Advances in Microbiology
Vol.3 No.1(2013), Article ID:29232,7 pages DOI:10.4236/aim.2013.31016
Improved Method for Isolation of Microbial RNA from Biofuel Feedstock for Metatranscriptomics*
1School of Molecular Biosciences, Washington State University, Richland, USA
2Pacific Northwest National Laboratory, Richland, USA
3Department of Animal Sciences, Institute for Genomic Biology and Energy Biosciences, University of Illinois, Urbana, USA
4DOE Joint Genome Institute, Walnut Creek, USA
Email: #matthias.hess@tricity.wsu.edu
Received November 28, 2012; revised December 29, 2012; accepted January 25, 2013
Keywords: RNA Extraction; Biofuels; CAZymes; Metatranscriptomics; Systems Microbiology; Microbial Ecology
ABSTRACT
Metatranscriptomics—gene express profiling via DNA sequencing—is a powerful tool to identify genes that are actively expressed and might contribute to the phenotype of individual organisms or the phenome (the sum of several phenotypes) of a microbial community. Furthermore, metatranscriptome studies can result in extensive catalogues of genes that encode for enzymes of industrial relevance. In both cases, a major challenge for generating a high quality metatranscriptome is the extreme lability of RNA and its susceptibility to ubiquitous RNAses. The microbial community (the microbiome) of the cow rumen efficiently degrades lignocelullosic biomass, generates significant amounts of methane, a greenhouse gas twenty times more potent than carbon dioxide, and is of general importance for the physiological wellbeing of the host animal. Metatranscriptomes of the rumen microbiome from animals kept under different conditions and from various types of rumen-incubated biomass can be expected to provide new insights into these highly interesting phenotypes and subsequently provide the framework for an enhanced understanding of this socioeconomically important ecosystem. The ability to isolate large amounts of intact RNA will significantly facilitate accurate transcript annotation and expression profiling. Here we report a method that combines mechanical disruption with chemical homogenization of the sample material and consistently yields 1 mg of intact RNA from 1 g of rumen-incubated biofuel feedstock. The yield of total RNA obtained with our method exceeds the RNA yield achieved with previously reported isolation techniques, which renders RNA isolated with the method presented here as an ideal starting material for metatranscriptomic analyses and other molecular biology applications that require significant amounts of starting material.
1. Introduction
The rumen is a complex ecosystem containing billions of microorganisms including archaea, bacteria, protozoa, and fungi [1,2]. Many of these microbes are tightly attached to solid rumen content [3] and produce biomassdegrading enzymes [1]. These naturally existing biomass degraders have attracted great interest by the scientific community due to their ability to convert complex lignocellulosic biomass into smaller molecules; and microbial enzymes from the cow rumen have been suggested for optimizing current industrial processes that produce lignocellulosic biofuels [4]. A major challenge in studying the microbial community (“the microbiome”) of the cow rumen and its biomass-degrading enzymatic machinery is the scarcity of appropriate isolation and cultivation techniques to obtain axenic cultures from environmental samples [5]. The consequential paucity of biomass-degrading isolates limits our ability to study the enzymatic machinery that underlies their economically important phenotype. Nucleotide sequencing using nextgeneration technologies is a powerful tool to overcome this cultivation bottleneck and has resulted in the discovery of thousands of new biomass-degrading genes and the assembly of several prokaryotic draft genomes from environmental DNA [6-11]. While these metagenomic studies provide insights into the genetic blueprint and the molecular machinery of microorganisms in a particular environment, metatranscriptomics can be used to identify those genes that are actively expressed under certain physiological conditions and to reveal transcripts from microbes that contribute to only a small fraction of the overall population, but which are of significance for the overall phenome of an ecosystem [12]. Despite the obvious benefits of metatranscriptomics, there is—to our knowledge—only one study that utilized next-generation sequencing (NGS) to obtain an expression profile of the microbiome associated with a ruminant animal [13]. This paucity might be partially due to the significant technical challenges that are associated with the isolation of RNA— a highly unstable molecule—and that the expensive and time-consuming RNA extraction techniques that are currently available still result in total RNA that contains significant fractions of undesirable rRNA [14]. Techniques for the extraction of total RNA have been optimized for microbial habitats as diverse as soil and ocean water and it is very likely that these and newly developed techniques will continuously be optimized according to the nature of the sample or the specific research objective [15- 18]. All successful RNA extraction techniques have in common that they are generating significant amounts of intact RNA at a relatively low cost and it can be hypothesized that the more distinct RNA extraction methods are publicly available the more likely it will be that a suitable protocol will be readily available for future metatranscriptome studies. As there has been an increased interest in generating an expression profile of the rumen microbiome, several protocols for extraction of total RNA from the liquid and solid phase of the rumen have recently been developed [19-21]. Average total RNA yields as high as ~200 μg of total RNA/(mL rumen fluid) and ~110 μg of total RNA/(g solid digesta) had been reported from a cannulated Holstein cow and a cannulated muskoxen respectively [20,21]. Here we report an extraction protocol that consistently yields ~1 mg of total RNA/(g rumen-incubated biomass) from a cannulated cow, which is a significant increase in RNA yield compared to previously reported methods. The technique reported here opens new perspectives in studying microbial gene expression in the rumen ecosystem using traditional molecular techniques and next generation sequencing technologies.
