Neuroscience & Medicine
Vol.3 No.1(2012), Article ID:17746,14 pages DOI:10.4236/nm.2012.31016
Valproic Acid, a Drug with Multiple Molecular Targets Related to Its Potential Neuroprotective Action
![]()
1Faculty of Medicine of Juazeiro do Norte (FMJ), Juazeiro do Norte, Brazil; 2Federal University of São Paulo (UNIFESP), São Paulo, Brazil.
Email: *gbviana@live.com
Received December 9th, 2011; revised January 14th, 2012; accepted February 15th, 2012
Keywords: Valproic Acid; Histone Deacetylases (HDACs) Inhibitors; Inflammation; Neuroprotection
ABSTRACT
Valproic acid (VA) is used worldwide as an antiepileptic drug and a mood stabilizer. Recently, VA was shown to act on cell growth, differentiation and apoptosis, by regulating gene expression at the molecular level, through epigenetic mechanisms. Thus, VA was demonstrated to act on the chromatin remodeling what is a consequence of the drug inhibition of histone deacetylases (HDACs) activity. Other studies uncovered the potential of VA to interfere with multiple regulatory mechanisms besides HDACs, as the GSK3 alpha and beta, Akt, ERK and phosphoinositol pathways, tricarboxylic acid cycle, GABA and OXPHOS system. The review focuses on the mechanisms of action of VA, showing that HDAC inhibitors, as VA, can be successfully used in the treatment of neurodegenerative disorders. This molecule, whose biological activities range from interactions with receptors and ion channels to the regulation of many catalytic reactions, has a central role in cellular cascades that regulate gene expression. Thus, inhibitors of HDACs, by positively affecting both neuronal degeneration and cognitive deficits, appear as promising drugs against various pathological conditions and neurodegenerative diseases. VA is known to present anti-inflammatory and antioxidative properties. And, since inflammation and oxidative stress are common links in neurodegeneration, VA is a drug that, from a clinical point of view, shows a great potential as a candidate for the treatment of neurodegenerative diseases related to excitotoxic events.
1. Introduction
Valproic acid (VA, 2-propylvaleric acid, 2-propylpentanoic acid or n-dipropylacetic acid) is a short-chain fatty acid (Figure 1) synthesized in 1882, as an analogue of valeric acid (present in the species Valeriana officinalis). VA was used as a metabolically inert solvent for organic compounds for eight decades. In 1962, Pierre Eymard, a research student at the University of Lyon (France) and colleagues, investigating potentially anticonvulsant drugs with low aqueous solubility, used VA as a solvent [1]. A strong anticonvulsant activity was observed in all solutions were VA was present, leading these authors to study VA as a potentially useful agent for epilepsy [2]. VA is rapidly metabolized in the liver, and the various resulting metabolites are also pharmacologically active [3].
VA bioavailability is attributed to the high permeability of the cell membrane to this compound and the virtual absence of first pass hepatic mechanism. In a previous work, the mechanisms of active transport of VA into the brain were demonstrated. This active transport results in high levels of VA in the CNS, minutes after its parenteral administration, in spite of its association with plasma proteins [4].
2. Clinical Uses of VA
VA is widely used in clinics as an anti-epileptic drug and for the therapy of bipolar disorders and migraine prophylaxis. The mechanism of VA action was initially related to its effects on amino acid neurotransmitters and modulation of intracellular pathways. Recently, VA showed pharmacological characteristics similar to those of other anticancer agents, acting on cell growth, differentiation and apoptosis. Other studies [5] showed that the drug exerts
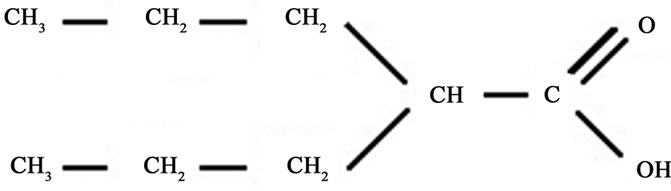
Figure 1. Molecular structure of Valproic acid (VA).
its effects by regulating gene expression at the molecular level, through epigenetic mechanisms. Thus, VA was demonstrated to act on the chromatin remodeling, a consequence of the drug inhibition of histone deacetylases (HDACs) activity, as it will be discussed later.
VA is generally regarded as a first choice agent for most forms of idiopathic and symptomatic generalized epilepsy. Many of these syndromes are associated with various types of seizures, including tonic-clonic, absence and myoclonic seizures. Thus, the prescription of broadspectrum antiepileptic drugs such as VA presents clear advantages. Evidences suggest that VA administered intravenously may be of value in the treatment of status epilepticus and non-convulsive seizures, but further studies are needed to determine its role in these clinical indications [6].
VA mechanism of action with respect to its antiepileptic effect is related to increased concentrations of the inhibitory neurotransmitter gamma-aminobutyric acid (GABA). In vitro studies showed that VA increases the activity of glutamic acid decarboxylase (GAD), an enzyme responsible for the synthesis of GABA, and inhibits the activity of the enzyme responsible for its degradation, GABA transaminase (GABA-T). Similarly to other anticonvulsants, such as carbamazepine and phenytoin, VA decreases the recovery time of the voltage-dependent sodium channel when in its inactivated state. In addition, VA limits the activity of calcium channels of type T, an action resembling that presented by ethosuximide, another anticonvulsant drug. It is accepted that reduction of the GABAergic activity leads to a pro-convulsant action, while its increase results in anticonvulsant effects. Moreover, differences of the VA effect on GABA concentrations in different brain areas were found [3].
There are at least four major mechanisms for increasing GABA concentrations in the brain: 1) inhibition of GABA degradation, 2) increase of GABA synthesis, 3) decrease in GABA turnover, and 4) reduction in the reuptake of GABA. The region with the greatest increase in VAinduced GABA production is the substantia nigra [3].
In the regulation of mood disorders, VA has been proven effective for decreasing the excitatory glutamatergic and increasing the GABAergic inhibitory neurotransmissions. Similarly to lithium, intracellular mechanisms of VA also include the regulation of various signaling pathways of protein kinases and gene expression. However, the effects of VA on the excitatory and inhibitory brain neurotransmitter systems, only partly justify the antiepileptic and neuroprotective effects of this drug. In patients, therapeutic doses of VA were observed to increase brain levels of GABA without affecting glutamate levels [7].
One of VA action mechanisms is to affect the neuronal function through modulation of ion channels. With respect to voltage-gated channels, VA reduces neuronal excitability, by acting on the conductances of sodium and potassium channels [8]. Data from the literature lead us to conclude that the direct effects of VA on voltage-gated ion channels are possibly less important for neuroprotective properties of antiepileptic drugs than the mechanisms involved in the regulation of cellular signaling pathways [5].
Recent studies have found beneficial effects of VA in the treatment of vascular and chronic daily headaches that may have components of migraine or tension headaches. Subsequently, placebo-controlled, randomized and prospective clinical trials demonstrated the efficacy of VA in the treatment of acute and chronic migraine. Thus, unlike what is already known in relation to neurological and psychiatric clinical uses of VA, its prescription to the therapy of migraine was proven to be effective in less than a decade [2].
Thus, sodium divalproex (a stable combination of sodium valproate and valproic acid in a 1:1 ratio) was approved by the Food and Drug Administration (FDA) for the prophylaxis of migraine, since the year 1996. A recent study revealed that 10% of American neurologists consider Depakote the first or second choice for migraine prophylaxis. According to a recent work, VA has proven to be efficacious in migraine prevention and is suitable for routine clinical use [9]. An important caveat must be taken into account, because this drug has a known teratogenic effect and its prescription to women in childbearing age should be done with caution [10].
Studies on mania and depression using VA started around the late nineties, when other antiepileptic agents were being tested in affective disorders. The drug’s approval by the FDA in 1995, for the treatment of mania, was based on randomized, prospective and blinded clinical studies by comparing VA with placebo or lithium. Overall, the kinetics of VA and its adverse effects appeared to be somehow different in observations of bipolar disorder and epilepsy. More recently, a controlled clinical trial found several aspects of the attenuation of depressive morbidity in patients with bipolar disorders chronically treated with VA, as compared to lithium.
3. VA and Neuroprotection
The term neuroprotection refers to a set of treatments or measures designed to protect nerve tissues against adverse cellular events induced by a reduced supply of oxygen, glucose or both to the brain [11]. The development of new drugs with a neuroprotective profile is necessary, due to the increasing number of diseases related to processes of neurodegeneration such as Alzheimer’s or Parkinson’s diseases and bipolar affective disorders. The further study of drugs already known is also of fundamental importance, since they may eventually be used in the treatment of neurodegenerative diseases [12]. Some anticonvulsants show neuroprotective effects and may be beneficial in reducing neuronal death. Thus, recently, the use of antiepileptic drugs as a possible neuroprotective strategy in brain ischemia is receiving increased attention, and many of them have been tested in animal models of stroke, providing encouraging results [13].
There is in the market a wide variety of pharmacological agents, used alone or in combination, which have the potential to offer protection to brain tissues over periods of hypoxia or ischemia. Nowadays, the use of neuroprotective agents in combination with hypothermia has supporters and also outspoken critics. There are several protocols in use and their advocates illustrate promising results, while other investigators point out to the ineffectiveness of their use. It is expected that, in the near future, pharmacological neuroprotection constitutes an important resource during procedures in which the cerebral circulation is interrupted or modified [11].
VA, with recognized properties of an antimania and antiepileptic drug, has recently been characterized as a neuroprotective drug. Some anticonvulsants show neuroprotective effects, and may be beneficial in reducing neuronal death resulting from cerebrovascular accidents (CVA) [14,15]. It is of interest to note that the excessive release of excitatory amino acids or reduction of the inhibitory amino acid GABA occur not only in epilepsy but also in cerebral ischemia [16,17]. The objective of the use of antiepileptic drugs as neuroprotective agents is to balance the abnormal brain excitability, by decreasing the excitatory transmission and/or increasing the neuronal inhibition [18,19].
It is known that antioxidant properties contribute to the neuroprotective effects of VA and lithium, and the promotion of antioxidant defense to the body may be in general the primary mechanism of neuroprotection by VA [5]. Recent evidence has demonstrated that VA reduces brain damage, leading to a functional improvement in a model of transient focal cerebral ischemia in rats, by modulating the activity of caspases [20]. This raises the possibility that the neuroprotective effect of VA requires a repeated treatment and probably involves apoptotic and not necrotic mechanisms [21,22]. It is known that the pathophysiology of cerebral ischemia is associated with multiple mechanisms, including neuroinflammation mediated by activated microglia and the infiltration of macrophages/monocytes.
In a recent work [23], the model of permanent occlusion of the middle cerebral artery (MCA) was used to study the effects of histone deacetylases (HDACs) inhibition in brain infarction induced by ischemia, neuroinflammation, gene expression and neurological deficits. The authors showed that inhibitors of HDACs as VA decreased the volume of cerebral infarction, suppressed the activation of microglia, reduced the number of these cells and inhibited other markers of ischemic brain, with no reduction on the levels of H3 acetylated histone. VA and other inhibitors of HDACs caused an increased expression of HSP70 (a 70 KDa inducible heat shock protein involved in excitatory neuropathologies), blocked the “downregulation” of phospho-Akt (being Akt a serine/threonine protein kinase that mediates growth factor associated with cell survival, and whose phosphorylation or phosphoAkt has been observed in human cancers) and “upregulation” of p53 (it is a protein crucial in multicellular organisms, where it regulates the cell cycle and, thus, functions as a tumor suppressor involved in preventing cancer), inducible nitric oxidase synthase (iNOS) and cyclooxygenase (COX-2), as observed in the model of permanent occlusion of MCA.
Other authors [20] showed, in a global model of transient focal ischemia in rats, that VA decreases the infarct size and reduces ischemia-induced neurological deficits. These authors also found that VA suppressed the activation of caspase-3 in the cerebral cortex induced by ischemia, increased levels of acetylated histone H3 in the cortex and striatum, and caused upregulation in the levels of HSP70 in both the ipsilateral and contralateral portions of cerebral hemispheres. These data indicate that VA has neuroprotective properties in cerebral ischemia models resulting from the inhibition of HDACs and induction of HSP70.
