S. Jiao et al. / J. Biomedical Science and Engineering 3 (2010) 317-321
Copyright © 2010 SciRes JBiSE
321
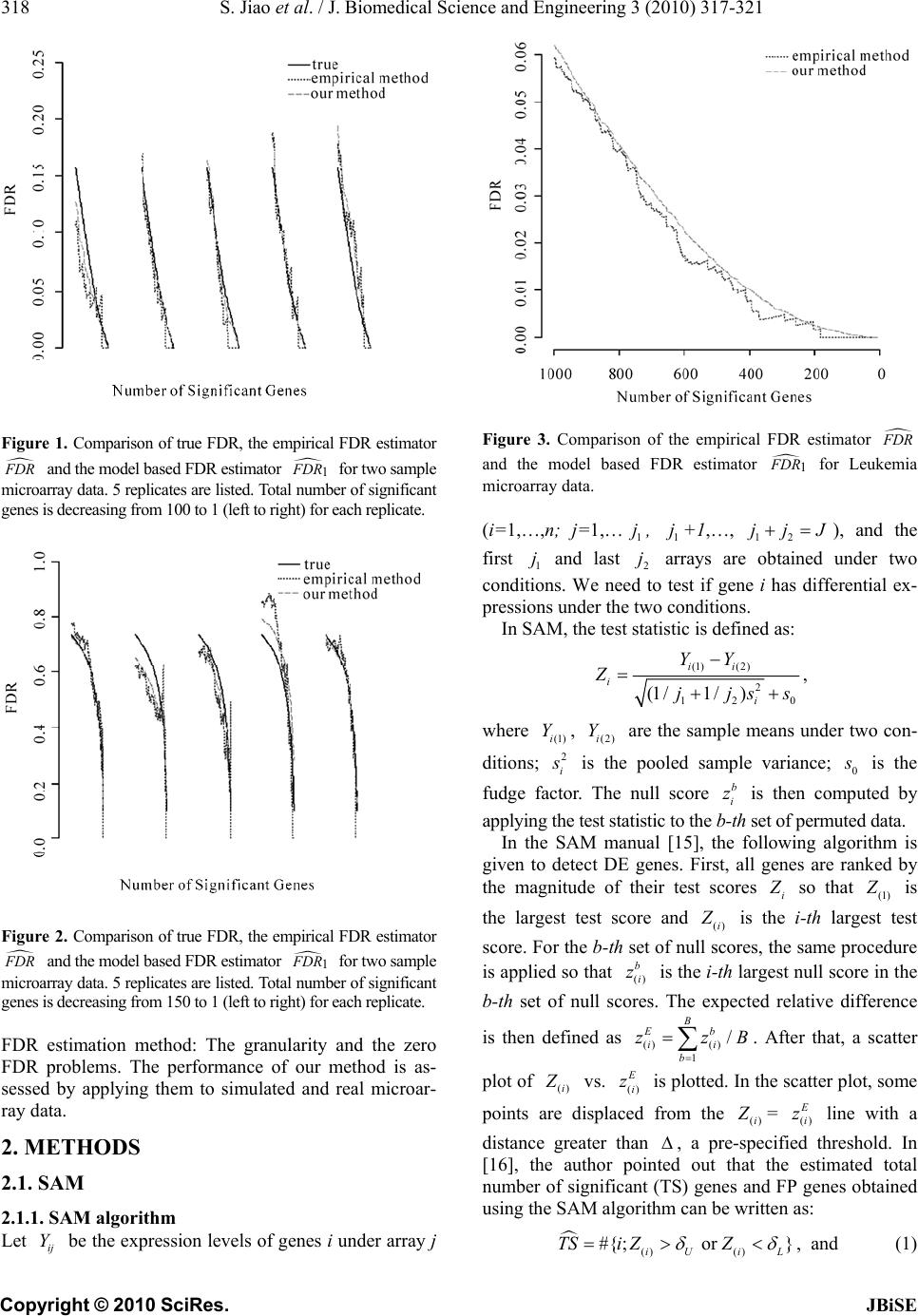
empirical method. The results also show that our esti-
mator provides more stable and accurate estimates of the
FDR. The advantage of our method is more evident in
the case when DE genes are not well separated with EE
genes and the variances of expression levels for every
gene are different. This is due to the fact that the permu-
tation FDR estimator is more easily affected by the sam-
pling variability.