2. Materials and Methods
2.1. Sample Collection
To enrich fiber adherent microbes, air-dried corn stover and switchgrass were ground into 2 mm pieces, weighed into individual in-situ nylon bags (50 μm pores; Ankom Technology, Macedon, NY, USA) and placed in the rumen of a cannulated cow [8]. The nylon bags were retrieved after 48 hrs, washed with H2O to remove loosely adherent microbes, and frozen immediately in liquid nitrogen. Samples were kept at −80˚C until RNA extraction. All procedures with the cow were carried out under an approved protocol with the University of Illinois Institutional Animal Care and Use of Animals Committee (IUCAC #06081).
2.2. Total RNA Isolation
The conventional TRIzol-based RNA isolation method [22], performed according to the manufacturer’s protocol after the samples had been manually ground in the presence of liquid nitrogen, served as the standard RNA isolation procedure and was used as a control method (MC). To optimize the procedure and increase RNA yield, we added a beat beading step. The individual steps of the new methods (MB) were as follows: 100 mg of frozen rumen-incubated biomass were ground to powder in liquid nitrogen using a pestle and mortar. One mL TRIzol reagent (Invitrogen, Carlsbad, CA) was added to the rumen-incubated biomass and the biomass/TRIzol mixture was transferred into a 2 mL microcentrifuge tube containing Lysing Matrix E (MP Biomedicals Solon, OH), followed by bead beating (3 × 1 min at room temperature, 2 min at 4˚C between individual beating steps) using a Mini-Beadbeater-16 (Biospec Products, Bartlesville OK). Homogenized samples were centrifuged (12,000 × g, 10 min at 4˚C); the supernatant was transferred to new tubes and incubated at room temperature for 5 min. Subsequent TRIzol-based RNA isolation was performed according to manufacturer’s instructions. To verify reproducibility each extraction was performed in biological and technical duplicates.
2.3. RNA Purification
To test the effect of an additional column purification step on RNA yield and integrity, total RNA was purified with the commercially available MEGAclear kit (Life Technologies, Grand Island, NY). The RNA purification procedures were performed according to the manufacturer’s instructions.
2.4. Analysis of RNA Quantity and Quality
RNA quantity and quality were analyzed with an Agilent 2100 bioanalyzer system (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s instructions.
2.5. Reverse Transcription and PCR
Total RNA (500 ng) was reverse-transcribed into cDNA using Invitrogen’s Superscript double-stranded cDNA Synthesis kit and random hexamer primers according to the manufacturer’s instructions. The synthesized cDNA was used to amplify fragments of three CAZyme genes (celF, xynD, and cel3) from Fibrobacter succinogenes S85 as previously described [19,21]. Subsequent PCR reactions were performed using 2x AmpliTag Gold PCR master mix (Invitrogen) with 1 µL first strand cDNA as template. The PCR conditions included: initial denaturation for 1 min at 94˚C, followed by 40 amplification cycles of (15 s at 94˚C, 30 s at 55˚C, and 45 s at 72˚C), and a final elongation for 10 min at 72˚C. A positive control PCR, with environmental DNA extracted from the rumen microbiome as template, and a negative control PCR (“no reverse transcriptase”) was performed for each target gene. The PCR products were visualized on a 1% agarose gel stained with ethidium bromide.
