IHC). Tissue sections were placed on silanizied slides, deparaffined and rehydrated in decreasing concentrations of ethanol. Antigen retrieval was performed by immersing the slides in 10 mM modified citrate buffer with pH 6.1 and heated in a microwave oven (700 W) 4 times for 5 min with an interval of 2 min for cooling the slides at room temperature. For Ki-67, the utilized target retrieval solution was with pH 9.1. All used solutions are produced by DAKOCytomation, Denmark. Endogenous peroxidase activity was blocked by 3% hydrogen peroxide (H2O2) for 5 min and slides were incubated for one hour at room temperature with monoclonal mouse antihuman antibodies against studied biomarkers. Detailed data for the used antibodies are presented in Table 2. LSAB+ visualization kit was used (DAKOCytomation, Denmark). The slides were incubated with a secondary biotinylated anti-mouse antibody for 15 min, followed by incubation with peroxidase-labelled streptavidin. The reaction was developed with 3,3’-diaminobenzidine chromogen solution that results in brown (nuclear or cytoplasmic) staining in the positive cells. Finally, slides were counterstained with hematoxylin. Negative controls were completed by omission of the primary antibody. Positive staining of normal, non-neoplastic epithelial cells, fibroblasts and lymphocytes within analyzed samples served as internal positive control for Ki-67, PCNA, pRb, p16, Bcl-2 and Bax immunostaining. Section of colon cancer with known high p53 expression was used as a positive control for p53. The expression was detected by light microscope (Olympus CX31) and the pictures were taken with a digital camera (Olympus C-5050). The slides were examined independently by two observers. More than 500 cells were counted in five representative high-power magnification fields (400×) for each sample. For Ki-67, PCNA, pRb, p16 and p53 only nuclear staining was determined as positive, and for the Bcl-2 and Bax only cytoplasmic staining was determined
Table 2. Primary antibody characteristics.
as positive. Samples were scored into four groups based on the percentage of positively stained tumor/dysplastic cells as follows: 0 (negative expression, 0% - 25%); 1+ (weak expression, 26% - 50%); 2+ (moderate expression, 51% - 75%) and 3+ (strong expression, 76% - 100%).
2.3. Statistical Analysis
Student’s t-test, Mann-Whitney test and ColmogorovSmirnov test were used to compare the mean level of expression of every separate marker between patients with EAC and patients with BE-associated dysplasia. The correlation between expression and clinicopathological features (T, N, M and G) in specimens with EAC was evaluated by Fisher’s Exact Test, Cramer’s V correlation coefficient, single-factor dispersion analysis and Bonferroni test. Single-factor logistic regression models were used to estimate if there any association between the altered expressions patterns of biomarkers and risk of developing EAC. Linear regression models were applied for survival analysis. All statistical analyses were performed with SPSS (version 13.0; SPSS, Chicago, IL). A p-value of <0.05 was considered statistically significant.
3. Results
The samples were suitable for interpretation in all cases. Expression of Ki-67, PCNA, p53 and Bcl-2 was observed in 15 (100%), 13 (87%), 15 (100%) and 1 (7%) of the EAC samples, respectively (Figures 1(a)-(e)). Loss of pRb, p16 and Bax expression was found in 6 (40%), 9 (60%) and 1 (7%) of the EAC samples, respectively. Expression of Ki-67, PCNA, p53 and Bcl-2 was found in 5 (100%), 4 (80%), 4 (80%) and 1 (20%) of the dysplasia samples, respectively. Loss of pRb and p16 expression was detected in 5 (100%) and 1 (20%) of the dysplasia samples, respectively. Loss of Bax expression was not observed in the dysplasia samples. Detailed data for the expression in EAC and dysplasia samples are presented in Table 3.
The expression of the markers was investigated in 2 cases with lymph-node metastases and 1 case with liver and omentum metastases from EAC (Figure 1(f)). PCNA, pRb, p53 and Bax expression, and loss of Ki-67, p16 and Bcl-2 was detected in one of the cases with lymph-node metastasis. In the other case with lymphnode metastasis, as well in the liver metastasis, only Bcl-2 was not expressed. In the omentum metastasis, expression of Ki-67, pRb, p16, p53 and Bax was coupled with PCNA and Bcl-2 loss (Table 4).