The neuroprotective effects, characteristic of mood stabilizers such as lithium and valproate, can be seen in conditions such as acute and chronic treatments with amphetamine [24], glutamatergic excitotoxicity [25,26] and neurotoxicity caused by accumulation of the amyloid protein [27]. In fact, studies have shown that lithium and valproate have the ability to mitigate certain neurodegenerative events, such as lipid peroxidation and protein oxidation induced by excitotoxicity [28,29], oxidative stress produced by FeCl3 [25], and brain damage caused by ischemia [30].
Despite their wide use in clinics, the mechanisms by which lithium and VA exert their effects are not yet completely understood [31]. It is known that lithium acts on a number of neurotransmitter systems and mechanisms for transmitting signals, as the hydrolysis of phosphoinositols, G proteins such as adenylate cyclase, glycogen synthase kinase (GSK-3) and protein kinase C (PKC) [26]. GSK-3 is a ubiquitous cellular serine/threonine protein kinase initially shown to regulate the glycogen metabolism and to inhibit the insulin signaling pathway [32-34]. It is involved in the formation of neuropathological lesions, cholinergic depletion and cell death associated with Alzheimer’s disease [35]. Such effects are believed to be responsible for long-term changes in neural transmission that lead to the prophylactic effects of lithium in the treatment of bipolar disorders. Through its effects on GSK-3 and PKC, lithium may also alter the phosphorylation of cytoskeletal proteins, which leads to plastic changes [36]. Lately, the in vivo inhibition of GSK-3 was shown to produce antidepressive-like behaviors [37], suggesting the potential of GSK-3 inhibitors as antidepressants.
The role of VA on kinase signaling pathways is well established and, in this respect, there are differences and similarities between the effect of VA and that produced by lithium [38]. In particular, VA activates protein kinases important for neuronal survival, such as Akt (also known as PKB) and mitogen-activated protein kinase (MAPKs), among others, and inhibits glycogen synthase kinase (GSK-3β). The cycle of phosphoinositol is particularly important to explain the effects of VA on neuroprotection, and is closely associated with the PI3K/PKB pathway. This pathway impinges upon a remarkable array of intracellular events, either directly or indirectly influencing whether or not a cell will undergo apoptosis [21,39,40], thus having a key role in neuronal survival [39,41,42]. VA causes an increase in the activation of Akt-dependent phosphorylation which seems to be mediated by PI3K pathway, as shown by in vitro and in vivo models [23, 43-49].
Other protein kinases involved in neuronal survival [50] and activated by VA are the mitogen-activated protein kinases (MAPKs). Aberrant or inappropriate functions of MAPKs have been identified in diseases such as cancer, diabetes and inflammatory processes [51]. Among the several MAPK enzymes regulating cell proliferation, differentiation, motility and survival, the best known are the extracellular signal-regulated kinases 1 and 2 (ERK1/ 2) [52]. In fact, VA was shown to activate ERKs in vivo and in vitro and, as a result, to regulate activation of transcription factors and gene expression [53,54]. These data support the fact that the neurotrophic effects of VA occur via ERK. VA shows opposite effects inhibiting GSK-3β which is also a serine/threonine protein kinase implicated in multiple cellular processes, including neuronal survival, and regulated through phosphorylation by several kinases, such as Akt, MAPKs, PKC and PKA. GSK-3β plays a fundamental role in the CNS by regulating various proteins in the cytoskeleton, as well as long-term nuclear events, and it is a common therapeutic target for lithium and VA that inhibit its activity, as raised before [32,55,56].
Both lithium and VA also inhibit the activity of PKC in vitro and in vivo [57]. VA decreased PKC activity [58] and expression of subunits alpha and epsilon, as well as the expression of the main substrate of PKC. In vivo, both lithium and VA prevent memory deficits and decrease the activity of PKCα-dependent phosphorylation [59]. Phosphorylation of substrates by kinases is an intermediate step, common to many signaling cascades that regulate important cellular functions. The involvement of VA, in many of these mechanisms regulated by kinases, indicates the enormous potential of this drug that, therefore, can interfere with the fundamental mechanisms of cellular function or dysfunction.
4. VA, Histone Deacetylase Inhibition and Cellular Gene Activity
The enzymes histone deacetylase (HDAC) and histone acetyltransferase (HAT) catalyze the acetylation and deacetylation, respectively, of lysine residues located at the NH2 end of histone and non-histone type proteins. The disturbance of this balance is often observed in cancers and, thus, the inhibition of HDAC appears as a new therapeutic strategy in cancer treatment [60]. The acetylation of lysine residues in the N-terminal of histone proteins removes positive charges of the molecule, thereby reducing the affinity between histones and DNA. This facilitates the access of RNA polymerase and transcription factors to the promoter region of DNA. Thus, histone acetylation increases gene transcription, while histone deacetylation suppresses the transcription process (Figure 2).
Therefore, inhibition of HDAC shifts the balance between the deacetylation activity of HDAC enzymes and the activity of histone acetylation acetyltransferases, resulting in histone core hyperacetilation [61]. Thus, HDACs remove acetyl groups from the ends of histone lysine residues present in the nuclear chromatin, and also acetylated sites in non-histone proteins [62]. Histones are proteins intimately associated with DNA molecules and are responsible for the structural integrity of chromatin, playing a key role in the regulation of gene expression (Figure 3).
On the other hand, nucleosomes (above) are subunits of chromatin, consisting of a piece of DNA coiled around a core of a histone type protein. In fact, the chromosomal DNA is compacted with the aid of such specialized proteins. Thus, histone proteins compact chromosomal DNA
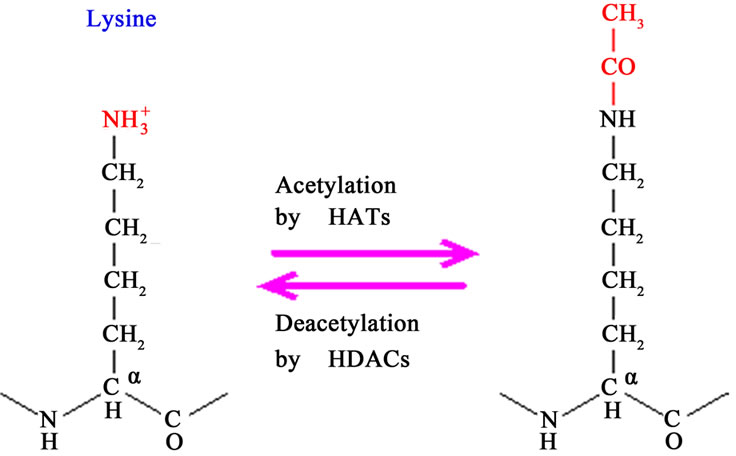
Figure 2. Acetylation and deacetylation of lysine residues (Source: http://www.web-books.com).
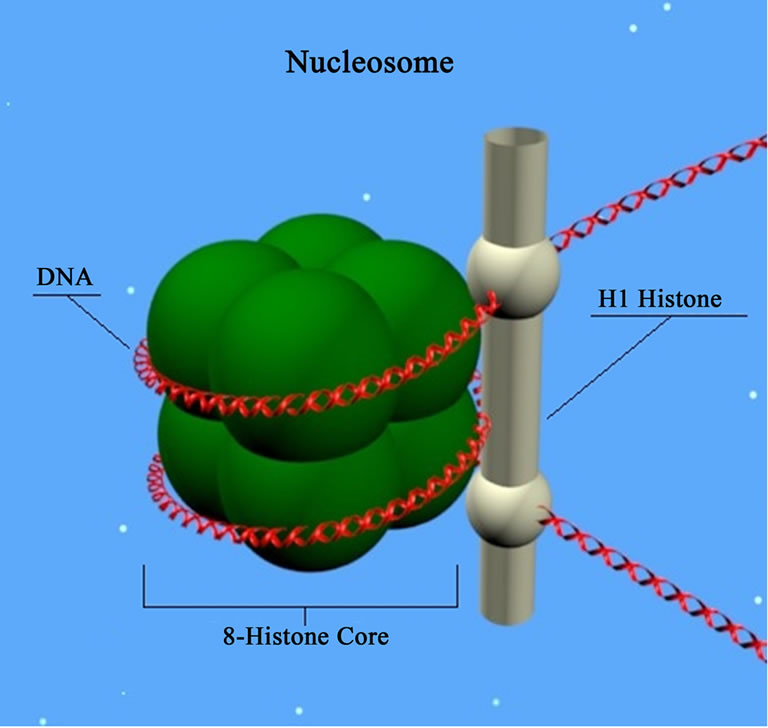
Figure 3. Nucleosome: basic unit of DNA packaging in eukaryotes, consisting of a segment of DNA wraped around a core of histone proteins (Source: http://www.molecularstation.com).
into the microscopic space of the eukaryotic nucleus, resulting in a DNA-protein complex called chromatin. Based on sequence phylogeny and function there are four distinct classes of HDACs: “class I (HDAC 1, 2, 3 and 8), class II (HDAC 4, 5, 6, 7, 9 and 10), class III is comprised by sirtuins (Sirt 1 - 7) and class IV (HDAC 11)” [63]. The HDAC inhibitors of diverse structures are capable of interrupting cell cycle progression, resulting in the stabilization of cell growth, differentiation and apoptosis [64]. Earlier studies have shown that some HDAC inhibitors exert neuroprotective effects in several neurodegenerative models [20, 65-71]. However, most of these reports focused on the direct effect of HDAC inhibitors in neurons, with little regard to the role of microglia in neuroprotection [72].
VA was demonstrated to exert its effects on the regulation of gene expression through two main mechanisms: on one hand, it regulates gene expression, acting on transcription factors, mainly by controlling protein phosphorylation, while on the other hand, it acts as an inhibitor of HDACs (from classes I and II), affecting epigenetic modulation of gene expression. Since the clinical performance of VA demands some time to occur, and its effects do not cease immediately after drug discontinuation, one may suggest that VA operates at the level of gene expression by regulating the activity of key transcription factors, as the transcription factor activator protein 1 (AP-1). It is known that AP-1 is a transcription factor for several important brain functions, such as development, plasticity and neurodegeneration [73].
Although there are several mechanisms of VA action, the current interest focuses on its use as a neuroprotective drug for the treatment of neurodegenerative diseases. The most likely way by which VA protects the brain against ischemia seems to be the inhibition of histone deacetylase (HDAC) [20,70,74-78]. The effects include, among others, protection against neuronal injury mediated by amino acids [3], as shown by in vitro and in vivo models of neuroprotection. VA produced an increased concentration of acetylated histone H3, accompanied by a decrease in cell damage and improved clinical outcomes [74].
Recent experiments have shown that VA can directly inhibit the HDAC activity, causing histone hyperacetilation. VA neuprotector activities appear to result from the activation of signaling pathways and also the inhibition of pro-apoptotic factors. Thus, in a model of intracerebral hemorrhage, VA activated not only histone H3 acetylation, but also pErk, pAkt, pCREB (cAMP response elementbinding, a transcription factor), and HSP70. In addition, it causes upregulation of Bcl-2 and Bcl-XL and downregulation of Bax (apoptotic regulatory proteins) and of the expression of Fas-L (Fas ligand; Fas is a protein belonging to the TNF family), IL-6 (interleukin-6), MMP-9 (matrix metallopeptidase 9), MIP-1 (macrophage inflammatory protein 1), MCP-1 (monocyte chemoattractant protein 1) and tPA (tissue plasminogen activator) [79].
New studies have shown that the neuroprotective effect of VA is related to the inhibition of some forms of apoptosis. The drug also protects against cell death induced by stress in the endoplasmic reticulum of glioma cells. In addition, VA induces axonal remodeling and clustering of synapses, probably through the indirect inhibition of GSK-3 [76]. This ability of VA to protect the body against apoptotic insults has been demonstrated in vitro and in vivo. A recent study [80] has shown that the pretreatment with VA, in association with lithium, protects brain neurons from glutamate-induced excitotoxicity in vitro. In addition, VA had beneficial effects in cellular and animal models of neurodegenerative diseases, such as Alzheimer’s, Parkinson’s and Huntington’s, spinal cord injury, spinal muscular atrophy, retinal degeneration, and infectious processes in patients with HIV-induced immunodeficiency [81]. The concentrations of native forms of alpha-synuclein, a protein marker for PD, were demonstrated to decrease in the substantia nigra and striatum of rats treated with rotenone, while nonubiquitinated forms of alphasynuclein increased in the same brain regions [5,82]. The treatment with VA reversed such changes.