3. Results
3.1. Isolation of Total RNA from Rumen-Incubated Biomass
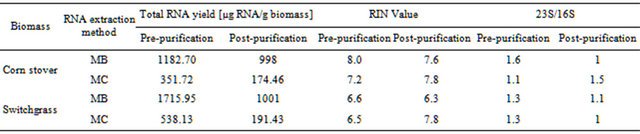
The ability to extract sufficient amounts of non-degraded RNA is crucial for the construction of cDNA libraries and subsequent expression profiling via nucleotide sequencing. To compare the efficiency of two different RNA extraction protocols, quantity and quality of RNA were determined using Agilent’s 2100 Bioanalyzer. Electropherograms (Figures 1(a)-(d)) of total RNA obtained with either method showed peaks specific for intact ribosomal RNA (rRNA) subunits (5S, 16S, and 23S) as well as additional RNA peaks between the 5S and 16S peaks, suggesting the presence of rRNAs, intact small mRNA and degraded mRNA. Our improved method (method MB) consistently yielded approximately 3-times more total RNA (~1 mg total RNA from one gram of rumenincubated biofuel feedstocks) than the conventional and widely used TRIzol-based method (method MC) (Table 1).
3.2. Effect of Column Purification on RNA Quality
To determine if the commercially available MEGAclear kit (Life Technologies, Grand Island, NY) is effective in removing a large fraction of small RNAs from total RNA extracted from rumen-incubated biomass, we analyzed the unpurified and purified RNA using Agilent’s Bioanalyzer
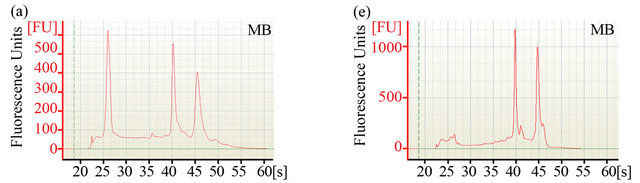
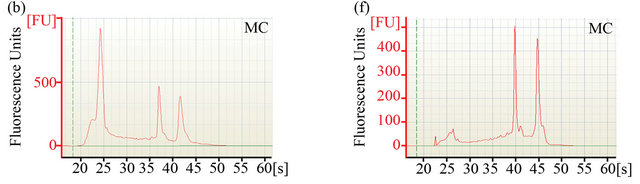
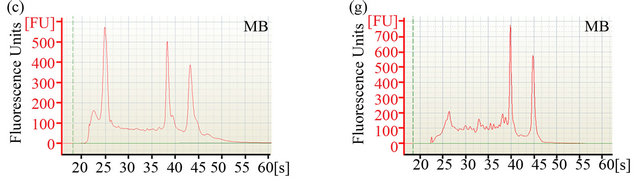
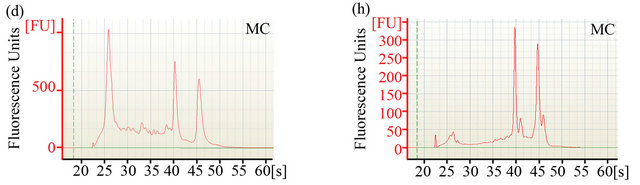
Figure 1. Analysis of RNA integrity. Integrity of pre-column purified ((a)-(d)) and post-column purified ((e)-(h)) RNA determined by Agilent 2100 bioanalyzer. (a), (b), (e) and (f): RNA from rumen-incubated corn stover; (c), (d), (g) and (h): RNA from rumen-incubated switchgrass; MB indicates method MB; MC indicates method MC.
Table 1. Comparison of different RNA extraction methods.
platform. The electropherograms (Figure 1) revealed a significant decrease in the size of the lower molecular weight RNA peaks, suggesting that the MEGAclear kit can be used to effectively remove a large fraction of the small RNAs from the pool of total RNA obtained with any of the methods tested during this study.