Statistically significant difference in the mean level of expression of the studied markers between patients with EAC and patients with BE-associated dysplasia was as-
 (a)
(a)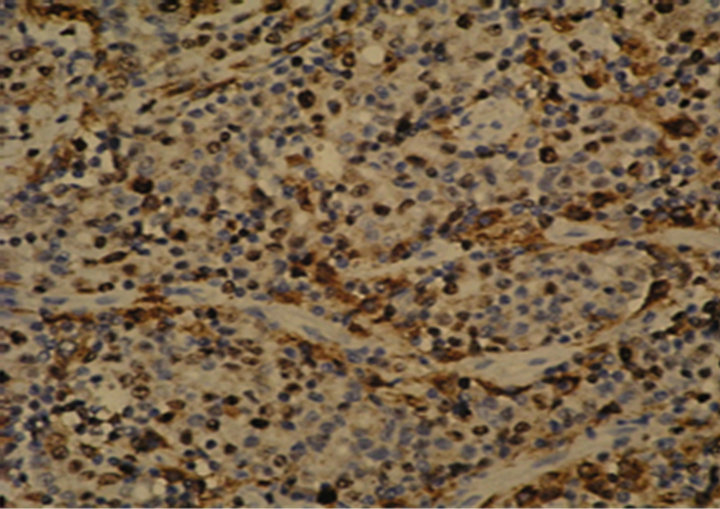 (b)
(b)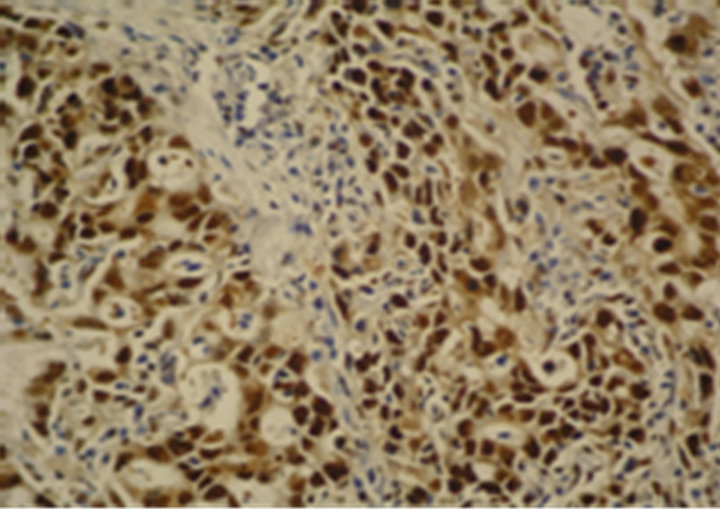 (c)
(c)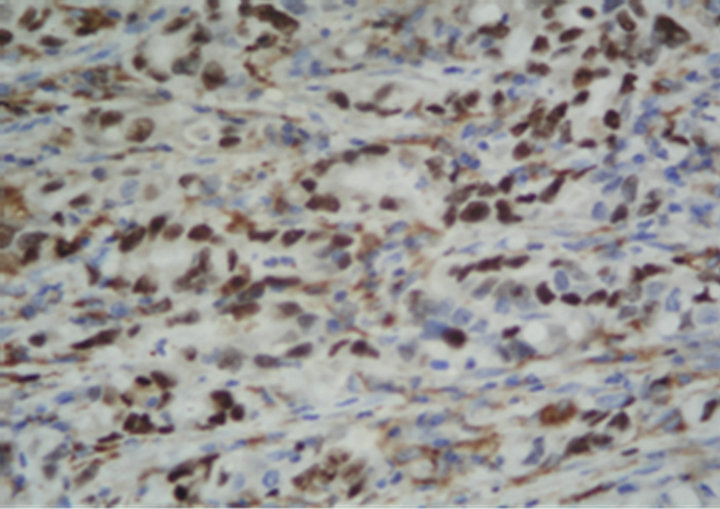 (d)
(d)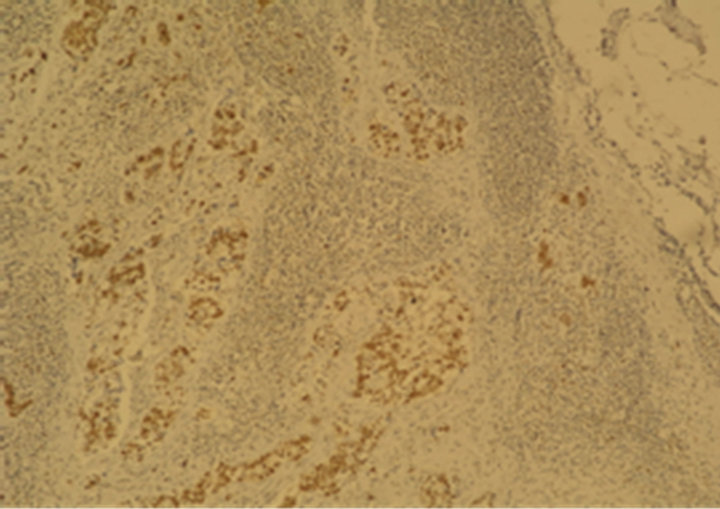 (e)
(e)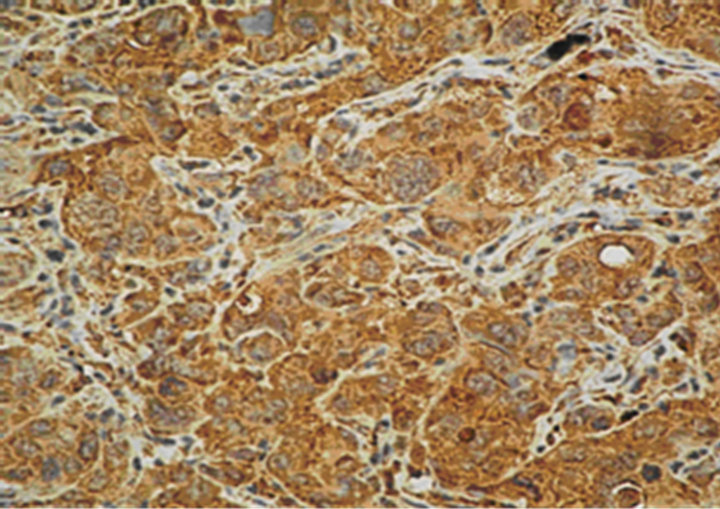 (f)
(f)
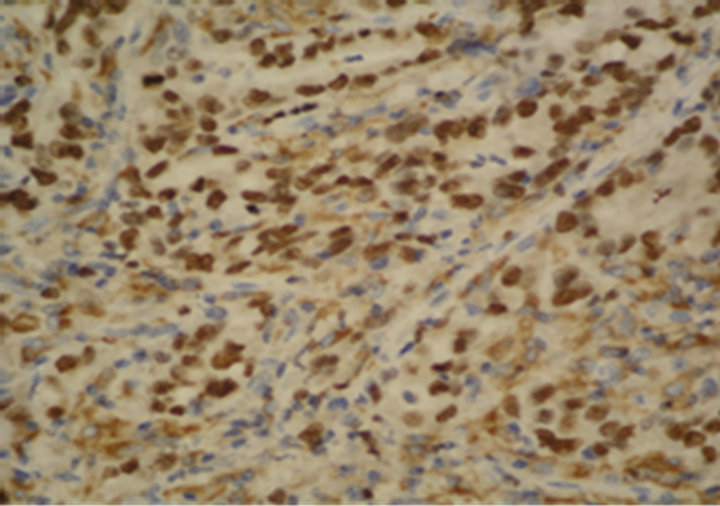
Figure 1. Immunostaining in esophageal adenocarcinoma (EAC) and lymph-node metastasis. (a) Ki-67 in EAC (400×); (b) pRb in EAC (400×); (c) p16 in EAC (400×); (d) p53 in EAC (400×); (e) Bax in EAC (400×); (f) PCNA in lymphnode metastasis from EAC (100×).