A current study showed that the efficacy of VA in cases of medulloblastomas correlates with the induction of H3 and H4 histones hyperacetilation, activation of p21 (a potent cyclin-dependent kinase-CDK inhibitor) via the restoration of p16/CDK4 (p16 plays an important role in regulating the cell cycle, and its mutations increase the risk of developing a variety of cancers), and suppression of the oncogene CMYC. Moreover, in vitro effects of VA were shown to be concentration and time-dependent, and the irreversible inhibition of cell growth could be achieved with a prolonged treatment. These data suggest, therefore, that VA has a great inhibitory activity against medulloblastoma tumor cells [83].
VA also has a role in the chromatin structure by altering gene expression and affecting other signaling systems, such as pathways regulated by Wnt/β-catenin (Wnt belongs to a family of secreted glycolipoproteins and, via the transcriptional co-activator β-catenin, controls the embryonic development and adult homeostasis). In addition, VA directly or indirectly alters Wnt/β-catenin signaling by inhibiting the activity of GSK-3β [84], as previously described. VA also increases the activity of transcription factors, in a family of activating proteins, as well as their DNA binding activity, through ERK. It is known that transcription factors, through receptor tyrosine kinases, recruit a large number of signaling proteins in order to execute cellular events. One of these is the signal transduction cascade of RAS-RAF-ERK (which has a role in regulating many physiological processes) [85,86]. Sustained signaling through this pathway often acts to promote cell differentiation [87]. The ERK pathway is used by several chemical mediators, whose effects include neuronal differentiation, neuronal survival, neuroplasticity, and potentially long-term learning and memory [88]. Aberrations in the ERK cascade are involved in various types of cancers and, thus, different proteins present in the cascade are potential therapeutic targets for the treatment of these pathologies.
VA promotes an increase in the number of neurites, the activation of ERKs [89], and an increase in the expression of genes related to the ERK pathway, such as the cone growth associated protein 43, and Bcl-2. It also affects nerve regeneration and, through its inhibition of the enzyme GSK-3, can block the synthesis of pro-apoptotic factors that contribute to neuronal loss, leading to cell survival and increased uptake and release of norepinephrine [75].
Until recently, the loss or atrophy of large numbers of neurons were regarded as inevitable facts, and neurogenesis in the adult human brain, as implausible. Several pathways that regulate cell survival are already known, and neurogenesis was, in fact, shown recently to occur in the adult human brain. In summary, VA has been reported to prolong the lifespan of neuronal cultures [66], activate Akt [90] and extracellular signal-regulated kinase [89,91,92], to stimulate neuronal differentiation [92], and to induce cytoprotective proteins such as Bcl-2 [93], glucose-regulated protein 78 [93,94], brain-derived neurotrophic factor [95] and HSP70 [20,66].
Similarly to lithium, VA in therapeutic doses has an inhibitory effect on GSK-3β, and its association with lithium results in synergistic effects on this enzyme inhibition [93,96]. In a study testing the mechanisms involved in the protection of cerebellar granule cells from neuronal apoptosis, lithium and VA, although having different chemical structures, were shown to present neuroprotective effects. However, the most characteristic protection mediated by VA occurs only in the presence of insulin or IGF-1, because under this condition GSK-3β would be inactive. When GSK-3 is active, VA is unable to induce the survival of cultures, ruling out the possibility of protection against apoptosis [97]. Active GSK-3 phosphorylates and promotes the degradation of substrates involved in molecular pathways, including Wnt/βcatenin in normal cells, thus blunting important oncogenic signaling.
VA at concentrations around 1 mM is able to induce differentiation and to inhibit proliferation of neural progenitor cells (NPCs), overcoming the effect of the basic fibroblast growth factor (bFGF), which inhibits the differentiation of NPCs. The activation of ERK-p21Cip/ WAF1 (p21Cip/WAF1 factor is an important regulator of cell proliferation induced by p53 and ERK pathways), produced by VA, does not occur through the common pathway involving the epidermal growth factor receptor (EGFR), a component above ERK in the normal sequence, due to significant reduction in the level of EGFR in the presence of VA. The level of the Ras protein (subfamily of small GTPases involved in cellular signal transduction), another component that precedes ERK, is significantly increased by the treatment with VA. Thus, ERK activation induced by VA occurs by an increased Ras stability, mediated by Wnt/β-catenin (which has an important role in both embryonic development and tumorigenesis). Also, the common path of Ras-ERK-p21CIP/ WAF1 is involved in mutual exclusive phenotypes of differentiation and proliferation in NPCs, and in the cerebral cortex of developing embryos [84]. The inhibitor of cyclin-dependent kinase, p21CIP/WAF1 (it negatively regulates the progression of cell cycle and has a potential usefulness in gene therapy), positively regulates cell proliferation induced by the insulin-growth factor (IGF-1) in cell lines of breast cancer [98].
VA has been recently described to be responsible for the phosphorylation of ERK in the induction of apoptosis, a statement that supports the Ca2+ hypothesis, since this cation is responsible for activating that pathway. Thus, VA seems to affect not only a transduction pathway (ERK), but also Ca2+, the main factor involved in neuronal transmission of electrical stimulation [97].
It is known that VA regulates the differentiation and proliferation of various cell types, such as mesenchymal and hematopoietic cells, neuroblastoma cells, primary neurons and neural progenitor cells (NPCs). Thus, it can reduce the proliferation of neuroblastoma cells by induction of the cell cycle regulator p21 Cip/WAF1, also important in the differentiation of hematopoietic cells induced by VA. However, the mechanism by which VA regulates differentiation and proliferation is not yet fully understood [84].
Furthermore, it has been recently shown that VA when systemically administered, reinforces the long-term memory for extinction of conditioned fear. Evidences showed that chromatin, a DNA-protein complex that packages the genomic DNA, has an important role in learning and memory. The modification of histones is a fundamental mechanism involved in epigenetic regulation, contributing to the consolidation of long-term memory. These histone modifications are mediated by direct enzymatic interactions with lysine residues, causing dynamic changes in the chromatin structure and helping to regulate gene expression induced by learning [99,100]. Changes in chromatin via inhibitors of histone acetyltransferases and HDACs can increase synaptic plasticity and hippocampus-dependent memory.
A useful paradigm for studying long-term memory is the Pavlovian fear conditioning, in which a conditioned stimulus is associated with an aversive unconditioned stimulus. It is known, however, that the repeated submission to a cue, in the absence of an associated stimulus, reduces the likelihood of expression of this memory, i.e., leads to its extinction. According to Cammarota et al., 2007 [101], this memory extinction is the inhibition of the original memory, and then does not involve forgetting, but instead a new learning.
Using a paradigm of contextual fear conditioning, Lattal et al. (2007) [102] showed that the systemic administration or direct infusion of a HDAC inhibitor, in the dorsal hippocampus, enhances long-term memory, reducing the likelihood of memory extinction. These results suggest that inhibitors of HDACs may even increase learning during extinction, being consistent with other studies showing the role of the hippocampus in the extinction context. Bredy and Barad (2008) [103] demonstrated that VA increases the processes of acquisition, extinction and reconsolidation of conditioned fear, that according to these authors have important implications for the use of HDACs inhibitors as co-adjuvants in the treatment of phobias and anxiety disorders. Recent findings [104] indicate that VA, given soon after a traumatic brain injury, can be neuroprotective, and improves the cognitive function in rats.
New evidence supports the idea that VA also has neuroprotective effects in vitro against glutamatergic excitotoxicity, endoplasmic reticulum stress, microglial activation induced by lipopolysaccharide (LPS) and death of dopaminergic neurons. In a model of cerebral ischemia in rats, the post-injury treatment with VA suppressed the ischemia-induced brain damage and neurological deficits. A large number of studies have suggested the potential of HDAC inhibitors, including VA, for the treatment of acute and chronic neurological diseases, such as amyotrophic lateral sclerosis and Alzheimer’s disease, among others [78,105,106].
Another study [107] with rats treated with VA for 14 days (300 mg/kg, twice a day), before an intrastriatal injection of 1.5 mM malonate (an inhibitor of the enzyme succinate dehydrogenase), showed that VA decreased the extracellular malonate-induced glutamate accumulation. This effect occurred in parallel with increased levels of the striatal glutamate transporter that, in turn, increased the high affinity glutamate uptake, suggesting this increase to contribute to the observed VA neuroprotection. The striatal levels of FOS (the FOS gene family encode proteins that can dimerize with proteins of the JUN family, forming the transcription factor complex AP-1, and are implicated in cell proliferation, differentiation, and transformation. On the other hand, c-fos and c-jun are protooncogenes of the immediate early genes-IEG class, that code FOS and JUN proteins, which in turn are inducible transcription factors-ITF controlling gene transcription) and HSP-70 were reduced, the levels of Bcl-2 and ERK remained unchanged, but an increase in histone acetylation was observed after VA treatment. These results suggest that the increase in glutamate uptake may contribute to the neuroprotection mediated by VA in the striatum [107].
While both lithium and VA regulate gene expression through transcription factors [108], VA also seems to work through distinct mechanisms involving epigenetic regulation of transcription. In fact, VA effectively inhibits HDACs, negative regulators of gene expression in various conditions, at therapeutic levels [109]. In addition, VA changes the structure of chromatin through the negative regulation of proteins associated with it, as already described. The downregulation of these proteins is related to the decondensation and increased sensitivity of DNA to nucleases. VA inhibits the activity of HDACs, possibly by binding to the enzyme’s catalytic center [110]. Literature data support the concept that the acetylation of chromatin and methylation of DNA are interrelated in a dynamic way, and that the effects of inhibitors of HDACs are not limited to changes in histone acetylation, but also cause changes in the state of the DNA, especially in its methylation [111,112].
The effects of VA as an inhibitor of HDACs could also be responsible for the increased expression of neurotrophins, such as BDNF and GDNF, related to neuroprotective and nootropic properties or yet stimulating memory processes [100,113,114]. All these effects of VA on cell development lead to the association of the drug-induced teratogenesis with its inhibitory effect on HDACs observed in vivo, explaining why VA causes spina bifida if taken during pregnancy [115]. In cell lines as neuroblastoma, VA attenuated the apoptosis induced by rotenone, a potent inhibitor of the mitochondrial respiratory chain complex 1, which induces neuronal death in a way similar to Parkinson’s disease. VA also protected cultures of dopaminergic neurons against the neurotoxicity induced by MPP+, another model of neurodegeneration in PD [47,77].
5. VA and Inflammation
Uncontrolled, chronic inflammation is now considered a major component of many widely occurring diseases, including asthma, atherosclerosis and peripheral vascular disease, Alzheimer’s disease, and cancer. These pathologies now join the group of disorders, such as arthritis and periodontitis, which have been well-recognized as being “inflammatory diseases”. While inflammation may not be the causative factor in these pathological conditions, it is now believed to contribute to their progression, tissue dysfunction, and ultimately to organ failure. Resolution of inflammation was once considered a passive process. In recent years, evidence has emerged that this is a biochemically active process, and more recent results have established some of the chemical mediators involved. Elucidation of biochemical pathways contributing to the resolution of inflammation has provided many new antiinflammatory targets and an opportunity for resolutionbased pharmacology for the treatment and prevention of inflammatory disorders.
An inflammatory response requires the recruitment of innate immune cells (e.g. neutrophils and monocyte/macrophages) and the induction of processes that allow them to engulf targets and to eliminate the aberrant stimuli. The resolution of inflammation involves apoptosis and subsequent clearance of activated inflammatory cells, a tightly regulated event. Furthermore, chronic inflammation is a characteristic feature in virtually all inflammatory diseases [116]. Epigenetic modifications occur in response to environmental changes, and play a fundamental role in gene expression following environmental stimuli. Among the several major epigenetic events are methylation and acetylation of histones and regulatory factors, DNA methylation and small non-coding RNAs. Despite evidences of the environmental role on gene expression, little is known about the epigenetic pathways involved in the modulation of inflammatory and antiinflammatory genes [117].
Although HDACs inhibitors (HDACsI) hyperacetylate histones, via the conserved N-terminal lysines present on histones, they also hyperacetylate cytoplasmic proteins, including transcription factors. Therefore unraveling chromatin and thus permitting transcription factors to bind to DNA is a prominent property of HDACsI, but not their only mechanism of action. Thus, they have been shown that, besides modulating gene expression, they can also modulate cytokine production after several stimuli [118].