3.3. RT-PCR and Amplification of Genes Coding for Carbohydrate Active Enzymes (CAZymes)
To determine whether the RNA, obtained from rumenincubated biomass using our improved method MB, was suitable for downstream molecular biology applications, we amplified fragments of three CAZymes (celF, xynD, and cel3) from Fibrobacter succinogenes S85 by reverse transcription PCR (RT-PCR). As shown in Figure 2, the internal fragments of celF, xynD, and cel3 genes were detected by RT-PCR, resulting in products of the same size as those obtained with the positive control PCR (contained environmental DNA extracted from the rumen microbiome). All negative controls (lacking reverse transcriptase) did not show an amplification product, confirming that positive amplicons were transcribed from mRNA.
4. Discussion & Conclusions
With the increasing interest of the scientific community in generating metatranscriptomes from rumen microbiomes it is essential to have a variety of optimized protocols for extracting microbial RNA directly from rumen-incubated biomass. Here we report a simple method that consistently yields large amounts of intact RNA from rumen-incubated biofuel feedstock and that does not require any step to separate microorganisms from the fiber material or to further purify the resulting RNA. This simplicity reduces the sample processing time and consequently the possibility of RNA degradation during the extraction procedure. The control method (MC) used to evaluate the efficiency of our new method was based on the conventional TRIzol-based RNA isolation technique that has been successfully used to extract RNA from various biological samples [22,23]. As undigested feed particle have been shown to prevent the TRIzol reagent from rapid sample penetration and to introduce a bias in the RNA recovered from microbial populations, samples are manually disrupted in liquid nitrogen using a mortar and pestle prior to exposing the samples to the TRIzol reagent, which enhances unbiased RNA recovery [21,24]. Using this control method we consistently obtained RNA yields >300 µg (352 µg and 538 µg total RNA from 1 gram rumen incubated corn stover and switchgrass, respectively), which is approximately four times more RNA than the 85 μg RNA from 1 gram of solid rumen content that was reported in a recent study in which the authors utilized a slightly altered version of this standard procedure [21]. However, as Wang and coauthors used a muskoxen and undefined solid rumen content with unknown rumen-incubation time, it is not certain if this difference in RNA yield is caused by a significantly lower microbial activity or lower density of the fiber-adherent microbial population. Furthermore, it cannot be excluded, although it seems very unlikely, that the microbial population within the muskoxen is more recalcitrant to this RNA extraction protocol than the population within the cow that was used in our experiment. Although the control method already yielded significant amounts of intact RNA, we were interested in optimizing this technique to ensure the availability of sufficient starting material for downstream procedures. To facilitate improved RNA isolation we subjected the samples to an additional manual disruption step and exposed them to a short, vigorous bead beading. These modifications resulted in a highly homogenized sample and an increased recovery of total non-degraded RNA. Using the modified method MB, we were able to isolate 1183 µg total RNA and 1716 µg total RNA from 1 gram of rumen incubated corn stover and switchgrass, respectively (Table 1), representing a 3-fold increase when compared to the control method. Assuming that a similar increase might be achieved from muskoxen, yields as high as 240 µg of intact RNA from 1 gram of muskoxen solid rumen fraction might be possible, which opens new possibilities in muskoxen metatransciptomics.
When using downstream techniques that rely on the presence of large mRNA fragments (e.g. Sanger sequencing, enrichment of full-length polyandenylated mRNA, construction of cDNA libraries for activity-based assays) a decrease in total RNA yield might be tolerable and an additional column cleanup step can be of great value and eventually essential for the ultimate success of a project. To determine the effect of an additional column purification step on the total RNA isolated with our improved method MB and the control method MC, we compared RNA yield, RNA integrity number (RIN), 23S/16S ratio and the electropherograms of pre-purified and post-purified samples.