certained only for the marker pRb (p = 0.006). Ki-67 expression was associated with T (p = 0.012; V = 0.585), but no association between the expression of the other markers and T, N, M or G categories was detected. Strong p53 expression was correlated with ninefold higher risk for the patient to develop EAC, than the patient with negative expression of p53. Patients who showed strong p53 expression lived 6.8 months longer than the patients with weak p53 expression and 8.6 months longer than the patients with moderate p53 expression. None of the rest of the analyzed markers was of prognostic value.
4. Discussion
Abnormal cell proliferation plays a decisive role in the development of human malignancies. Ki-67 expression is successfully used for estimation of the proliferation in BE evolution. The results from various studies showed that Ki-67 proliferation index is increasing progressively from BE to EAC [20-23]. The ubiquitous expression of Ki-67 in this study confirms that the elevated proliferation is an inherent molecular alteration in EAC. In accordance with the results reported by Feith et al. [24], we
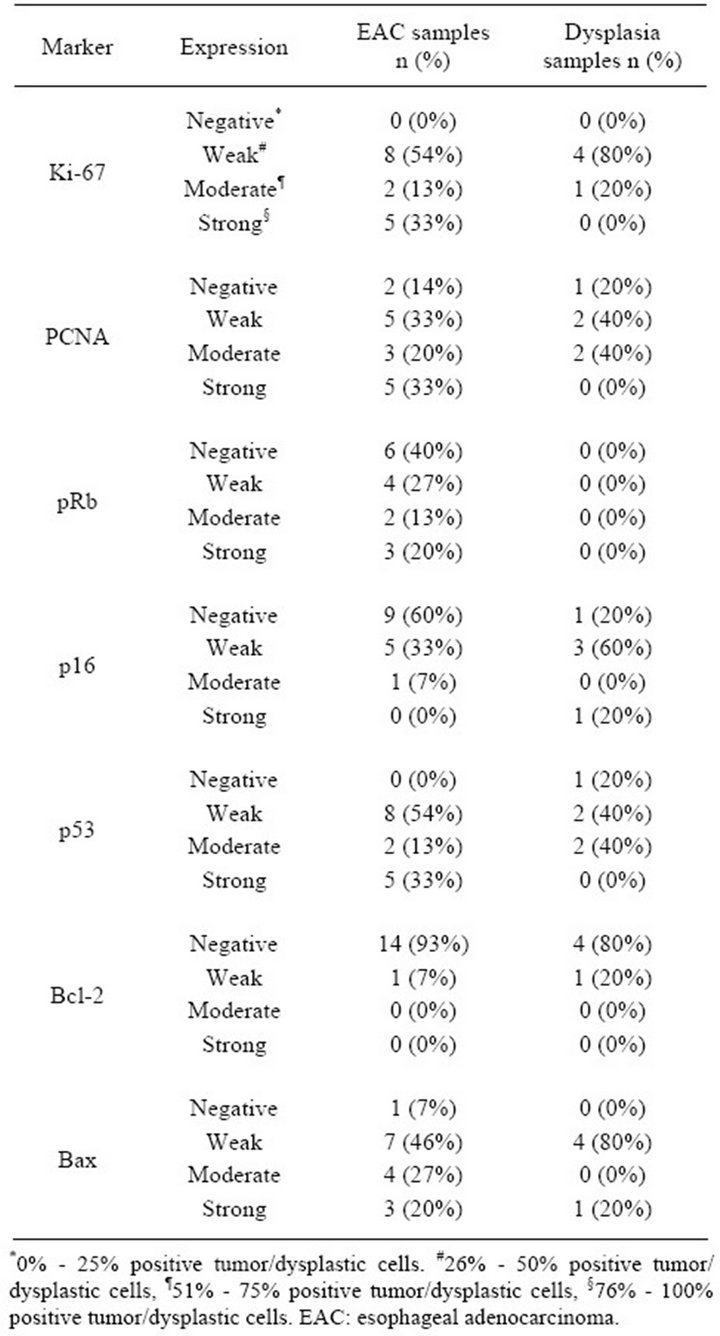
Table 3. Immunostaining of Ki-67, PCNA, pRb, p16, p53, Bcl-2 and Bax in EAC and BE-associated dysplasia samples.
detected abundant number of positive Ki-67 nuclei at the luminal surface. Ki-67 expression also demonstrated association with T category. The results suggest that high proliferation in EAC, measured by Ki-67, could predict the extent of the primary tumor, but these findings require further studies.