Recent evidences have shown that HDACsI, besides their potential as anticancer agents, could also be used for the treatment of chronic immune and inflammatory disorders. Thus, although the majority of drug development in these areas has focused on the treatment of cancer, a number of HDACsI have emerged as promising anti-inflammatory agents [119].
We recently showed that VA reduces polymerphonuclear cells migration and myeloperoxidase (a biomarker of inflammation) release, as evaluated by in vivo and in vitro models [120]. Besides exerting an anti-inflammatory activity, VA also presents a potent antinociceptive effect as recently demonstrated by us [121,122]. Other in vitro and in vivo data indicate that HDACsI may be antiinflammatory, due to their effects on cell death, acting through acetylation of non-histone proteins [123]. As discussed earlier, HDACsI have potent in vivo and in vitro anticancer actions. They also exhibit strong anti-inflammatory effects, as demonstrated in several experimental models, what makes HDACsI a promising new class of drugs for the treatment of various diseases where the inflammatory component is associated. Besides, they are being studied in several disease models not related to their pro-apoptotic properties used to treat cancer.
The spectrum of these diseases responsive to HDACsI is mainly due to their anti-inflammatory properties, observed in vitro as well as in animal models. A unifying property is the reduction in cytokine production and inhibition of cytokine post-receptor signaling. Distinctly from their use in cancer, the reduction in inflammation by HDACsI is consistently observed at low concentrations, if compared to higher concentrations required for killing tumor cells [124]. Recent data have shown that glial cells of mice present histone deacetylase activities, and the inhibition of this enzyme suppresses the neuroinflammatory response, due to changes in the transcriptional machinery [125].
Evidences have demonstrated immunomodulatory effects of HDACs on microglia [126,127], and previous studies reported that VA attenuated neuronal damage induced by lipopolysaccharide (LPS), in mixed cultures of mesencephalic, neuronal and glial cells. According to Peng et al. (2005) [70], this neuroprotection is partly associated with the suppression of inflammation, mediated by microglia, and the decrease in the number of microglial cells. Another study [128] showed that VA induces apoptosis in microglial BV-2 cell lines, however, the mechanism responsible for the decreased cell numbers or the process of apoptotic microglia is not yet fully understood.
It is worthwhile knowing that granulocytes are important in the pathogenesis of various inflammatory diseases. Apoptosis is crucial in resolving the inflammatory process and, in cancer cells, apoptosis is induced by HDACsI as VA, not occuring in normal cells. Therefore, the inhibition of histone deacetylase by VA makes this drug a promising new pharmacological strategy in the treatment of cancer and neurodegenerative diseases, among others, where the inflammatory process is directly or indirectly involved in the pathogenesis [129].
Figure 4 shows that neurological and psychiatric disorders involve epigenetic modifications of key neuronal genes and the possible intervention of HDACsI. Neurodegenerative diseases, as well as psychiatric and neurodevelopmental disorders can involve aberrant acetylation and methylation of histones and/or DNA methyllation. These epigenetic modifications can determine the transcriptional state of regulatory genes crucial to synaptic plasticity, cognition, and mood. VA by inhibiting the activity of HDACs shifts the balance toward the active transcription of neuronal genes and the amelioration of plasticity and cognitive deficits.
6. Conclusions
The literature review shows that valproic acid is a versatile anticonvulsant drug, worldwide used in the treatment of epilepsy and prophylaxis of migraine. With respect to its neuroprotective effect, the majority of data, although sometimes controversial, come from studies performed in the last decade. The inhibition of HDACs enhances the acetylation of chromatin, and promotes the reduction of neuronal damage. Its effects against apoptotic insults, induced by both endoplasmic reticulum stress and glutamatergic excitotoxicity, as well as by the inhibition of the enzyme GSK-3 lead to axonal remodeling and blockade of the synthesis of pro-apoptotic factors.
Furthermore, some of VA actions result from its inhibitory properties on the enzyme histone deacetylase (HDAC), and therefore the drug can specifically modulate gene expression. However, many questions concerning the role of inhibitors of HDACs, in the regulation of epigenetic mechanisms in the brain, are still open. Firstly, the incomplete knowledge on chromatin remodeling complexes in the brain, and how its components are dynamically regulated according to cells development and type or brain area. Secondly, the target genes for HDACsI that are involved in the mediation and formation of memory should be better known. Thirdly, the potential role of non-histone protein substrates for HDACs, in neuroplasticity and brain functions, needs to be better known, beyond the classical view on the role presented by inhibitors of HDACs in the regulation of epigenetic mechanisms via histone acetylation. Fourthly, the needed identification of new molecules and allosteric inhibitors of HDACs with an increased selectivity may help to a better understanding of the different modes of enzyme regulation. Although various non-selective HDACsI, including VA, are generally well tolerated by animals and humans, the search for more selective compounds, acting on different HDAC family members, is of significant importance. Surely, this may lead to drugs with fewer side effects and greater efficacy, as potential therapeutic agents for the treatment of neuropsychiatric and neurodegenerative diseases [131-133]. It has been shown that the combination of antipsychotic drugs with VA, increasing DNA methylation by an unknown mechanism, causes a chromatin remodeling that brings about a beneficial
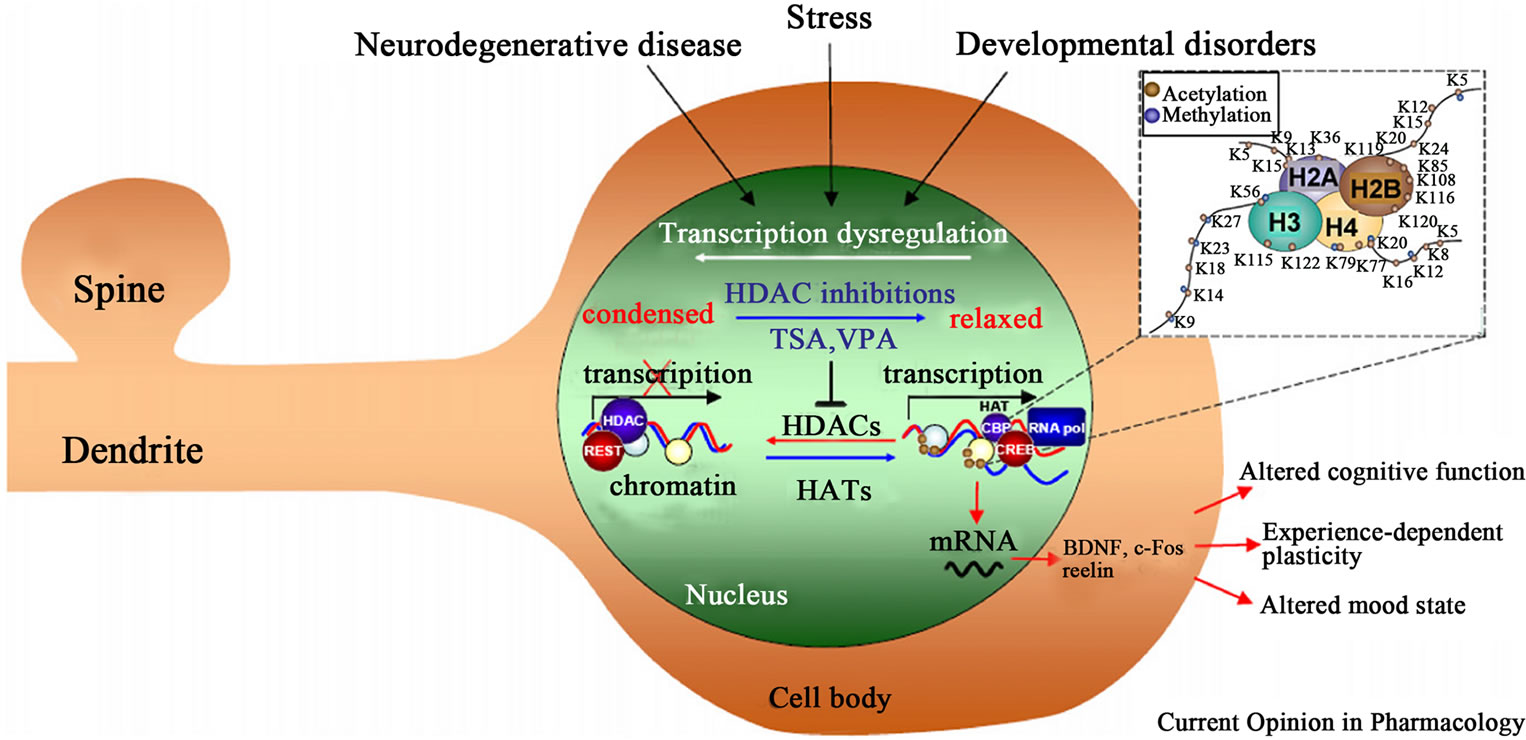
Figure 4. Shows the possible intervention of HDACsI, including valproic acid, on neurological and psychiatric disorders. Source: Abel and Zukin (2008) [130], with permission from the publishers of Current Opinion in Pharmacology.
change in the epigenetic GABAergic dysfunction, typical of schizophrenic and bipolar disorder patients [134]. It is believed that the identification of a brain enzyme facilitating DNA demethylation, and the understanding of how drugs induce DNA demethylation are crucial for the advancement of a new line of pharmacological interventions to treat neurodevelopmental, neuropsychiatric, and neurodegenerative diseases.
In recent years, more and more evidence has been accumulated showing that HDACsI, such as VA, can be successfully used in the treatment of neurodegenerative disorders [106]. However, the results are not conclusive, since lately some studies showed that the chronic treatment with valproate did not delay agitation or the functional decline in Alzheimer’s disease patients, and was even associated with accelerated brain volume loss [135, 136].
Finally, the literature shows a plethora of molecular interactions and cellular processes affected by VA, besides HDACs, as the GSK3 alpha and beta, Akt, ERK and phosphoinositol pathways, tricarboxylic acid cycle, GABA and the OXPHOS system (comprised of five enzyme complexes implicated in various functions, in addition to their primary role in energy generating processes) [48, 137]. This system, whose biological activities range from interactions with receptors and ion channels to the regulation of many catalytic reactions, has a central role in cellular cascades that regulate gene expression. Such studies have stimulated a vast array of investigators, pharmacologists, molecular biologists, physiologists and clinicians.
Thus, inhibitors of HDACs, positively affecting both neuronal degeneration and cognitive deficits, appear as promising drugs against various pathological conditions and neurodegenerative diseases [130]. It is also important to emphasize that many overlapping events occur in neurodegenerative diseases. VA is a pleitropic drug acting on multiple molecular targets, and thus may interfere directly or indirectly with all the common links involved with neurodegenerative diseases, as shown above (Tables 1 and 2). In particular, from a clinical point of view, although VA is a drug that shows a great potential as a

Table 1. Common links in part or totally involved with neurodegenerative diseases.
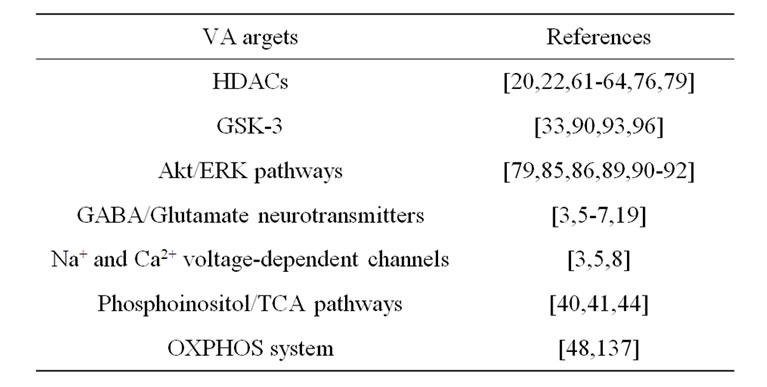
Table 2. Main molecular targets involved in VA neuroprotection.
candidate for the treatment of neurodegenerative diseases related to excitotoxic events, more and well designed clinical studies are greatly needed in order to clarify better and safer conditions for its chronic use.
REFERENCES
- H. Meunier, G. Carraz, Y. Meunier, P. Eymard and P. Aimard, “Propriétés Pharmacodynamiques de L`acide nDipropylacétique,” Therapie, Vol. 18, 1963, pp. 435-438.
- T. R. Henry, “The History of Valproate in Clinical Neuroscience,” Psychopharmacology Bulletin, Vol. 37, Suppl. 2, 2003, pp. 5-16.