Regardless of the extraction method, the measured RNA yield decreased after column purification (Table 1), which suggests that an additional column purification step might be impractical if the RNA yield is the limiting factor. Furthermore, column purification seemed to remove the majority of 5S rRNA and other small mRNAs as indicated by the disappearance of the 5S rRNA-specific and other peaks that are associated with small mRNAs (Figures 1(e)-(f)). Removal of small RNAs might not seem to be a major problem at first, but it has to be taken into consideration that these fragments could represent valuable sequence information encoded on small RNAs that might be essential for some biological processes. Considering that these small RNAs can be successfully sequenced and analyzed with recently developed sequencing platforms and analysis tools [25,26],
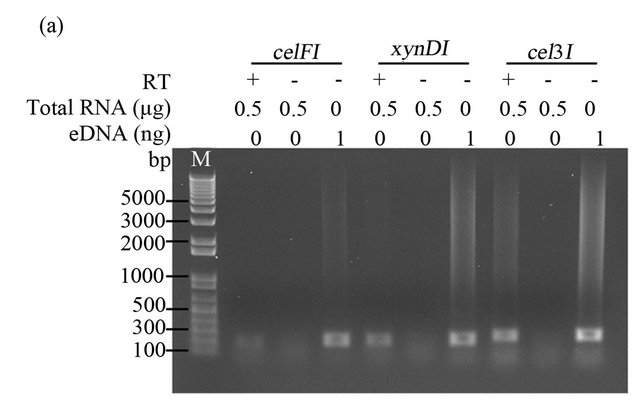
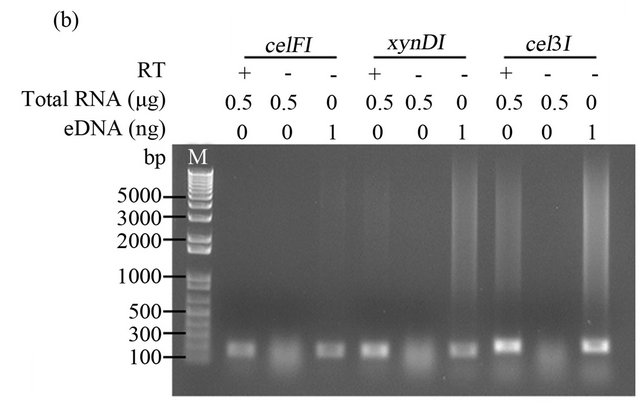
Figure 2. RT-PCR amplification of Fibrobacter succinogenes S85 transcripts. cDNA synthesized with 0.5 µg total RNA extracted from rumen-incubated corn stover (a) and switchgrass (b), with (+) or without (-) 1 µl of reverse transcriptase (RT). eDNA indicates environmental DNA extracted from rumen-incubated corn stover (a) or swich grass (b), M indicates DNA ladder. PCR products were loaded on a 1% agarose TAE gel.
it might be useful to avoid removal of these small molecules. Preservation of small RNAs and RNA fragments seems in particular logical as current sequencing platforms generate extremely large numbers of short reads in the range of 50 - 250 base pairs and non-degraded RNA will be sheared mechanically or computationally during library construction or the sequencing process, respectively. In light of the fact that small RNA fragments might hold insights into microbial processes that are currently poorly understood [27,28], it is our opinion that utilization of a column cleanup step is obsolete when highdepth next generation sequencing platforms are employed for data generation.
The RNA integrity number (RIN) calculated by Agilent’s Bioanalyzer software takes the ratio of the ribosomal bands and the complete electrophoretic trace into consideration and has been proposed as a convenient technique for determining the integrity of total RNA [29]. Despite its convenience it has been suggested to use the RIN and the rRNA ratios rather as approximate values and not as accurate measures as they have not been validated thoroughly for total RNA from bacteria and inconsistent results were obtained from mixed RNA samples [21]. In theory, fully intact RNA is indicated by a RIN of 10 with decreasing RINs indicating lower RNA integrity. We used the RINs to determine the impact of the different extraction methods on the overall integrity of the extracted RNA. RINs determined for our improved method (MB) and the control method (MC) were comparable to each other (Table 1) and to the RINs reported for RNA isolated from rumen content using the method developed recently by Wang and colleagues [21]. This suggests that our improved method, the control method and its slightly modified version used by Wang et al. are producing total RNA of similar quality. The fact that method MB and MC yield RNA of similar quality is further supported by their consistent 23S/16S rRNA ratios. If increased amounts of RNA are not crucial for downstream processing all three methods seem to provide a reliable and reproducible method for the extraction of sufficient intact RNA from rumen-incubated solids. Although it was not possible to detect significant RNA degradation during the purification process, it cannot be excluded that it occurs due to the extremely sensitive nature of RNA.