Similar to Ki-67 staining, high expression of the proliferative marker PCNA was found in the analyzed samples. Salient PCNA expression in EAC progression was also observed in previous studies [25-27]. Our results corroborate the data that PCNA is expressed at high levels in EAC and BE-associated dysplasia. PCNA, however, has role not only in the DNA replication, but also in the post-replicative processing, DNA repair and apop-
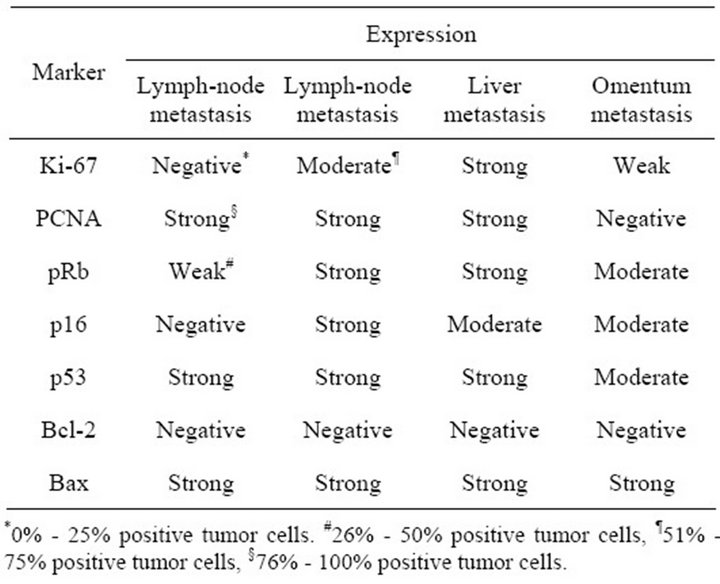
Table 4. Immunostaining of Ki-67, PCNA, pRb, p16, p53, Bcl-2 and Bax in lymph-node, liver and omentum metastases from esophageal adenocarcinoma.
tosis. Thus, the versatile nature of PCNA impedes the unambiguous interpretation of its expression. Additionally, the PCNA expression could be overestimated, because PCNA has long half-life and could be found in post-proliferative cell state. Nevertheless, the observed enhanced PCNA expression is most likely a sign of prominent proliferation.
Limited and controversial data exist about the expression of the cell cycle regulator and tumor suppressor retinoblastoma protein (pRb) in EAC pathogenesis. Some studies showed that loss of heterozygosity (LOH) at 13q, the pRb locus, is a common event in EAC [14,28,29]. In contrast, other studies have reported rarer loss of Rb gene in patients with HGD and EAC [30,31]. Using IHC, Sarbia et al. found enhanced expression of pRB in HGD and EAC [31]. Conversely, Coppola et al. observed negative pRb staining in 39% of the EAC cases, focal/weak pRb staining in 41% of the EAC cases and gradual pRb loss in the progression to invasive EAC [32]. Similarly, we detected lack of pRb in 6 (40%) of the EAC samples and pRb loss in the all dysplasia samples. The absence of pRb suggests that the tumor suppressor function of pRb is switched off in these samples and the esophageal tissue is predisposed to a high cell turnover. We found pRb expression in 9 (60%) of the samples with EAC, but manifested homogeneous pRb staining (>50%) was considered as abnormal, while nonhomogeneous, weak expression of pRb was considered as normal. Our hypothesis was based on the characteristic of the used anti-human pRb antibody, which clone G3-245 recognizes different forms of pRb (underphosphorylated, phosphorylated and hyperphosphorylated). Thus, the moderate and strong pRb expression in 5 (33%) of the EAC samples is probably due to the non-active hyperphosphorylated form of pRb (ppRb) and indicates an increased proliferation, whereas the weak pRb expression in 4 (27%) of the EAC samples is associated with the active, underphosphorylated pRb. The single appreciable difference in the expression between the tumor and dysplasia samples was found for pRb, but this result should be interpreted cautiously due to the small number of samples. Nevertheless, the lack of pRb expression in the dysplasia samples suggests the existence of diverse molecular pathways in the progression of BE.
Another very important molecular player in the carcinogenesis of EAC is a cyclin-dependent kinases (cdk) inhibitor p16. Tumor suppressor p16 blocks cdk4/6 phosphorylation of pRb and G1/S phase cell cycle transition. Inactivation of p16 is often observed in EAC. In the present study, expression of p16 was lost in 9 (60%) of the EAC samples and in 1 (20%) of the dysplasia samples. Our data are concordant with the reports of former studies [33-35]. Loss of p16 contributes to the derailment of the cell cycle and permits an uncontrolled proliferation in neoplastic esophageal tissue. Observed simultaneous and noticeable expression of Ki-67 and PCNA in all p16 immunostained samples, however, presumes that the antiproliferative protective mechanisms of p16 could be accidentally surmounted in EAC carcinogenesis.
The tumor suppressor protein p53 is a crucial factor for the maintenance of the genomic stability. The wild type p53 is usually not detectable in the normal cells, but abnormal truncated p53 protein could be detected by IHC. Mutations and LOH of p53 are reported in the majority of esophageal adenocarcinomas [36,37]. The rates of immunohistochemical expression of p53 in EAC range from 37% to 90% [26,38,39]. We observed p53 expression in all 15 (100%) esophageal cancer samples. In agreement with the data presented by Krishnadath et al. [40], significant p53 expression was also found in dysplasia samples. Our findings suggest that p53 is implicated in the carcinogenesis of EAC. It must be pointed, however, that the relation between p53 gene mutation and p53 protein expression is questionable [41]. Nonetheless, we speculate that p53 is very important molecule in EAC pathogenesis and the loss of functional p53 is a prerequisite for apoptotic disorders in EAC. Data concerning the prognostic role of p53 in EAC are conflicting. Some studies support the prognostic impact of p53 in patients with EAC [38]. Other studies deny any connection between prognosis and EAC [33,41]. In our study, p53 expression was correlated with substantial risk of developing EAC, which suggests that p53 could be used as prognostic marker. Patients with strong p53 expression, however, showed slightly better survival rate than the patients with weak and moderate p53 expression. This surprising result could be explained by the small number of investigated patients. It is also possible that the observed strong p53 expression is partially connected with the accumulation of wild type p53. Additional studies should be performed to verify the prognostic role of p53 in EAC pathogenesis.