- M. J. Owens and C. B. Nemeroff, “Pharmacology of Valproate,” Psychopharmacology Bulletin, Vol. 37, Suppl. 2, 2003, pp. 17-24.
- E. M. Cornford, C. P. Diep and W. M. Pardridge, “BloodBrain Barrier Transport of Valproic Acid,” Journal of Neurochemistry, Vol. 44, No. 5, 1985, pp. 1541-1550. doi:10.1111/j.1471-4159.1985.tb08793.x
- B. Monti, E. Polazzi and A. Contestabile, “Biochemical, Molecular and Epigenetic Mechanisms of Valproic Acid Neuroprotection,” Current Molecular Pharmacology, Vol. 2, No. 1, 2009, pp. 95-109. doi:10.2174/1874467210902010095
- E. Perucca, “Pharmacological and Therapeutic Properties of Valproate: A Summary after 35 Years of Clinical Experience,” CNS Drugs, Vol. 16, No. 10, 2002, pp. 695- 714. doi:10.2165/00023210-200216100-00004
- O. A. Petroff, D. L. Rothman, K. L. Behar, F. Hyder and R. H. Mattson, “Effects of Valproate and Other Antiepileptic Drugs on Brain Glutamate, Glutamine and GABA in Patients with Refractory Complex Partial Seizures,” Seizure, Vol. 8, No. 2, 1999, pp. 120-127. doi:10.1053/seiz.1999.0267
- A. M. Vandongen, M. G. Vanerp and R. A. Voskuyl, “Valproate Reduces Excitability by Blockade of Sodium and Potassium Conductance,” Epilepsia, Vol. 27, No. 3, 1986, pp. 177-182. doi:10.1111/j.1528-1157.1986.tb03525.x
- E. P. Chronicl and W. M Mulleners, “Anticonvulsant Drugs for Migraine Prophylaxis,” Cochrane Database of Systematic Reviews, No. 3, 2009, p. CD003226.
- M. J. Morrell, “Reproductive and Metabolic Disorders in Women with Epilepsy,” Epilepsia, Vol. 44, No. S4, 2003, pp. 11-20. doi:10.1046/j.1528-1157.44.s4.2.x
- R. Sreedhar and S. V. Gadhinglajkar, “Pharmacological Neuroprotection,” Indian Journal of Anaesthesia, Vol. 47, No. 1, 2003, pp. 8-22.
- E. C. Lauterbach, J. Victoroff, K. L. Coburn, S. D. Shillcutt, et al., “Psychopharmacological Neuroprotection in Neurodegenerative Disease: Assessing the Preclinical Data,” The Journal of Neuropsychiatry & Clinical Neurosciencesi, Vol. 22, No. 1, 2010, pp. 8-18. doi:10.1176/appi.neuropsych.22.1.8
- C. Costa, G. Martella, B. Picconi, C. Prosperetti, et al., “Multiple Mechanisms Underlying the Neuroprotective Effects of Antiepileptic Drugs against in Vitro Ischemia,” Stroke, Vol. 37, 2006, pp. 1319-1326. doi:10.1161/01.STR.0000217303.22856.38
- P. Calabresi, L. M. Cupini, D. Centonze, F. Pisani and G. Bernardi, “Antiepileptic Drugs as a Possible Neuroprotective Strategy in Brain Ischemia,” Annals of Neurology, Vol. 53, No. 6, 2003, pp. 693-702. doi:10.1002/ana.10603
- R. R. Leker and M. Y. Neufeld, “Anti-Epileptic Drugs as Possible Neuroprotectants in Cerebral Ischemia,” Brain Research Reviews, Vol. 42, No. 3, 2003, pp. 187-203. doi:10.1016/S0165-0173(03)00170-X
- D. Centonze, G. A. Marfia, A. Pisani and B. Picconi, “Ionic Mechanisms Underlying Differential Vulnerability to Ischemia in Striatal Neurons,” Progress in Neurobiology, Vol. 63, No. 6, 2001, pp. 687-696. doi:10.1016/S0301-0082(00)00037-X
- P. Calabresi, E. Saulle, D. Centonzed and A. Pisani, “Post-Ischaemic Long-Term Synaptic Potentiation in the Striatum: A Putative Mechanism for Cell Type-Specific Vulnerability,” Brain, Vol. 125, No. 4, 2002, pp. 844-860. doi:10.1093/brain/awf073
- M. A. Rogawski and W. Loscher, “The Neurobiology of Antiepileptic Drugs,” Nature Reviews Neuroscience, Vol. 5, 2004, pp. 553-564. doi:10.1038/nrn1430
- P. Calabresi, B. Picconi, E. Saulle and D. Centonze, “Is Pharmacological Neuroprotection Dependent on Reduced Glutamate Release?” Stroke, Vol. 31, 2000, pp. 766-772. doi:10.1161/01.STR.31.3.766
- M. Ren, Y. Leng, M. Jeong, P. R. Leeds and D. M. Chuang, “Valproic Acid Reduces Brain Damage Induced by Transient Focal Cerebral Ischemia in Rats: Potential Roles of Histone Deacetylase Inhibition and Heat Shock Protein Induction,” Journal of Neurochemistry, Vol. 89, No. 6, 2004, pp. 1358-1367. doi:10.1111/j.1471-4159.2004.02406.x
- A. Mora, R. A. González-Polo, J. M. Fuentes, G. Soler and F. Centeno, “Different Mechanisms of Protection against Apoptosis by Valproate and Li+,” European Journal of Biochemistry, Vol. 266, No. 3, 1999, pp. 886-891. doi:10.1046/j.1432-1327.1999.00919.x
- S. Eyal, B. Yagen, E. Sobol, Y. Altschuler, M. Shmuel and M. Bialer, “The Activity of Antiepileptic Drugs as Histone Deacetylase Inhibitors,” Epilepsia, Vol. 45, No. 7, 2004, pp. 737-744. doi:10.1111/j.0013-9580.2004.00104.x
- H. J. Kim, M. Rowe, M. Ren, J. S. Hong, P. S. Chen and D. M. Chuang, “Histone Deacetylase Inhibitors Exhibit Anti-Inflammatory and Neuroprotective Effects in a Rat Permanent Ischemic Model of Stroke: Multiple Mechanisms of Action,” Journal of Pharmacology and Experimental Therapeutics, Vol. 321, No. 3, 2007, pp. 892-901. doi:10.1124/jpet.107.120188
- B. N. Frey, S. S. Valvassori, G. Z. Réus and M. R. Martins, “Changes in Antioxidant Defense Enzymes after dAmphetamine Exposure: Implications as an Animal Model of Mania,” Neurochemical Research, Vol. 31, No. 5, 2006, pp. 699-703. doi:10.1007/s11064-006-9070-6
- J. F. Wang, J. E. Azzam and L. T. Young, “Valproate Inhibits Oxidative Damage to Lipid and Protein in Primary Cultured Rat Cerebrocortical Cells,” Neuroscience, Vol. 116, No. 2, 2003, pp. 485-489. doi:10.1016/S0306-4522(02)00655-3
- R. Hashimoto, N. Takei, K. Shimazu, L. Christ, B. Lu and D. M. Chuang, “Lithium Induces Brain-Derived Neurotrophic Factor and Activates TrkB in Rodent Cortical Neurons: An Essential Step for Neuroprotection against Glutamate Excitotoxicity,” Neuropharmacology, Vol. 43, No. 7, 2002, pp. 1173-1179. doi:10.1016/S0028-3908(02)00217-4
- G. Alvarez, J. R. Muñoz-Montaño, J. Satrústegui, J. Avila, E. Bogónez and J. Díaz-Nido, “Lithium Protects Cultured Neurons against Beta-Amyloid-Induced Neurodegeneration,” FEBS Letters, Vol. 453, No. 3, 1999, pp. 260-264. doi:10.1016/S0014-5793(99)00685-7
- S. Jakopec, D. Karlovic, K. Dubravcic and D. Batinic, “Lithium Effect on Glutamate Induced Damage in Glioblastoma Cells,” Collegium Antropologicum, Vol. 32, Suppl. 1, 2008, pp. 87-91.
- L. Shao, L. T. Young and J. F. Wang, “Chronic Treatment with Mood Stabilizers Lithium and Valproate Prevents Excitotoxicity by Inhibiting Oxidative Stress in Rat Cerebral Cortical Cells,” Biological Psychiatry, Vol. 58, No. 11, 2005, pp. 879-884. doi:10.1016/j.biopsych.2005.04.052
- Q. Bian, T. Shi, D. M. Chuang and Y. Qian, “Lithium Reduces Ischemia-Induced Hippocampal CA1 Damage and Behavioral Deficits in Gerbils,” Brain Research, Vol. 1184, 2007, pp. 270-276. doi:10.1016/j.brainres.2007.09.054
- M. K. Rowe and D. M. Chuang, “Lithium Neuroprotection: Molecular Mechanisms and Clinical Implications,” Expert Reviews in Molecular Medicine, Vol. 6, 2004, pp. 1-18. doi:10.1017/S1462399404008385
- H. Eldar-Filkelman, “Glycogen Synthase Kinase 3: An Emerging Therapeutic Target,” Trends in Molecular Medicine, Vol. 8, No. 3, 2002, pp.126-132. doi:10.1016/S1471-4914(01)02266-3
- C. A. Grimes and R. S. Jope, “The Multifaceted Roles of Glycogen Synthase Kinase 3Beta in Cellular Signaling,” Progress in Neurobiology, Vol. 65, No. 4, 2001, pp. 391- 426. doi:10.1016/S0301-0082(01)00011-9
- J. R. Woodgett, “Judging a Protein by More than Its Name: GSK-3,” Science’s STKE, Vol. 2011, No. 100, 2001, p. RE12.
- G. N. Bijur, P. De Sarno and R. S. Jope, “Glycogen Synthase Kinase-3b Facilitates Staurosporine and Heat ShockInduced Apoptosis: Protection by Lithium,” Journal of Biological Chemistry, Vol. 275, 2000, pp. 7583-7590. doi:10.1074/jbc.275.11.7583
- R. H. Lenox and C. Hahn, “Overview of the Mechanism of Action of Lithium in the Brain: Fifty Year Update,” Journal of Clinical Psychiatry, Vol. 61, Suppl. 9, 2000, pp. 5-15.
- O. Kaidanovich-Beilin, A. Milman, A. Weizman, C. G. Pick and H. Eldar-Finkelman, “Rapid AntidepressiveLike Activity of Specific Glycogen Synthase Kinase-3 Inhibitor and Its Effect on β-Catenin in Mouse Hippocampus,” Biological Psychiatry, Vol. 55, No. 8, 2004, pp. 781-784. doi:10.1016/j.biopsych.2004.01.008
- N. Gurvich and P. S. Klein, “Lithium and Valproic Acid: Parallels and Contrasts in Diverse Signaling Contexts,” Pharmacology & Therapeutics, Vol. 96, No. 1, 2002, pp. 45-66. doi:10.1016/S0163-7258(02)00299-1
- A. Brunet, S. R. Datta and M. E. Greenberg, “Transcription-Dependent and-Independent Control of Neuronal Survival by the PI3K-Akt Signaling Pathway,” Current Opinion in Neurobiology, Vol. 11, No. 3, 2001, pp. 297- 305. doi:10.1016/S0959-4388(00)00211-7
- V. Duronio, “The Life of a Cell: Apoptosis Regulation by the PI3K/PKB Pathway,” Biochemical Journal, Vol. 415, 2008, pp. 333-344. doi:10.1042/BJ20081056
- T. K. Creson, P. Yuan, H. K. Manji and G. Chen, “Evidence for Involvement of ERK, PI3K and RSK in Induction of Bcl-2 by Valproate,” Journal of Molecular Neuroscience, Vol. 37, No. 2, 2008, pp. 123-134. doi:10.1007/s12031-008-9122-2
- C. A. Bondy and C. M. Cheng, “Signaling by InsulinLike Growth Factor 1 in Brain,” European Journal of Pharmacology, Vol. 490, No. 1-3, 2004, pp. 25-31. doi:10.1016/j.ejphar.2004.02.042
- E. Chalecka-Franaszek and De-Maw Chuang, “Lithium Activates the Serine/Threonine Kinase Akt-1 and Suppresses Glutamate-Induced Inhibition of Akt-1 Activity in Neurons,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 96, No. 15, 1999, pp. 8745-8750. doi:10.1073/pnas.96.15.8745
- A. Mora, G. Sabio, J. C. Alonso, G. Soler and F. Centeno, “Different Dependence of Lithium and Valproate on PI3K/ PKB Pathway,” Bipolar Disorders, Vol. 4, No. 3, 2002, pp. 195-200. doi:10.1034/j.1399-5618.2002.40301.x
- J. Chen, F. M. Ghazawi, W. Bakkar and Q. Li, “Valproate Acid and Butyrate Induce Apoptosis in Human Cancer Cells through Inhibition of Gene Expression of Akt/Protein Kinase B,” Molecular Cancer, Vol. 5, 2006, p. 71. doi:10.1186/1476-4598-5-71
- J. M. Beaulieu, T. D. Sotnikova, W. D. Yao, L. Kockeritz, et al., “Lithium Antagonizes Dopamine-Dependent Behaviors Mediated by na AKT/Glycogen Synthase Kinase 3 Signaling Cascade,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 101, No. 14, 2004, pp. 5099-5104. doi:10.1073/pnas.0307921101
- T. Pan, X. Li, W. Xie, J. Jankovic and W. Le, “Valproic Acid Mediated Hsp70 Induction and Antiapoptotic Neuroprotection in SH-SY5Y Cells,” FEBS Letters, Vol. 579, No. 30, 2005, pp. 6716-6720. doi:10.1016/j.febslet.2005.10.067
- M. Kostrouchová, Z. Kostrouch and M. Kostrouchová, “Valproic Acid, a Molecular Lead to Multiple Regulatory Pathways,” Folia Biologica (Praha), Vol. 53, 2007, pp. 37-49.