To determine if our improved method was suitable for the isolation of RNA fragments with a size similar to the size of the fragments that are commonly generated in the library construction process and to the read length that is typical for sequence reads generated on NGS platforms, we designed primers targeting an internal region (ranging between 186 bp and 240 bp) of three CAZyme genes (celF, xynD, and cel3) from the fibrolytic rumen bacterium Fibrobacter succinogenes S85 [19]. The three fragments were detected by RT-PCR, suggesting that our method can be used, similar to the methods reported by Béra-Mailett et al. (2009) and Wang et al. (2011), to isolate ≤200 bp fragments of functional mRNAs. The extracted RNA can subsequently be used to rapidly quantify transcripts of interest using RT-PCR or for metatranscriptomic analysis in which short sequence reads are assembled computationally into scaffolds and full-length genes.
In summary, we developed a simple and cost efficient technique that requires only basic equipment and complements already existing protocols for the extraction of intact RNA from rumen-incubated biomass for subsequent metatranscriptomic analysis and other molecular biology applications. The improved MB method presented here allows the rapid isolation of three times more intact RNA compared to other extraction protocols, which opens new perspectives in the field of rumen metatranscripomics and rumen ecology. Additional methods optimized for the extraction of high-quality RNA from other ruminants and other rumen-incubated biomass will be of great value for future studies aimed at enhancing our understanding of the microbial processes that occur in the rumen ecosystem and that have significant global socio-economical bearings.
5. Acknowledgements
Work performed by LMM was conducted at EMSL, a national scientific user facility sponsored by DOE’s Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory. PNNL is operated by Battelle for the US DOE under contract AC06-76RLO 1830. MH, HP and the work performed in the laboratory of MH were funded through MH’s start-up funding provided by Washington State University. Cannulated cows used in this research were provided by the Department of Animal Sciences, University of Illinois and procedures with the cow were carried out under a protocol approved by the University of Illinois Institutional Animal Care and Use of Animals Committee (IUCAC #06081).
REFERENCES
- P. N. Hobson and C. S. Stewart, “The Rumen Microbial Ecosystem,” 2nd Edition, Blackie Academic & Professional, New York, 1997. doi:10.1007/978-94-009-1453-7
- A. N. Hristov, T. R. Callaway, C. Lee and S. E. Dowd, “Rumen Bacterial, Archaeal, and Fungal Diversity of Dairy Cows in Response to Ingestion of Lauric or Myristic Acid,” Journal of Animal Science, Vol. 90, No. 12, 2012, pp. 4449-4457. doi:10.2527/jas.2011-4624
- T. A. McAllister, H. D. Bae, G. A. Jones and K. J. Cheng, “Microbial Attachment and Feed Digestion in the Rumen,” Journal of Animal Science, Vol. 72, No. 11, 1994, pp. 3004-3018.
- E. M. Rubin, “Genomics of Cellulosic Biofuels,” Nature, Vol. 454, No. 7026, 2008, pp. 841-845.
- D. B. Wilson, “Microbial Diversity of Cellulose Hydrolysis,” Current Opinion in Microbiology, Vol. 14, No. 3, 2011, pp. 259-263. doi:10.1016/j.mib.2011.04.004
- R. Mackelprang, M. P. Waldrop, K. M. DeAngelis, M. M. David, K. L. Chavarria, S. J. Blazewicz, E. M. Rubin and J. K. Jansson, “Metagenomic Analysis of a Permafrost Microbial Community Reveals a Rapid Response to Thaw,” Nature, Vol. 480, No. 7377, 2011, pp. 368-371. doi:10.1038/nature10576
- M. Allgaier, A. Reddy, J. I. Park, N. Ivanova, P. D’Haeseleer, S. Lowry, R. Sapra, T. C. Hazen, B. A. Simmons, J. S. VanderGheynst and P. Hugenholtz, “Targeted Discovery of Glycoside Hydrolases from a Switchgrass—Adapted Compost Community,” PloS One, Vol. 5, No. 1, 2010, pp. 1-9.
- M. Hess, A. Sczyrba, R. Egan, T. W. Kim, H. Chokhawala, G. Schroth, S. Luo, D. S. Clark, F. Chen, T. Zhang, R. I. Mackie, L. A. Pennacchio, S. G. Tringe, A. Visel, T. Woyke, Z. Wang and E. M. Rubin, “Metagenomic Discovery of Biomass-Degrading Genes and Genomes from Cow Rumen,” Science, Vol. 331, No. 6010, 2011, pp. 463- 467.