The significance of the antiapoptotic oncoprotein Bcl-2 in EAC pathogenesis is still disputable. Shimizu et al. found increasing expression of Bcl-2 mRNA in the progression of BE [42]. Soslow et al., using IHC, reported a linear association between Bcl-2/Bax ratio and increasing histological severity during the evolution of BE [43]. In contrast, more studies identified a significant reduction and loss of Bcl-2 in the advancement of BE to EAC [23,44-50]. We observed weak expression of Bcl-2 only in 1 (7%) of the EAC samples and in 1 (20%) of the dysplasia samples, while Bcl-2 staining was found predominantly in the lymphocytes. Similarly, Goldblum et al. did not find Bcl-2 expression in any of the cases with EAC and BE-associated dysplasia, but Bcl-2 was detected only in the proliferative zone of non-neoplastic tissue and in the adjacent lymph follicles [47]. In our study, the negligible Bcl-2 expression could be an evidence of the minor antiapoptotic role, which Bcl-2 plays in EAC and BE-associated dysplasia. The intriguing data that Bcl-2 availability, one of the most important antiapoptotic molecules, is not indispensable for BE-associated dysplasia and EAC, deserve attention and require further research.
The ratio of proapoptotic to antiapoptotic proteins, including Bax/Bcl-2 could trigger the apoptosis. Using a semiquantitative RT-PCR, Bax et al. found increased Bax/Bcl-2 ratio in HGD samples of patients with BE [51]. Bax immunostaining is studied more intensively in esophageal squamous cell carcinoma with conflicting data [52,53]. In the present study, almost all of the analyzed samples showed Bax immunopositivity. Our data yield results similar to previous studies [44,49]. By contrast with these reports, van der Woude et al. reported a decreased Bax expression in the evolution of BE [48]. The expression of Bax in our study most probably is not prompted by p53-dependent signaling pathway due to the presumed inactivation of p53 in the analyzed samples. Myashita et al. have shown that Bax expression could be induced and by p53-independent mechanisms [54]. The remarkable immunostaining pattern of proapoptotic Bax in the malignant tissue is a paradoxical molecular event to a certain extent. The observed Bax expression could be a cellular response to the high proliferation rate, although this supposed safety mechanism probably is not working in EAC and BE-associated dysplasia.
Despite improvements in surgical and neoadjuvant therapy, dissemination of malignant cells is one of the major reasons for dismal outcome in patients with EAC. Immunohistochemical expression of metastases from EAC is not sufficiently studied. The overall staining of the markers in the analyzed metastases samples was similar, although not completely equal to the staining in the corresponding esophageal adenocarcinomas. Nevertheless, the expression in the metastases most likely is a proof for the aggressive behavior of the primary EAC and mirrors the injured equilibrium between the proliferation and apoptosis in the secondary lesions.
Limitations of this study include the small number of selected patients and natural heterogeneity of the tissue specimens. Technical pitfalls of IHC technique should also be considered, because differences in antigen retrieval procedure, numerous scoring systems and diverse antibody clones are sources for various interpretations of protein expression.
In summary, this study indicates that Ki-67, PCNA, pRb, p16, p53 and Bax are involved in the carcinogenesis of EAC. The Bcl-2 influence upon the cell fate in the end of Barrett’s M-D-A sequence is insignificant. Disturbed balance between cell proliferation and apoptosis is necessary for the development of EAC and BE-associated dysplasia. Immunostaining pattern of p53 could be used as predictive and prognostic factor in EAC. More studies should be done to validate our data. Future larger studies will clarify the precise role of proliferation and apoptosis in EAC carcinogenesis.
5. Acknowledgements
This study was supported by the National Science Fund, Ministry of Education and Science.
