Journal of Cancer Therapy
Vol.2 No.4(2011), Article ID:7797,7 pages DOI:10.4236/jct.2011.24073
Current Perspectives on Sunitinib Targeted Therapy for Tumors
![]()
Faculty of Science, Damanhour University, Damanhour, Egypt.
Email: k.kamel2002@yahoo.com
Received February 27th, 2011; revised July 20th, 2011; accepted July 31st, 2011.
Keywords: Platelet-Derived Growth Factor (PDGF), Cytochrome P450 Enzyme (CYP3A4), Dose-Limiting Toxicities (DLTs), Hepatocyte Growth Factor (HGF), Tyrosine Kinases (TKs)
ABSTRACT
This review highlights therapeutic agents from recent cancer therapeutic trials showing the greatest potential for further clinical use for sunitinib in the near future. In fact, sunitinib is one of multi-tyrosine kinase inhibitors; tyrosine kinases are enzymes, which transfer phosphate groups from ATP to the hydroxyl group of tyrosine residues on signal transduction molecules. Phosphorylation of signal transduction molecules, in turn, induces dramatic changes in tumor growth, including activation of angiogenesis and DNA synthesis. Therefore, sustain efforts have been directed for developing inhibitors for angiogenesis, which is the marginal process for tumor growth and development through targeting TKs. Almost if not all angiogenesis inhibitors target the vascular endothelial growth factor (VEGF) signaling pathway.
1. Mechanism of Action
The accumulation of hypoxia inducible factor induces the expression of hypoxia-inducible genes, e.g. VEGF and platelet-derived growth factor (PDGF) [1-3] . Angiogenesis inhibitors, such as the monoclonal antibody bevacizumab (Avastin, Genentech/Roche) and two kinase inhibitors sunitinib (SU11248, Sutent, Pfizer) and sorafenib (BAY43-9006, Nexavar, Bayer) [4] target the vascular endothelial growth factor (VEGF) signaling pathway. Upon binding of these growth factors to their respective tyrosine kinase receptors, the cell migration, proliferation and survival take place [5]. Since, it acts as a multi-targeted tyrosine kinase inhibitor, sunitinib has widely recommended in the clinic (Figure 1).
Due to its stringent ability to inhibit the activity of VEGF receptors 1, 2, and 3; PDGF receptors a and b; KIT; Fms-like TK-3 (FLT3); colony-stimulating factor receptor type 1; and neurotrophic factor receptor [6,7] . Since, the small molecule receptor tyrosine kinase inhibitors (RTKs), including VEGFR, PDGFR and KIT, are expressed on many if not all tumors [8,9] , sunitinib may have potential for use in several tumors types.
2. Doses of Sunitinib
A recent study has evaluated sunitinib effects on 637 tumor cell lines, only two tumor cell lines among others, were highly sensitive to sunitinib (at 1 µM) due to activated PDGFRα signaling, implicating resistance to the 635 others [10]. However, almost all studies used sunitinib in vitro at higher concentrations up to 10 µM [6]. Since, based on animal data and its high volume of distribution, target tissues are expected to have higher sunitinib concentrations than plasma [11]. In xenografted renal cell cancers in mice when treated with a non-toxic dose of sunitinib, the intratumoral concentrations were found to be in the range of 10 µM [4] . In human studies, sunitinib could induce apoptosis in renal cell cancer cell lines in a concentration-dependent way from 1.25 to 10 µM [12]. Of note the incidence of apoptosis in cancer cells as a result of exposed sunitinib is correlated with reduced activation of STAT3, which has adverse effects on chemotherapy sensitivity even in the case of solid tumors [13].
3. Sunitinib Metabolism and Pharmacokinetics
Sunitinib malate is described chemically as Butanedioic acid, hydroxy-, (2S)-, compound with N-[2-(diethylamino)ethyl]-5-[(Z)-(5-fluoro-1,2-dihydro-2-oxo-3H-indol-3- ylidine)methyl]-2,4-dimethyl-1H-pyrrole-3-carboxamide
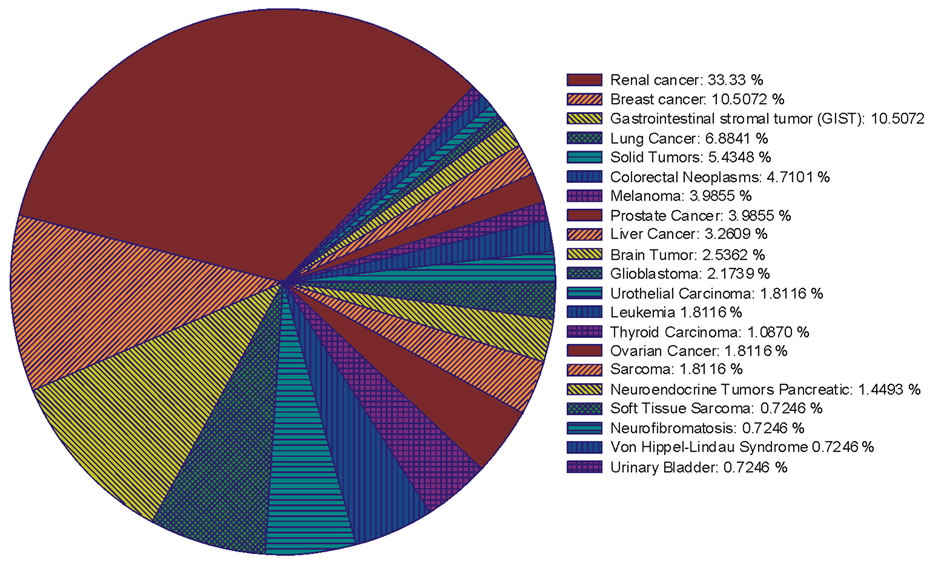
Figure 1. The percentage ratio of the clinical trials that implicated sunitinib alone or in combination with other chemotherapy drugs for treating different cancer types.