- P. B. Pope, S. E. Denman, M. Jones, S. G. Tringe, K. Barry, S. A. Malfatti, A. C. McHardy, J. F. Cheng, P. Hugenholtz, C. S. McSweeney and M. Morrison, “Adaptation to Herbivory by the Tammar Wallaby Includes Bacterial and Glycoside Hydrolase Profiles Different from Other Herbivores,” Proceedings of the National Academy of Sciences of the USA, Vol. 107, No. 33, 2010, pp. 14793- 14798. doi:10.1073/pnas.1005297107
- G. Suen, J. J. Scott, F. O. Aylward, S. M. Adams, S. G. Tringe, A. A. Pinto-Tomas, C. E. Foster, M. Pauly, P. J. Weimer, K. W. Barry, L. A. Goodwin, P. Bouffard, L. Li, J. Osterberger, T. T. Harkins, S. C. Slater, T. J. Donohue and C. R. Currie, “An Insect Herbivore Microbiome with High Plant Biomass-Degrading Capacity,” PLoS Genetics, Vol. 6, No. 9, 2010, Article ID: e1001129.
- P. Narasingarao, S. Podell, J. A. Ugalde, C. BrochierArmanet, J. B. Emerson, J. J. Brocks, K. B. Heidelberg, J. F. Banfield and E. E. Allen, “De Novo Metagenomic Assembly Reveals Abundant Novel Major Lineage of Archaea in Hypersaline Microbial Communities,” The ISME Journal, Vol. 6, No. 1, 2012, pp. 81-93. doi:10.1038/ismej.2011.78
- F. Warnecke and M. Hess, “A Perspective: Metatranscriptomics as a Tool for the Discovery of Novel Biocatalysts,” Journal of Biotechnology, Vol. 142, No. 1, 2009, pp. 91- 95. doi:10.1016/j.jbiotec.2009.03.022
- M. Qi, P. Wang, N. O’Toole, P. S. Barboza, E. Ungerfeld, M. B. Leigh, L. B. Selinger, G. Butler, A. Tsang, T. A. McAllister and R. J. Forster, “Snapshot of the Eukaryotic gene Expression in Muskoxen Rumen—A Metatranscriptomic Approach,” PLoS One, Vol. 6, No. 5, 2011, p. e20521.
- K. C. McGrath, S. R. Thomas-Hall, C. T. Cheng, L. Leo, A. Alexa, S. Schmidt and P. M. Schenk, “Isolation and Analysis of mRNA from Environmental Microbial Communities,” Journal of Microbiological Methods, Vol. 75, No. 2, 2008, pp. 172-176.
- J. A. Gilbert, D. Field, Y. Huang, R. Edwards, W. Li, P. Gilna and I. Joint, “Detection of Large Numbers of Novel Sequences in the Metatranscriptomes of Complex Marine Microbial Communities,” PLoS One, Vol. 3, No. 8, 2008, p. e3042. doi:10.1371/journal.pone.0003042
- J. Frias-Lopez, Y. Shi, G. W. Tyson, M. L. Coleman, S. C. Schuster, S. W. Chisholm and E. F. Delong, “Microbial Community Gene Expression in Ocean Surface Waters,” Proceedings of the National Academy of Sciences of the USA, Vol. 105, No. 10, 2008, pp. 3805-3810. doi:10.1073/pnas.0708897105
- R. S. Poretsky, I. Hewson, S. Sun, A. E. Allen, J. P. Zehr and M. A. Moran, “Comparative Day/Night Metatranscriptomic Analysis of Microbial Communities in the North Pacific Subtropical Gyre,” Environmental Microbiology, Vol. 11, No. 6, 2009, pp. 1358-1375. doi:10.1111/j.1462-2920.2008.01863.x
- R. S. Poretsky, S. Gifford, J. Rinta-Kanto, M. Vila-Costa and M. A. Moran, “Analyzing Gene Expression from Marine Microbial Communities Using Environmental Transcriptomics,” Journal of Experimental Medicine, Vol. 18, No. 24, 2009, p. 1086.