REFERENCES
- D. A. Corley, T. R. Levin, L. A. Habel, N. S. Weiss and P. A. Buffler, “Surveillance and Survival in Barrett’s Adenocarcinomas: A Population-Based Study,” Gastroenterology, Vol. 122, No. 3, 2002, pp. 633-640. doi:10.1053/gast.2002.31879
- E. Bollschweiler, E. Wolfgarten, C. Gutschow and A. H. Hölscher, “Demographic Variations in the Rising Incidence of Esophageal Adenocarcinoma in White Males,” Cancer, Vol. 92, No. 3, 2001, pp. 549-555. doi:10.1002/1097-0142(20010801)92:3<549::AID-CNCR1354>3.0.CO;2-L
- J. Lagergren, R. Bergström, A. Lindgren and O. Nyrén, “Symptomatic Gastroesophageal Reflux as a Risk Factor for Esophageal Adenocarcinoma,” The New England Journal of Medicine, Vol. 340, No. 11, 1999, pp. 825-831. doi:10.1056/NEJM199903183401101
- A. J. Cameron, B. J. Ott and W. S. Payne, “The Incidence of Adenocarcinoma in Columnar-Lined (Barrett’s) Esophagus,” The New England Journal of Medicine, Vol. 313, No. 14, 1985, pp. 857-859. doi:10.1056/NEJM199903183401101
- M. Lindblad, L. A. Rodríguez and J. Lagergren, “Body mass, Tobacco and Alcohol and Risk of Esophageal, Gastric Cardia, and Gastric Non-Cardia Adenocarcinoma among Men and Women in a Nested Case-Control Study,” Cancer Causes and Control, Vol. 16, No. 3, 2005, pp. 285-294. doi: 10.1007/s10552-004-3485-7
- M. B. Cook, F. Kamangar, D. C. Whiteman, et al., “Cigarette Smoking and Adenocarcinomas of the Esophagus and Esophagogastric Junction: A Pooled Analysis from the International BEACON Consortium,” Journal of the National Cancer Institute, Vol. 102, No. 17, 2010, pp. 1344-1353. doi:10.1093/jnci/djq289
- N. D. Freedman, L. J. Murray, F. Kamangar, et al., “Alcohol Intake and Risk of Oesophageal Adenocarcinoma: A Pooled Analysis from the BEACON Consortium,” Gut, Vol. 60, No. 8, 2011, pp. 1029-1037. doi:10.1136/gut.2010.233866
- C. A. González, G. Pera, A. Agudo, et al., “Fruit and Vegetable Intake and the Risk of Stomach and Oesophagus Adenocarcinoma in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST),” International Journal of Cancer, Vol. 118, No. 10, 2006, pp. 2559-2566. doi:10.1002/ijc.21678
- P. Terry, J. Lagergren, H. Hansen, A. Wolk and Nyrén O, “Fruit and Vegetable Consumption in the Prevention of Oesophageal and Cardia Cancers,” European Journal of Cancer Prevention, Vol. 10, No. 4, 2001, pp. 365-369.
- N. Shaheen and D. F. Ransohoff, “Gastroesophageal ReFlux, Barrett Esophagus, and Esophageal Cancer: Scientific Review,” Journal of the American Medical Association, Vol. 287, No. 15, 2002, pp. 1972-1981. doi:10.1001/jama.287.15.1972
- J. Jankowski, D. Provenzale and P. Moayyedi, “Esophageal Adenocarcinoma Arising from Barrett’s Metaplasia Has Regional Variations in the West,” Gastroenterology, Vol. 122, No. 2, 2002, pp. 588-590.
- W. Meyer, F. Vollmar and W. Bär, “Barrett-Esophagus Following Total Gastrectomy. A Contribution to It’s Pathogenesis,” Endoscopy, Vol. 11, No. 2, 1979, pp. 121- 126.
- M. T. Barrett, C. A. Sanchez, P. C. Galipeau, K. Neshat, M. Emond and B. J. Reid, “Allelic Loss of 9p21 and Mutation of the CDKN2/P16 Gene Develop as Early Lesions during Neoplastic Progression in Barrett’s Oesophagus,” Oncogene, Vol. 13, No. 9, 1996, pp. 1867-1873.
- M. T. Barrett, C. A. Sanchez, L. J. Prevo, et al., “Evolution of Neoplastic Cell Lineages in Barrett Oesophagus,” Nature Genetics, Vol. 22, No. 1, 1999, pp. 106-109. doi: 10.1038/8816
- D. J. Wong, T. G. Paulson, L. J. Prevo, et al., “pl6INK4a Lesions Are Common, Early Abnormalities That Undergo Clonal Expansion in Barrett’s Metaplastic Epithelium,” Cancer Research, Vol. 61, No. 22, 2001, pp. 8284-8289.
- P. L. Blount, P. C. Galipeau, C. A. Sanchez, et al., “17p Allelic Losses in Diploid Cells of Patients with Barrett’s Esophagus Who Develop Aneuploidy,” Cancer Research, Vol. 54, No. 9, 1994, pp. 2292-2295.
- P. C. Galipeau, L. J. Prevo, C. A. Sanchez, G. M. Longton and B. J. Reid, “Clonal Expansion and Loss of Heterozygosity at Chromosomes 9p and 17p in Premalignant Esophageal (Barrett’s) Tissue,” Journal of the National Cancer Institute, Vol. 91, No. 24, 1999, pp. 2087-2095. doi: 10.1093/jnci/91.24.2087
- J. A. Jankowski, N. A. Wright, S. J. Meltzer, et al., “Molecular Evolution of the Metaplasia-Dysplasia-Adenocarcinoma Sequence in the Esophagus,” The American Journal of Pathology, Vol. 154, No. 4, 1999, pp. 965-973. doi:10.1016/S0002-9440(10)65346-1
- C. C. Maley, P. C. Galipeau, J. C. Finley, et al., “Genetic Clonal Diversity Predicts Progression to Esophageal Adenocarcinoma,” Nature Genetics, Vol. 38, No. 4, 2006, pp. 468-473. doi:10.1038/ng1768
- M. K. Hong, W. B. Laskin, B. E. Herman, et al., “Expansion of the Ki-67 Proliferative Compartment Correlates with Degree of Dysplasia in Barrett’s Esophagus,” Cancer, Vol. 75, No. 2, 1995, pp. 423-429. doi:10.1002/1097-0142(19950115)75:2<423::AID-CNCR2820750202>3.0.CO;2-5
- L. Yacoub, H. Goldman and R. Odze, “Transforming Growth Factor-Alpha, Epidermal Growth Factor Receptor, and MiB-1 Expression in Barrett’s-Associated Neoplasia: Correlation with Prognosis,” Modern Pathology, Vol. 10, No. 2, 1997, pp. 105-112.