- M. H. Liang, J. R. Wendland and D. M. Chuang, “Lithium Inhibits Smad3/4 Transactivation via Increased CREB Activity induced by PKA and Akt Signaling,” Molecular and Cellular Neuroscience, Vol. 37, No. 3, 2008, pp. 440- 453. doi:10.1016/j.mcn.2007.10.017
- M. Miloso, A. Scuteri, D. Foudah and G. Tredici, “MAPKs as Mediators of Cell Fate Determination: An Approach to Neurodegenerative Diseases,” Current Medicinal Chemistry, Vol. 15, No. 6, 2008, pp. 538-548. doi:10.2174/092986708783769731
- M. C. Lawrence, A. Jivan, C. Shao and L. Duan, “The Roles of MAPKs in Diseases,” Cell Research, Vol. 18, 2008, pp. 436-442. doi:10.1038/cr.2008.37
- M. Cargnello and P. P. Roux, “Activation and Function of MAPs and Their Substrates, the MAPK-Activated Protein Kinases,” Microbiology and Molecular Biology Reviews, Vol. 75, No. 1, 2011, pp. 50-83. doi:10.1128/MMBR.00031-10
- F. Zhang, C. J. Phiel, L. Spece, N. Gurvich and P. S. Klein, “Inhibitory Phosphorylation of Glycogen Synthase Kinase-3 (GSK-3) in Response to Lithium. Evidence for Autoregulation of GSK-3,” Journal of Biological Chemistry, Vol. 278, 2003, pp. 33067-33077. doi:10.1074/jbc.M212635200
- H. Einat, P. Yuan, T. D. Gould, J. Li, et al., “The Role of the Extracellular Signal-Regulated Kinase Signaling Pathway in Mood Modulation,” Journal of Neuroscience, Vol. 23, No. 19, 2003, pp. 7311-7316.
- X. Li, G. N. Bijur and R. S. Jope, “Glycogen Synthase Kinase-3beta, Mood Stabilizers, and Neuroprotection,” Bipolar Disorders, Vol. 4, No. 2, 2002, pp. 137-144. doi:10.1034/j.1399-5618.2002.40201.x
- T. D. Gould and H. K. Manji, “Glycogen Synthase Kinase-3: A Putative Molecular Target for Lithium Mimetic Drugs,” Neuropsychopharmacology, Vol. 30, No. 7, 2005, pp. 1223-1237.
- J. T. Coyle and H. K. Manji, “Getting Balance: Drugs for Bipolar Disorder Share Target,” Nature Medicine, Vol. 8, 2002, pp. 557-558. doi:10.1038/nm0602-557
- G. Chen, H. K. Manji, D. B. Hawver, C. B. Wright and W. Z. Potter, “Chronic Sodium Valproate Selectively Decreases Protein Kinase C Alpha and Epsilon in Vitro,” Journal of Neurochemistry, Vol. 63, No. 6, 1994, pp. 2361- 2364. doi:10.1046/j.1471-4159.1994.63062361.x
- S. G. Birbaum, P. X. Yuan, M. Wang and S. Vijayraghavan, “Protein Kinase C Overactivity Impairs Prefrontal Cortical Regulation of Working Memory,” Science, Vol. 306, No. 5697, 2004, pp. 882-884. doi:10.1126/science.1100021
- S. Chateauvieux, F. Morceau, M. Dicato and M. Diederich, “Molecular and Therapeutic Potential and Toxicity of Valproic Acid,” Journal of Biomedicine and Biotechnology, Vol. 2010, 2010, p. 479364. doi:10.1155/2010/479364
- D. Marchion and P. Münster, “Development of Histone Deacetylase Inhibitors for Cancer Treatment,” Expert Review of Anticancer Therapy, Vol. 7, No. 4, 2007, pp. 583- 598. doi:10.1586/14737140.7.4.583
- C. M. Marson, “Histone Deacetylase Inhibitors: Design, Structure-Activity Relationships and Therapeutic Implications for Cancer,” Anti-Cancer Agents in Medicinal Chemistry, Vol. 9, No. 6, 2009, pp. 661-692.
- M. Bantscheff, C. Hopf, M. M. Savitski, A. Dittmann, et al., “Chemoproteomics Profiling of HDAC Inhibitors Reveals Selective Targeting of HDAC Complexes,” Nature Biotechnology, Vol. 29, 2011, pp. 255-265. doi:10.1038/nbt.1759
- J. E. Bolden, M. J. Peart and R. W. Johnstone, “Anticancer Activities of Histone Deacetylase Inhibitors,” Nature Reviews Drug Discovery, Vol. 5, 2006, pp. 769-784. doi:10.1038/nrd2133
- R. J. Ferrante, J. K. Kubilus, J. Lee and H. Ryu, “Histone Deacetylase Inhibition by Sodium Butyrate Chemotherapy Ameliorates the Neurodegenerative Phenotype in Huntington’s Disease Mice,” Journal of Neuroscience, Vol. 23, No. 28, 2003, pp. 9418-9427.
- M. R. Jeong, R. Hashimoto, W. Senatorov, K. Fujimakik, et al., “Valproic Acid, a Mood Stabilizer and Anticonvulsant, Protects Rat Cerebral Cortical Neurons from Spontaneous Cell Death: A Role of Histone Deacetylase Inhibition,” FEBS Letters, Vol. 542, No. 1, 2003, pp. 74-78. doi:10.1016/S0014-5793(03)00350-8
- G. Gardian, L. Yang, C. Cleren and N. Y. Calingasan, “Neuroprotective Effects of Phenylbutyrate AGAINST MPTP Neurotoxicity,” Neuromolecular Medicine, Vol. 5, No. 23, 2004, pp. 235-241. doi:10.1385/NMM:5:3:235
- G. Gardian, S. E. Browne, D. K. Choi and P. Klivenyi, “Neuroprotective Effects of Phenylbutyrate in the N171- 82Q Transgenic Mouse Model of Huntington’s Disease,” Journal of Biological Chemistry, Vol. 280, No. 1, 2005, pp. 556-563.
- M. Minamiyama, M. Katsuno, H. Adachi and M. Waza, “Sodium Butyrate Ameliorates Phenotypic Expression in a Transgenic Mouse Model of Spinal and Bulbar Muscular Atrophy,” Human Molecular Genetics, Vol. 13, No. 11, 2004, pp. 1183-1192. doi:10.1093/hmg/ddh131
- G. S. Peng, G. Li, N. S. Tzengc and P. S. Chen, “Valproate Pretreatment Protects Dopaminergic Neurons from LPS-Induced Neurotoxicity in Rat Primary Midbrain Cultures: Role of Microglia,” Molecular Brain Research, Vol. 134, No. 1, 2005, pp. 162-169. doi:10.1016/j.molbrainres.2004.10.021
- S. Petri, M. Kiaei, K. Kipian and J. Chen, “Additive Neuroprotective Effects of a Histone Deacetylase Inhibitor and a Catalytic Antioxidant in a Transgenic Mouse Model of Amyotrophic Lateral Sclerosis,” Neurobiology of Disease, Vol. 22, No. 1, 2006, pp. 40-49. doi:10.1016/j.nbd.2005.09.013
- P. S. Chen, C. C. Wang, C. D. Bortner and G. S. Peng, “Valproic Acid and Other HDAC Inhibitors Induce Microglial Apoptosis and Attenuate Lipopolysaccharide-Induced Dopaminergic Neurotoxicity,” Neuroscience, Vol. 149, No. 1, 2007, pp. 203-212. doi:10.1016/j.neuroscience.2007.06.053
- G. Raivich and A. Behrens, “Role of the AP-1 Transcription Factor c-JUN in Developing, Adult and Injured Brain,” Progress in Neurobiology, Vol. 78, No. 6, 2006, pp. 347-363. doi:10.1016/j.pneurobio.2006.03.006
- J. A. Williams, C. J. Barreiro, L. U. Nwakanma and M. S. Lange, “Valproic Acid Prevents Brain Injury in a Canine Model of Hypothermic Circulatory Arrest: A Promising New Approach to Neuroprotection during Cardiac Surgery,” The Annals of Thoracic Surgery, Vol. 81, No. 6, 2006, pp. 2235-2242. doi:10.1016/j.athoracsur.2005.12.060
- H. Dou, K. Birusingh, J. Faraci, S. Gorantia, et al., “Neuroprotective Activities of Sodium Valproate in a Mu-rine Model of Human Immunodeficiency Virus-1 Encephalitis,” Journal of Neuroscience, Vol. 23, No. 27, 2003, pp. 9162- 9170.
- H. Kanai, A. Sawa, R. W. Chen, P. Leeds and D. M. Chuang, “Valproic Acid Inhibits Histone Deacetylase Activity and Suppresses Excitotoxicity-Induced GAPDH Nuclear Accumulation and Apoptotic Death in Neurons,” The Pharmacogenomics Journal, Vol. 4, 2004, pp. 336- 344. doi:10.1038/sj.tpj.6500269
- P. S. Chen, G. S. Peng, G. Li, S. Yang, et al., “Valproate Protects Dopaminergic Neurons in Midbrain Neuron/Glia Cultures by Stimulating the Release of Neurotrophic Factors from Astrocytes,” Molecular Psychiatry, Vol. 11, No. 12, 2006, pp. 1116-1125. doi:10.1038/sj.mp.4001893
- Y. Leng and De-Maw Chuang, “Endogenous α-Synuclein Is Induced by Valproic Acid through Histone Deacetylase Inhibition and Participates in Neuroprotection against Glutamate-Induced Excitotoxicity,” Journal of Neuroscience, Vol. 26, No. 28, 2006, pp. 7502-7512. doi:10.1523/JNEUROSCI.0096-06.2006
- D. I. Sinn, S. J. Kim, K. H. Jung and S. T. Lee, “Valproic Acid-Mediated Neuroprotection in Intracerebral Hemorrhage via Histone Deacetylase Inhibition and Transcriptional Activation,” Neurobiology of Disease, Vol. 26, No. 2, 2007, pp. 464-472. doi:10.1016/j.nbd.2007.02.006
- Y. Leng, M. H. Liang, M. Ren, et al., “Synergistic Neuroprotective Effects of Lithium and Valproic Acid or other Histone Deacetylase Inhibitors in Neurons: Roles of Glycogen Synthase Kinase-3 Inhibition,” Journal of Neuroscience, Vol. 28, No. 10, 2008, pp. 2576-2588. doi:10.1523/JNEUROSCI.5467-07.2008
- S. R. D’Mello, “Histone Deacetylases as Targets for the Treatment of Human Neurodegenerative Diseases,” Drug News & Perspectives, Vol. 22, No. 9, 2009, pp. 513-524. doi:10.1358/dnp.2009.22.9.1437959
- B. Monti, V. Gatta, F. Piretti, S. S. Rafaelli, M. Virgili and A. Contestabile, “Valproic Acid Is Neuroprotective in the Rotenone Rat Model of Parkinson’s Disease: Involvement of Alpha-Synuclein,” Neurotoxicity Research, Vol. 17, No. 2, 2010, pp. 130-141. doi:10.1007/s12640-009-9090-5
- X. N. Li, Q. Shu, J. M. F. Su, L. Perlaky, S. M. Blaney and C. C. Lau, “Valproic Acid Induces Growth Arrest, Apoptosis, and Senescence in Medulloblastomas by Increasing Histone Hyperacetylation and Regulating Expression of p21Cip1, CDK4, and CMYC,” Molecular Cancer Therapeutics, Vol. 4, No. 12, 2005, pp. 1912- 1922. doi:10.1158/1535-7163.MCT-05-0184
- T. Akiama, “Wnt/Beta-Catenin Signaling,” Cytokine Growth Factor Review, Vol. 11, No. 4, 2000, pp. 273-282. doi:10.1016/S1359-6101(00)00011-3
- M. Barbacid, “Ras Genes,” Annual Review of Biochemistry, Vol. 56, 1987, pp. 779-827. doi:10.1146/annurev.bi.56.070187.004023
- G.-A. Jung, J.-Y. Yoon, B.-S. Moon, D.-H. Yang, et al., “Valproic Acid Induces Differentiation and Inhibition of Proliferation in Neural Progenitor Cells via the Beta-Catenin-Ras-ERK-p21Cip/WAF1 Pathway,” BMC Cell Biology, Vol. 9, 2008, p. 66. doi:10.1186/1471-2121-9-66
- D. Bar-Sagi and A. Hall, “Ras and Rho GRTases: A Family Reunion,” Cell, Vol. 103, No. 2, 2000, pp. 227- 238. doi:10.1016/S0092-8674(00)00115-X
- P. V. Nunes, P. Wacker, O. V. Forlenza and W. F. Gattaz, “O Uso do Lítio em Idosos: Evidências de Sua Ação Neuroprotetora,” Revista de Psiquiatria Clínica, Vol. 29, 2002, pp. 248-255.