- C. Bera-Maillet, P. Mosoni, A. Kwasiborski, F. Suau, Y. Ribot and E. Forano, “Development of a RT-qPCR Method for the Quantification of Fibrobacter Succinogenes S85 Glycoside Hydrolase Transcripts in the Rumen Content of Gnotobiotic and Conventional Sheep,” Journal of Microbiological Methods, Vol. 77, No. 1, 2009, pp. 8-16. doi:10.1016/j.mimet.2008.11.009
- S. Kang, S. E. Denman, M. Morrison, Z. Yu and C. S. McSweeney, “An Efficient RNA Extraction Method for Estimating Gut Microbial Diversity by Polymerase Chain Reaction,” Current Microbiology, Vol. 58, No. 5, 2009, pp. 464-471. doi:10.1007/s00284-008-9345-z
- P. Wang, M. Qi, P. Barboza, M. B. Leigh, E. Ungerfeld, L. B. Selinger, T. A. McAllister and R. J. Forster, “Isolation of High-Quality Total RNA from Rumen Anaerobic Bacteria and Fungi, and Subsequent Detection of Glycoside Hydrolases,” Analytical Biochemistry, Vol. 57, No. 7, 2011, pp. 590-598. doi:10.1139/w11-048
- P. Chomczynski and N. Sacchi, “Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate-Phenol-Chloroform Extraction,” Analytical Biochemistry, Vol. 162, No. 1, 1987, pp. 156-159.
- P. Chomczynski and N. Sacchi, “The Single-Step Method of RNA Isolation by Acid Guanidinium ThiocyanatePhenol-Chloroform Extraction: Twenty-Something Years On,” Nature Protocols, Vol. 1, No. 2, 2006, pp. 581-585. doi:10.1038/nprot.2006.83
- S. Ramos, M. L. Tejido, M. J. Ranilla, M. E. Martinez, C. Saro and M. D. Carro, “Influence of Detachment Procedure and Diet on Recovery of Solid-Associated Bacteria from Sheep Ruminal Digesta and Representativeness of Bacterial Isolates as Assessed by Automated Ribosomal Intergenic Spacer Analysis-Polymerase Chain Reaction,” Journal of Dairy Science, Vol. 92, No. 11, 2009, pp. 5659- 5668. doi:10.3168/jds.2009-2273
- N. Xu, Y. Li, Y. T. Zhao, L. Guo, Y. Y. Fang, J. H. Zhao, X. J. Wang, L. Huang and H. S. Guo, “Identification and Characterization of Small RNAs in the Hyperthermophilic Archaeon Sulfolobus Solfataricus,” PLoS One, Vol. 7, No. 4, 2012, p. e35306. doi:10.1371/journal.pone.0035306
- S. C. Li, W. C. Chan, M. R. Ho, K. W. Tsai, L. Y. Hu, C. H. Lai, C. N. Hsu, P. P. Hwang and W. C. Lin, “Discovery and Characterization of Medaka miRNA Genes by Next Generation Sequencing Platform,” BMC Genomics, Vol. 11, No. S4, 2010, p. S8. doi:10.1186/1471-2164-11-S4-S8
- M. A. Hernandez-Prieto, V. Schon, J. Georg, L. Barreira, J. Varela, W. R. Hess and M. E. Futschik, “Iron Deprivation in Synechocystis: Inference of Pathways, Non-Coding RNAs, and Regulatory Elements from Comprehensive Expression Profiling,” G3 (Bethesda), Vol. 2, No. 12, 2012, pp. 1475-1495.
- Q. Zhou, Z. Wang, J. Zhang, H. Meng and B. Huang, “Genome—Wide Identification and Profiling of MicroRNALike RNAs from Metarhizium Anisopliae during Development,” Fungal Biology, Vol. 116, No. 11, 2012, pp. 1156- 1162. doi:10.1016/j.funbio.2012.09.001
- A. Schroeder, O. Mueller, S. Stocker, R. Salowsky, M. Leiber, M. Gassmann, S. Lightfoot, W. Menzel, M. Granzow and T. Ragg, “The RIN: An RNA Integrity Number for Assigning Integrity Values to RNA Measurements,” BMC Molecular Biology, Vol. 7, No. 3, 2006. doi:10.1186/1471-2199-7-3
NOTES
*Authors’ Contribution: Conceived and designed the experiments: MH, HP, RM. Performed the experiments: MH, HP, LM. Analyzed the data: MH, HP, LMM, DC. Wrote the paper: MH, HP, DC, RM.
#Corresponding author.