- M. Binato, R. R. Gurski, R. B. Fagundes, L. Meurer and M. I. Edelweiss, “p53 and Ki-67 Overexpression in Gastroesophageal Reflux Disease—Barrett’s Esophagus and Adenocarcinoma Sequence,” Diseases of the Esophagus, Vol. 22, No. 7, 2009, pp. 588-595. doi:10.1111/j.1442-2050.2009.00953.x
- N. Rioux-Leclercq, B. Turlin, F. Sutherland, et al., “Analysis of Ki-67, p53 and Bcl-2 Expression in the Dysplasia-Carcinoma Sequence of Barrett’s Esophagus,” Oncology Reports, Vol. 6, No. 4, 1999, pp. 877-882.
- M. Feith, H. J. Stein, J. Mueller and J. R. Siewert, “Malignant Degeneration of Barrett’s Esophagus: The Role of the Ki-67 Proliferation Fraction, Expression of E-Cadherin and p53,” Diseases of the Esophagus, Vol. 17, No. 4, 2004, pp. 322-327. doi:10.1111/j.1442-2050.2004.00434.x
- J. Jankowski, R. McMenemin, C. Yu, D. Hopwood and K. G. Wormsley, “Proliferating Cell Nuclear Antigen in Oesophageal Diseases; Correlation with Transforming Growth Factor Alpha Expression,” Gut, Vol. 33, No. 5, 1992, pp. 587-591. doi:10.1136/gut.33.5.587
- R. Kim, M. R. Clarke, M. F. Melhem, et al., “Expression of p53, PCNA, and C-erbB-2 in Barrett’s Metaplasia and Adenocarcinoma,” Digestive Diseases and Sciences, Vol. 42, No. 12, 1997, pp. 2453-2462. doi:10.1023/A:1018891923998
- P. Gillen, M. McDermott, D. Grehan, D. O. Hourihane and T. P. Hennessy, “Proliferating Cell Nuclear Antigen in the Assessment of Barrett’s Mucosa,” British Journal of Surgery, Vol. 81, No. 12, 1994, pp. 1766-1768. doi:10.1002/bjs.1800811219
- Y. Huang, R. F. Boynton, P. L. Blount, et al., “Loss of Heterozygosity Involves Multiple Tumor Suppressor Genes in Human Esophageal Cancers,” Cancer Research, Vol. 52, No. 23, 1992, pp. 6525-6530.
- R. F. Boynton, Y. Huang, P. L. Blount, et al., “Frequent Loss of Heterozygosity at the Retinoblastoma Locus in Human Esophageal Cancer,” Cancer Research, Vol. 51, No. 20, 1991, pp. 5766-5769.
- S. H. Doak, G. J. Jenkins, E. M. Parry, et al., “Chromosome 4 Hyperploidy Represents an Early Genetic Aberration in Premalignant Barrett’s Oesophagus,” Gut, Vol. 52, No. 5, 2003, pp. 623-628. doi:10.1136/gut.52.5.623
- M. Sarbia, U. Tekin, M. Zeriouh, A. Donner and H. E. Gabbert, “Expression of the Rb Protein, Allelic Imbalance of the RB Gene and Amplification of the CDK4 Gene in Metaplasias, Dysplasias and Carcinomas in Barrett’s Oesophagus,” Anticancer Research, Vol. 21, No. 1A, 2001, pp. 387-392.
- D. Coppola, R. H. Schreiber, L. Mora, W. Dalton and R. C. Karl, “Significance of Fas and Retinoblastoma Protein Expression during the Progression of Barrett’s Metaplasia to Adenocarcinoma,” Annals of Surgical Oncology, Vol. 6, No. 3, 1999, pp. 298-304. doi:10.1007/s10434-999-0298-7
- R. Langer, B. H. A. Von Rahden, J. Nahrig, et al., “Prognostic Significance of Expression Patterns of cerbB-2, p53, p16 INK4A, p27KIP1, cyclin D1 and Epidermal Growth Factor Receptor in Oesophageal Adenocarcinoma: A Tissue Microarray Study,” Journal of Clinical Pathology, Vol. 59, No. 6, 2006, pp. 631-634. doi:10.1136/jcp.2005.034298
- L. J. Hardie, S. J. Darnton, Y. L. Wallis, et al., “p16 Expression in Barrett’s Esophagus and Esophageal Adenocarcinoma: Association with Genetic and Epigenetic Alterations,” Cancer Letters, Vol. 217, No. 2, 2005, pp. 221-230. doi:10.1016/j.canlet.2004.06.025
- Y. S. Bian, M. C. Osterheld, C. Fontolliet, F. T. Bosman and J. Benhattar, “p16 Inactivation by Methylation of the CDKN2A Promoter Occurs Early during Neoplastic Progression in Barrett’s Esophagus,” Gastroenterology, Vol. 122, No. 4, 2002, pp. 1113-1121. doi:10.1053/gast.2002.32370
- R. Hamelin, J. F. Flejou, F. Muzeau, et al., “TP53 Gene Mutations and p53 Protein Immunoreactivity in Malignant and Premalignant Barrett’s Esophagus,” Gastroenterology, Vol. 107, No. 4, 1994, pp. 1012-1018.
- P. L. Blount, S. J. Meltzer, J. Yin, Y. Huang, M. J. Krasna and B. J. Reid, “Clonal Ordering of 17p and 5q Allelic Losses in Barrett Dysplasia and Adenocarcinoma,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 90, No. 8, 1993, pp. 3221-3225.
- A. G. Casson, S. C. Evans, A. Gillis, et al., “Clinical Implications of p53 Tumor Suppressor Gene Mutation and Protein Expression in Esophageal Adenocarcinomas: Results of a Ten-Year Prospective Study,” Journal of Thoracic and Cardiovascular Surgery, Vol. 125, No. 5, 2003, pp. 1121-1131. doi:10.1067/mtc.2003.176
- M. Younes, R. M. Lebovitz, L. V. Lechago and J. Lechago, “p53 Accumulation in Barrett’s Metaplasia, Dysplasia and Carcinoma: A Follow-Up Study,” Gastroenterology, Vol. 105, No. 6, 1993, pp. 1637-1642.