- P. X. Yuan, L. D. Huang, Y. M. Jiang, J. S. Gutkind, et al., “The Mood Stabilizer Valproic Acid Activates Mitogen-Activated Protein Kinases and Promotes Neurite Growth,” Journal of Biological Chemistry, Vol. 276, 2001, pp. 31674-31683. doi:10.1074/jbc.M104309200
- P. De Sarno, X. Li and R. S. Jope, “Regulation of Akt and Glycogen Synthase Kinase-3 Beta Phosphorylation by Sodium Valproate and Lithium,” Neuropharmacology, Vol. 43, No. 7, 2002, pp. 1158-1164. doi:10.1016/S0028-3908(02)00215-0
- Y. Hao, T. Creson, L. Zhang, P. Li, et al., “Mood Stabilizer Valproate, Promotes ERK Pathway-Dependent Cortical Neuronal Growth and Neurogenesis,” Journal of Neuroscience, Vol. 24, No. 29, 2004, pp. 6590-6599. doi:10.1523/JNEUROSCI.5747-03.2004
- J. Hsieh, K. Nakashima, T. Kuwabara, E. Mejia and F. H. Gage, “Histone Deacetylase Inhibition-Mediated Neuronal Differentiation of Multipotent Adult Neural Progenitor Cells,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 101, No. 47, 2004, pp. 16659-16664. doi:10.1073/pnas.0407643101
- G. Chen, L. D. Huang, Y. M. Jiang and H. K. Manji, “The Mood Stabilizing Agent Valproate Inhibits the Activity of Glycogen Synthase Kinase-3,” Journal of Neurochemistry, Vol. 72, No. 3, 1999, pp. 1327-1330. doi:10.1046/j.1471-4159.2000.0721327.x
- J. W. Kim, J. E. Lee. M. J. Kim, E. G. Cho, et al., “Glycogen Synthase Kinase 3 Beta Is a Natural Activator of Mitogen-Activated Protein Kinase/Extracellular SignalRegulated Kinase Kinase 1 (MEKK1),” Journal of Biological Chemistry, Vol. 278, 2003, pp. 13995-14001. doi:10.1074/jbc.M300253200
- V. Stambolic, L. Ruel and J. R. Woodgett, “Lithium Inhibits Glycogen Synthase Kinase-3 Activity and Mimics Wingless Signalling in Intact Cells,” Current Biology, Vol. 6, No. 12, 1996, pp. 1664-1668. doi:10.1016/S0960-9822(02)70790-2
- D. M. Chuang, Z. Wang and C. T. Chiu, “GSK-3 as a Target for Lithium-Induced Neuroprotection against Excitotoxicity in Neuronal Cultures and Animal Models of Ischemic Stroke,” Frontiers in Molecular Neuroscience, Vol. 4, 2011, p. 15. doi:10.3389/fnmol.2011.00015
- A. M. Corral, “Mecanismos Implicados en la Protección de la Apoptosis Neuronal en céLulas Granulares de Cerebelo por Litio y Valproato,” Tesis Doctoral Dirigida por Francisco Centeno Velázquez, Universidad de Extremadura, Mérida, 2001.
- J. Dupont, M. Karas and D. Leroith, “The Cyclin-Dependent Kinase Inhibitor p21CIP/WAF Is a Positive Regulator of Insulin-Like Growth Factor 1-Induced Cell Proliferation in MCF-7 Human Breast Cancer Cells,” Journal of Biological Chemistry, Vol. 278, 2003, pp. 37256-37264. doi:10.1074/jbc.M302355200
- J. M. Levenson, K. J. O’Riordan, K. D. Brown, M. A. Trinh, et al., “Regulation of Histone Acetylation during Memory Formation on the Hippocampus,” Journal of Biological Chemistry, Vol. 279, 2004, pp. 40545-40559. doi:10.1074/jbc.M402229200
- T. W. Bredy, H. Wu, C. Crego, J. Zellhoefer, Y. E. Sun and M. Barad, “Histone Modifications around Individual BDNF Gene Promotors in Prefrontal Cortex Are Associated with Extinction of Conditioned Fear,” Learning & Memory, Vol. 14, 2007, pp. 268-276. doi:10.1101/lm.500907
- M. Cammarota, L. R. M. Bevilaqua, M. R. M. Vianna, J. H. Medina and I. Izquierdo, “The Extinction of Conditioned Fear: Structural and Molecular Basis and Therapeutic Use,” Revista Brasileira de Psiquiatria, Vol. 29, No. 1, 2007, pp. 80-85. doi:10.1590/S1516-44462006005000022
- K. M. Lattal, R. M. Barrett and M. A. Wood, “Systemic or Intrahippocampal Delivery of Histone Deacetylase Inhibitors Facilitates Fear Extinction,” Behavioral Neuroscience, Vol. 121, No. 5, 2007, pp. 1125-1131. doi:10.1037/0735-7044.121.5.1125
- G. Raivich and A. Behrens, “Role of the AP-1 Transcription Factor c-Jun in Developing, Adult and Injured Brain,” Progress in Neurobiology, Vol. 78, No. 6, 2006, pp. 347- 363. doi:10.1016/j.pneurobio.2006.03.006
- P. K. Dash, S. A. Orsi, M. Zhang, R. J. Grill, et al., “Valproate Administered after Traumatic Brain Injury Provides Neuroprotection and Improves Cognitive Function in Rats,” Plos ONE, Vol. 5, No. 6, 2010, p. e11383.
- E. A. Thomas, “Focal Nature of Neurological Disorders Necessitates Isotype-Selective Histone Deacetylase (HDAC) Inhibitors,” Molecular Neurobiology, Vol. 40, No. 1, 2009, pp. 33-45. doi:10.1007/s12035-009-8067-y
- N. N. Nalivaeva, N. D. Belyaev and A. J. Turner, “Sodium Valproate: An Old Drug with New Roles,” Trends in Pharmacological Sciences, Vol. 30, No. 10, 2009, pp. 509-514. doi:10.1016/j.tips.2009.07.002
- C. Morland, K. A. Boldingh, E. G. Iversen and B. Hassel, “Valproate Is Neuroprotective against Malonate Toxicity in Rat Striatum: An Association with Augmentation of High-Affinity Glutamate Uptake,” Journal of Cerebral Blood Flow and Metabolism, Vol. 24, 2004, pp. 1226- 1234. doi:10.1097/01.WCB.0000138666.25305.A7
- R. S. Williams, L. Cheng, A. W. Mudge and A. J. Harwood, “A Common Mechanism of Action for Three MoodStabilizing Drugs,” Nature, Vol. 417, 2002, pp. 292-295. doi:10.1038/417292a
- C. J. Phiel, F. Zhang, E. Y. Huang and M. G. Guenther, “Histone Deacetylase Is a Direct Target of Valproic Acid, a Potent Anticonvulsant, Mood Stabilizer, and Teratogen,” Journal of Biological Chemistry, Vol. 276, 2001, pp. 36734-36741. doi:10.1074/jbc.M101287200
- M. Gottlicher, S. Minucci, P. Zhu and O. H. Kramer, “Valproic Acid Defines a Novel Class of HDAC Inhibitors Inducing Differentiation of Transformed Cells,” The EMBO Journal, Vol. 20, 2001, pp. 6969-7698. doi:10.1093/emboj/20.24.6969
- N. Detich, V. Bovenzi and M. Szyf, “Valproate Induces Replication-Independent Active DNA Demetylation,” Journal of Biological Chemistry, Vol. 278, 2003, pp. 27586- 27592. doi:10.1074/jbc.M303740200
- S. Milutinovic, A. C. Dálessio, N. Detich and M. Szyf, “Valproate Reduces Widespread Epigenetic Reprogramming Which Involves Demethylation of Specific Genes,” Carcinogenesis, Vol. 28, No. 3, 2007, pp. 560-571. doi:10.1093/carcin/bgl167
- L. M. Castro, M. Gallant and L. P. Niles, “Novel Targets for Valproic Acid and Upregulation of Melatonin Receptors and Neurotrophic Factors in C6 Glioma Cells,” Journal of Neurochemistry, Vol. 95, No. 5, 2005, pp. 1227- 1236. doi:10.1111/j.1471-4159.2005.03457.x
- X. Wu, P. S. Chen, S. Dallas and B. Wilson, “Histone Deacetylase Inhibitors Upregulate Astrocyte GDNF and BDNF Gene Transcription and Protect Dopaminergic Neurons” International Journal of Neuropsychopharmacology, Vol. 9, No. 3, 2008, pp. 1-12.
- N. Gurvich, M. G. Berman, B. S. Wittner and R. C. Gentleman, “Association of Valproate-Induced Teratogenenesis with Histone Deacetylase Inhibition in Vivo,” FASEB Journal, Vol. 19, No. 9, 2005, pp. 1166-1168.
- B. H. Maskrey, I. L. Megson, P. D. Whitfield and A. G. Rossi, “Mechanisms of Resolution of Inflammation, a Focus on Cardiovascular Disease,” Arteriosclerosis, Thrombosis and Vascular Biology, Vol. 31, 2011, pp. 1001- 1006. doi:10.1161/ATVBAHA.110.213850
- D. Bayarsaihan, “Epigenetic Mechanisms in Inflammation,” Journal of Dental Research, Vol. 90, No. 1, 2011, pp. 9-17. doi:10.1177/0022034510378683
- L. A. B. Joosten, F. Leoni, S. Meghji and P. Mascagni, “Inhibition of HDAC Activity by ITF2357 Ameliorates Joint Inflammation and Prevents Cartilage and Bone Destruction in Experimental Arthritis,” Molecular Medicine, Vol. 17, No. 5-6, 2011, pp. 391-396. doi:10.2119/molmed.2011.00058
- S. Shuttlewort and S. Kerry, “HDAC Inhibitors: New Promise in the Treatment of Immune and Inflammatory Disease,” Innovations in Pharmaceutical Technology, 2011. http://www.Iptonline.com/articles/public/pg/nonprint.pdf
- J. C. Ximenes, D. O. Gonçalves, R. Siqueira and L. K. A. M. Leal, “Valproic Acid Reduces Polymorphonuclear Cell Migration and Myeloperoxidase Release: An in Vivo and in Vitro Study,” XXVI Annual Meeting of the Brazilian Federation of Biological Societies (FESBE), Rio de Janeiro, Brazil, 2011.