REFERENCES
[1] Tusher, V.G., Tibshirani, R. and Chu, G. (2001) Signifi-
cant analysis of microarrays applied to the ionizing ra-
diation response. PNAS, 98, 5116-5121.
[2] Long, A. D., Mangalam, H. J., Chan, B. Y. P., Tolleri, L.,
Hatfield, W. G. and Baldi, P. (2001) Improved statistical
inference from DNA microarray data using analysis of
variance and a Bayesian statistical frame work, 276,
19937-19944.
[3] Kerr, M.K., Martin, M. and Churchill, G. (2000) Analysis
of variance for gene expression microarray data, Journal
of Computational Biology, 7, 819-837.
[4] Thomas, J.G., Olson, J.M., Tapscott, S.J. and Zhao, L. P.
(2001) An efficient and robust statistical modelling ap-
proach to discover differentially expressed genes using
genomic expression profiles. Genome Research, 11,
1227-1236.
[5] Baldi, P. L. and Long, A. D. (2001) A Bayesian frame-
work for the analysis of microarray expression data:
Regularized t-test and statistical inference of gene
changes. Bioinformatics, 17, 509-519.
[6] Kendziorski, C. M., Newton, M. A., Lan, H. And Gould,
M. N. (2003) On parametric empirical Bayes methods for
comparing multiple groups using replicated gene expres-
sion profiles. Statistics in Medi ci ne, 22, 819-837.
[7] Newton, M., Noueiry, A., Ahlquist, P., Sarkar, D. (2004)
Detecting differential gene expression with a semi-
parametric hierarchical mixture method. Biostatistics,
5(2), 155-176.
[8] Smyth, G. K. (2004) Linear models and empirical Bayes
methods for assessing differential expression in microar-
ray experiments. Statistical Applications in Genetics and
Molecular Bi ology, 3(1).
[9] Efron, B., Tibshirani R., Storey, J. D., Tusher, V. (2001)
Empirical Bayes analysis of a microarray experiment.,
Journal of the American Statistical Association, 96,
1151-1160.
[10] Efron, B., Tibshirani, R., Gross, V. and Chu, G. (2000)
Microarrays and their use in a comparative experiment,
Technical report, Statistics Department, Standard Uni-
versity.
[11] Dudoit, S., Yang, H. Y., Callow, J. M. and Speed, P. T.
(2002) Statistical methods for identifying differentially
expressed genes in replicated cDNA microarray experi-
ments. Statistica Sinica, 111-139.
[12] Troyanskaya, O. G., Garber, M. E., Brown, P. O., Bot-
stein, D. and Altman, R. B. (2002) Nonparametric meth-
ods for identifying differentially expressed genes in mi-
croarray data. Bioinformatics, 18, 1454-1461.
[13] Broberg, P. (2003) Ranking genes with respect to differ-
ential expression , Genome Biology, 4, R41.
[14] Pan, W., Lin, J. and Le, C. (2003) A mixture model ap-
proach to detecting differentially expressed genes with
microarray data. Functional & integrative genomics, 3,
117-124.
[15] Chu, G., Narasimhan, B., Tibshirani, R. and Tusher, V.
SAM “significance analysis” of microarrays-users guide and
technical document, http://www-stat.stanford.edu/~tibs/
SAM/sam.pdf.
[16] Zhang, S. (2007) A comprehensive evaluation of SAM,
the SAM R-package and a simple modification to im-
prove its performance, BMC Bio inform atics , 8, 230.
[17] Benjamini, Y. and Hochberg, Y. (1995) Controlling the
false discovery rate: a practical and powerful approach to
multiple testing. Journal of the Royal Statistical Society,
57, 289-300.
[18] Storey, J.D. and Tibshirani, R. (2003) Statistical signifi-
cance for genomewide studies. PNAS, 100, 9440-9445.
[19] Liu, C. and Rubin, D. (1995) ML estimation of the t dis-
tribution using EM and its extensions ECM and ECME.
Statistica Sinica, 5, 19-39.
[20] Jiao, S. and Zhang, S. (2008) The t-mixture model ap-
proach for detecting differentially expressed genes in
microarrays. Functional & Integrative Genomics, 8,
181-186.
[21] Golub, T. R., Slonim, D. K., Tamayo, P., Huard, C.,
Gaasenbeek, M., Mesirov, J. P., Coller, H., Loh, M. L.,
Downing, J.R. and Caligiuri, M. (1999) Molecular clas-
sification of cancer: class discovery and class prediction
by gene expression monitoring. Science, 285, 531-537.