- K. K. Krishnadath, H. W. Tilanus, M. van Blankenstein, F. T. Bosman and A. H. Mulder, “Accumulation of p53 Protein in Normal, Dysplastic and Neoplastic Barrett’s Esophagus,” Journal of Pathology, Vol. 175, No. 2, 1995, pp. 175-180. doi:10.1002/path.1711750204
- G. Coggi, S. Bosari, M. Roncalli, et al., “p53 Protein Accumulation and p53 Gene Mutation in Esophageal Carcinoma. A Molecular and Immunohistochemical Study with Clinicopathologic Correlations,” Cancer, Vol. 79, No. 3, 1997, pp. 425-432. doi:10.1002/(SICI)1097-0142(19970201)79:3<425::AID-CNCR1>3.0.CO;2-H
- D. Shimizu, D. Vallböhmer, H. Kuramochi, et al., “Increasing Cyclooxygenase-2 (cox-2) Gene Expression in the Progression of Barrett’s Esophagus to Adenocarcinoma Correlates with That of Bcl-2,” International Journal of Cancer, Vol. 119, No. 4, 2006, pp. 765-770. doi:10.1002/ijc.21922
- R. A. Soslow, H. Remotti, R. N. Baergen and N. K. Altorki, “Suppression of Apoptosis Does Not Foster Neoplastic Barrett’s Esophagus,” Modern Pathology, Vol. 12, No. 3, 1999, pp. 239-250.
- A. A. Raouf, D. A. Evoy, E. Carton, E. Mulligan, M. M. Griffin and J. V. Reynolds, “Loss of Bcl-2 Expression in Barrett’s Dysplasia and Adenocarcinoma Is Associated with Tumor Progression and Worse Survival but Not with Response to Neoadjuvant Chemoradiation,” Diseases of the Esophagus, Vol. 16, No. 1, 2003, pp. 17-23. doi:10.1046/j.1442-2050.2003.00281.x
- P. Bhargava, G. M. Eisen, D. A. Holterman, et al., “Endoscopic Mapping and Surrogate Markers for Better Surveillance in Barrett Esophagus. A Study of 700 Biopsy Specimens,” American Journal of Clinical Pathology, Vol. 114, No. 4, 2000, pp. 552-563. doi:10.1309/93WG-ERRB-PN57-C15A
- G. Y. Lauwers, O. Kandemir, P. S. Kubilis and G. V. Scott, “Cellular Kinetics in Barrett’s Epithelium Carcinogenic Sequence: Role of Apoptosis, Bcl-2 Protein and Cellular Proliferation,” Modern Pathology, Vol. 10, No. 12, 1997, pp. 1201-1208.
- J. R. Goldblum and T. W. Rice, “Bcl-2 Protein Expression in the Barrett’s Metaplasia-Dysplasia-Carcinoma Sequence,” Modern Pathology, Vol. 8, No. 8, 1995, pp. 866-869.
- C. J. van Der Woude, P. L. Jansen, A. T. Tiebosch, et al., “Barrett’s Metaplasia-Dysplasia-Carcinoma Sequence: A Switch to a More Resistant Phenotype,” Human Pathology, Vol. 33, No. 7, 2002, pp. 686-692. doi:10.1053/hupa.2002.124908
- T. A. Woodward, P. D. Klinger, P. V. Genko and J. T. Wolfe, “Barrett’s Esophagus, Apoptosis and Cell Cycle Regulation: Correlation of p53 with Bax, Bcl-2, and p21 Protein Expression,” Anticancer Research, Vol. 20, No. 4, 2000, pp. 2427-2432.
- N. Katada, R. A. Hinder, T. C. Smyrk, et al., “Apoptosis Is Inhibited Early in the Dysplasia-Carcinoma Sequence of Barrett’s Oesophagus,” Archives of Surgery, Vol. 132, No. 7, 1997, pp. 728-733. doi:10.1001/archsurg.1997.01430310042007
- D. A. Bax, J. Haringsma, A. W. Einerhand, et al., “MUC4 Is Increased in High Grade Intraepithelial Neoplasia in Barrett’s Oesophagus and Is Associated with a Proapoptotic Bax to Bcl-2 Ratio,” Journal of Clinical Pathology, Vol. 57, No. 12, 2004, pp. 1267-1272. doi:10.1136/jcp.2004.017020
- I. Sturm, H. Petrowsky, R. Volz, et al., “Analysis of p53/BAX/pl6ink4a/CDKN2 in Esophageal Squamous Cell Carcinoma: High BAX and p16 ink4a/CDKN2 Identifies Patients with Good Prognosis,” Journal of Clinical Oncology, Vol. 19, No. 8, 2001, pp. 2272-2281.
- A. Kurabayashi, M. Furihata, M. Matsumoto, Y. Ohtsuki, S. Sasaquri and S. Ogoshi, “Expression of Bax and Apoptosis-Related Proteins in Human Esophageal Squamous Cell Carcinoma Including Dysplasia,” Modern Pathology, Vol. 14, No. 8, 2001, pp. 741-747. doi:10.1038/modpathol.3880383
- T. Miyashita, S. Krajewski, M. Krajewska, et al., “Tumor Suppressor p53 Is a Regulator of Bcl-2 and Bax Gene Expression in Vitro and in Vivo,” Oncogene, Vol. 9, No. 6, 1994, pp. 1799-1805.