- J. C. Ximenes, E. C. Lima-Verde, R. S. Pinheiro, K. R. T. Neves, D. O. Gonçalves and G. S. B. Viana, “Avaliação dos Efeitos Anti-Inflamatórios e Antinociceptivos do Ácido valpróIco (AV) em Modelos Experimentais de Edema de pata e no Teste da Formalina, em Roedores,” XXXIV Annual Congress of the Brazilian Society of Neuroscience and Behavior, Caxambu, Brazil, 2010.
- K. S. Figueiredo, M. L. A. Oliveira Sales, J. B. Fontenele, G. S.B.Viana and F. H. C. Félix, “Valproate Effect in Carrageenan-Induced Thermal Hyperalgesia in Female Rats Has A Bimodal Dose-Response Curve,” to be submitted.
- I. M. Adcock, “Histone Deacetylase Inhibitors as Novel Anti-Inflammatory Agents,” Current Opinion in Investigational Drugs, Vol. 7, No. 11, 2006, pp. 966-973.
- C. A. Dinarello, G. Fossati and P. Mascagni, “Histone Deacetylase Inhibitors for Treating a Spectrum of Diseases Not Related to Cancer,” Molecular Medicine, Vol. 17, No. 5-6, 2011, pp. 333-352. doi:10.2119/molmed.2011.00116
- G. Faraco, M. Pittelli, L. Cavone and S. Fossati, “Histone Deacetylase (HDAC) Inhibitors Reduce the Glial Inflammatory Response in Vitro and in Vivo,” Neurobiology of Disease, Vol. 36, No. 2, 2009, pp. 269-279. doi:10.1016/j.nbd.2009.07.019
- T. Suuronen, J. Huuskonen, R. Pihlaja, S. Kyrylenko and A. Salminen, “Regulation of Microglial Inflammatory Response by Histone Deacetylase Inhibitors,” Journal of Neurochemistry, Vol. 87, No. 2, 2003, pp. 407-416. doi:10.1046/j.1471-4159.2003.02004.x
- J. Huuskonen, T. Suuronen, T. Nuutinen, S. Kyrylenko and A. Salminen, “Regulation of Microglial Inflammatory Response by Sodium Butyrate and Short-Chain Fatty Acids,” Pharmacology & Pharmaceutical Medicine, Vol. 141, No. 5, 2004, pp. 874-880. doi:10.1038/sj.bjp.0705682
- M. Dragunow, J. M. Greenwood, R. E. Cameron, P. J. Narayan, et al., “Valproic Acid Induces Caspase 3-Mediated Apoptosis in Microglial Cells,” Neuroscience, Vol. 140, No. 4, 2006, pp. 1149-1156. doi:10.1016/j.neuroscience.2006.02.065
- H. Kankaanranta, M. Janka, P. Ilmarinen-Salo and K. Ito, “Histone Deacetylase Inhibitors Induce Apoptosis in Human Eosinophils and Neutrophils,” Journal of Inflammation, Vol. 7, 2010, pp. 1-15. doi:10.1186/1476-9255-7-9
- T. Abel and S. Zukin, “Epigenetic Targets of HDAC Inhibition in Neurodegenerative and Psychiatric Disorders,” Current Opinion in Pharmacology, Vol. 8, No. 1, 2008, pp. 57-64. doi:10.1016/j.coph.2007.12.002
- S. J. K. M. Haggarty, J. C. Koeler, J. C. Wong, C. M. Grozinger, et al., “Domain-Selective Small Molecule Inhibitor of Histone Deacetylase 6 (HDAC6)-Mediated Tubulin Deacetylation,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 100, No. 8, 2003, pp. 4389-4394. doi:10.1073/pnas.0430973100
- J. S. Guan, S. J. Haggart, E. Giacometti and J. H. Dannenberg, “HDAC2 Negatively Regulates Memory Formation and Synaptic Plasticity,” Nature, Vol. 459, 2009, pp. 55-60. doi:10.1038/nature07925
- M. Kilgore, C. A. Miller, D. M. Fass and K. M. Henning, “Inhibitors of Class 1 Histone Deacetylases Reverse Contextual Memory Deficits in a Mouse Model of Alzheimer’s Disease,” Neuropsychopharmacology, Vol. 35, 2010, pp. 870-880. doi:10.1038/npp.2009.197
- E. Costa, Y. Chen, E. Dong and D. R. Grayson, “GABAergic Promoter Hypermethylation as a Model to Study the Neurochemistry of Schizophrenia Vulnerability,” Expert Review of Neurotherapeutics, Vol. 9, No. 1, 2009, pp. 87-98. doi:10.1586/14737175.9.1.87
- P. N. Tariot, L. S. Schneider, J. Cummings, R. G. Thomas, et al., “Chronic Divalproex Sodium to Attenuate Agitation and Clinical Progression of Alzheimer Disease,” Archives of General Psychiatry, Vol. 68, No. 8, 2011, pp. 853-861. doi:10.1001/archgenpsychiatry.2011.72
- A. S. Fleischer, D. Truran, J. T. Mai and J. B. Langbaum, “Chronic Divalproex Sodium Use and Brain Atrophy in Alzheimer Disease,” Neurology, Vol. 77, No. 13, 2011, pp. 1263-1271. doi:10.1212/WNL.0b013e318230a16c
- M. K. Tripathy and D. Mitra, “Differential Modulation of Mitochondrial OXPHOS System during HIV-1-Induced T Cell Apoptosis: Up Regulation of Complex-IV Subunit and Its Possible Implications,” Apoptosis, Vol. 15, No. 1, 2010, pp. 28-40. doi:10.1007/s10495-009-0408-9
- T. Nonaka and M. Hasegawa, “In Vitro Recapitulation of Aberrant Protein Inclusions in Neurodegenerative Diseases,” Communicative & Integrative Biology, Vol. 4, No. 4, 2011, pp. 501-502.
- S. A. Lipton, Z. Gu and T. Nakamura, “Inflammatory Mediators Leading to Protein Misfolding and Uncompetitive/Fast Off-Rate Drug Therapy for Neurodegenerative Disorders,” International Review of Neurobiology, Vol. 82, 2007, pp. 1-27. doi:10.1016/S0074-7742(07)82001-0
- A. T. Welzel and D. M. Walsh, “Aberrant Protein Structure and Diseases of the Brain,” Irish Journal of Medical Science, Vol. 180, No. 1, 2011, pp. 15-22. doi:10.1007/s11845-010-0606-z
- S. Oddo, “The Ubiquitin-Proteosome System in Azheimer’s Diseases,” Journal of Cellular and Molecular Medicine, Vol. 12, No. 2, 2008, pp. 363-373. doi:10.1111/j.1582-4934.2008.00276.x
- C. W. Olanow and K. S. McNaught, “Ubiquitin-Proteosome System and Parkinson’s Disease,” Movement Disorders, Vol. 21, No. 11, 2006, pp. 1006-1023.
- C. A. Ross and C. M. Pickar, “The Ubiquitin-Proteosome Pathway in Parkinson’s Disease and Other Neurodegenerative Diseases,” Trends in Cell Biology, Vol. 14, No. 12, 2004, pp. 703-711. doi:10.1016/j.tcb.2004.10.006
- B. J. Tabner, S. Tumbull, O. El-Agnaf and D. Allsop, “Production of Reactive Oxygen Species from Aggregating Proteins Implicated in Alzheimer’s Disease, Parkinson’s Disease and Other Neurodegenerative Diseases,” Current Topics in Medicinal Chemistry, Vol. 1, 2001, pp. 507-517. doi:10.2174/1568026013394822
- K. Sas, H. Robotka, J. Toldi and L. Vécsei, “Mitochondria, Metabolic Disturbances, Oxidative Stress and the Kinure-Nine System, with Focus on Neurodegenerative Diseases,” Journal of Neuroscience, Vol. 257, 2007, pp. 221-239.
- J. Emerit, M. Edeas and F. Bricaire, “Neurodegenerative Diseases and Oxidative Stress,” Biomedicine & Pharmacotherapy, Vol. 58, No. 1, 2004, pp. 39-46. doi:10.1016/j.biopha.2003.11.004
- N. Shibata and M. Kobayashi, “The Role for Oxidative Stress in Neurodegenerative Diseases,” Brain Nerve, Vol. 60, No. 2, 2008, pp. 157-170.
- M. M. Esiri, “The Interplay between Inflammation and Neurodegeneration in CNS Disease,” Journal of Neuroimmunology, Vol. 184, No. 1-2, 2007, pp. 4-16.
- T. Wyss-Coray and L. Mucke, “Inflammation in Neurodegenerative Disease: A Doble-Edged Sword,” Neuron, Vol. 35, No. 3, 2002, pp. 419-432. doi:10.1016/S0896-6273(02)00794-8
- F. Zipp and O. Aktas, “The Brain as Target of Inflammation: Common Pathways Link Inflammatory and Neurodegenerative Diseases,” Trends in Biochemical Sciences, Vol. 29, No. 9, 2010, pp. 518-527.
- G. C. Brown and J. J. Neher, “Inflammatory Neurodegeneration and Mechanisms of MicróGlial Killing of Neurons,” Molecular Neurobiology, Vol. 41, No. 2-3, 2010, pp. 242-247. doi:10.1007/s12035-010-8105-9
- B. Liu and J.-S. Hong, “Role of Microglia in Inflammation-Mediated Neurodegenerative Diseases: Mechanisms and Strategies for Therapeutic Intervention,” Journal of Pharmacology and Experimental Therapeutics, Vol. 304, No. 1, 2003, pp. 1-7. doi:10.1124/jpet.102.035048
- A. Cagnin, M. Kassiou, S. R. Meikle and R. B. Banati, “In Vivo Evidence for MicróGlial Activation in Neurodegenerative Dementia,” Acta Neurologica Scandinavica, Vol. 114, No. S185, 2006, pp. 107-114. doi:10.1111/j.1600-0404.2006.00694.x
- L.-F. Lue, Y.-M. Kuo, T. Beach and D. G. Walker, “Microglia Activation and Anti-Inflammatory Regulation in Alzheimer’s Disease,” Molecular Neurobiology, Vol. 41, No. 2-3, 2010, pp. 115-128. doi:10.1007/s12035-010-8106-8
- M. A. Burguillos, T. Deierborg, E. Kavanagh, A. Persson, et al., “Caspase Signalling Controls Microglia Activation and Neurotoxicity,” Nature, Vol. 472, No. 7343, 2011, pp. 319-324. doi:10.1038/nature09788
- J. A. Beller, G. G. Gurkoff, R. F. Berman and B. G. Lyeth, “Pharmacological Enhancement of Glutamate Transport Reduces Excitotoxicity in Vitro,” Restorative Neurology and Neuroscience, Vol. 29, No. 5, 2011, pp. 331-346.
- K. Kim, S. G. Lee, T. P. Kegelman, Z. Z. Su, et al., “Role of Excitatory Amino Acid Transporter-2 (EAAT2) and Glutamate in Neurodegeneration: Opportunities for Developing Novel Therapeutics,” Journal of Cellular Physiology, Vol. 226, No. 10, 1011, pp. 2484-2493.
- S. K. Kidd and J. S. Schneider, “Protective Effects of Valproic Acid on the Nigrostriatal Dopamine System in a 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson’s Disease,” Neuroscience, Vol. 194, 2011, pp. 189-194. doi:10.1016/j.neuroscience.2011.08.010
- M. T. Lin and M. F. Beal, “Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases,” Nature, Vol. 443, 2006, pp. 787-795. doi:10.1038/nature05292
- M. B. Moura, L. S. Santos and B. V. Houten, “Mitochondrial Dysfunction in Neurodegenerative Diseases and Cancer,” Environmental and Molecular Mutagenesis, Vol. 51, No. 5, 2010, pp. 395-405.
- S. Hammed and G.-Y. R. Hsung, “The Role of Mitochondria in Aging, Neurodegenerative Disease, and Future Therapeutic Options,” BC Medical Journal, Vol. 53, No. 4, 2011, pp. 188-192.
- H. Du, L. Guo and S. S. Yan, “Synaptic Mitochondrial Pathology in Alzheimer’s Disease,” Antioxidants & Redox Signaling, 2011, Ahead of Print.
NOTES
*Corresponding author.