(1:1). The molecular formula is C22H27FN4O2·C4H6O5 and the molecular weight is 532.6 daltons. To date, it is not completely clear which patient characteristics render an individual patient at risk for sunitinib-induced toxicity [14] . Therefore, sunitinib is used as palliative therapy with no standard therapeutic options available after failure of the therapy (Figure 2).
Following oral administration, sunitinib is well absorbed from the gastrointestinal tract with maximum plasma concentrations observed between 6 and 12 h after

Figure 2. The percentage ratio for the application of sunitinib in cancer therapy in different clinical trails.
administration. Bioavailability is not affected by food intake [15] and no significant changes in pharmacokinetics are observed with repeat versus single dosing [16]. Sunitinib is metabolized primarily in the liver by the cytochrome P450 enzyme CYP3A4 [17] . Both parent compound and an active N-desethyl metabolite (SU12662) [16] have similar biochemical activity and potency. The primary metabolite is further metabolized by CYP3A4 to its secondary inactive metabolite [18] . The primary active metabolite comprises 23% to 37% of the total exposure. In a human mass balance study of 14C-labeled sunitinib, 61% of the dose was eliminated in feces, with renal elimination accounting for 16% of the administered dose [17] . Total oral clearance (CL/F) ranged from 34 to 62 L/h with an interpatient variability of 40%. The terminal half-lives of sunitinib and its active metabolite are 40 h to 60 h and 80 h to 110 h, respectively. Steady-state conditions of sunitinib and its active metabolite are reached in 2 weeks. Polymorphisms in specific genes CYP1A1, ABCB1, ABCG2, NR1I3, VEGFR2, and FLT3 encoding for metabolizing enzymes, efflux transporters, and drug targets are associated with sunitinibrelated toxicities ADDIN EN.CITE ADDIN EN.CITE.DATA [14] . The pharmacokinetics of sunitinib and sunitinib malate have been evaluated in 135 healthy volunteers and in 266 patients with solid tumors [17,19] . The study have shown that following a single oral dose, peak plasma sunitinib concentrations occur between 6 and 12 hours post-dose [16,20] . In addition, sunitinib and SU12662 have previously been shown to display linear pharmacokinetics and have prolonged half-lives of 40 and 80 hours, respectively [21].
4. Sunitinib-Drug-Interactions
4.1. Coadministration of Sunitinib with Strong Cytochrome P450 Enzyme (CYP3A4) Inhibitors
The coadministration of sunitinib with strong inhibitors of the CYP3A4 family (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, voriconizole) may increase sunitinib concentrations. For instance, coadministration of sunitinib with strong CYP3A4 inhibitor, ketoconazole, resulted in 49% and 51% increases in the combined AUC of sunitinib and its active metabolite. Therefore, the administration of sunitinib along with the strong CYP3A4 inhibitors should be avoided to prevent an increased risk of toxicity due to increased drug exposure. In the contrary, concurrent administration of SUTENT with the strong CYP3A4 inducer, rifampin, resulted in a 23% and 46% reduction in the combined AUC of sunitinib and its active metabolite, respectively, after a single dose of SUTENT in healthy volunteers.
4.2. Coadministration of Sunitinib with Strong Cytochrome P450 Enzyme (CYP3A4) Inducers
Coadministration of SUTENT with inducers of the CYP3A4 family (e.g., dexamethasone, phenytoin, carbamazepine, rifampin, rifabutin, rifapentin, phenobarbital, St. John’s Wort) may decrease sunitinib concentrations. A dose increase for SUTENT should be considered when it must be co-administered with CYP3A4 inducers. There is no specific antidote for overdosage with SUTENT. A few cases of accidental overdose have been reported; these cases were associated with adverse reactions consistent with the known safety profile of SUTENT, or without adverse reactions. A case of intentional overdose involving the ingestion of 1500 mg of SUTENT in an attempted suicide was reported without adverse reaction. In non-clinical studies mortality was observed following as few as 5 daily doses of 500 mg/kg (3000 mg/m2) in rats. At this dose, signs of toxicity included impaired muscle coordination, head shakes, hypoactivity, ocular discharge, piloerection and gastrointestinal distress. Mortality and similar signs of toxicity were observed at lower doses when administered for longer durations. In a study to determine the effect of GJ, which is a potent intestinal cytochrome P450 (CYP)3A4 inhibitor, on steady state sunitinib pharmacokinetics (PK) [22]. In this study concomitant use of grapefruit juice (GJ) and sunitinib resulted in an 11% increase of the relative bioavailability of sunitinib (P < 0.05). The effect of GJ on CYP3A4 activity was confirmed by an increase of ~50% of mean midazolam exposure (AUC0-24 h) from 122.1 to 182.0 ng·h/mL (P = 0.034).
5. Clinical Trials of Sunitinib
Sunitinib, in phase II and III trials was associated with durable clinical benefit in nearly 25% of 147 patients with advanced GIST resistant/intolerant to imatinib [23]. Furthermore, sunitinib is therapeutically effective for both on imatinib-resistant GIST and advanced renal cell carcinoma with modest tolerability [24]. Interestingly, a recent case-report confirmed clinical benefit of sunitinib in a chemo-refractory adrenocortical carcinoma patient, suggesting that it may be active in a highly Pgp overexpressing tumor type [25] . Furthermore, depending on the resistance or sensitivity of specific vessels, which may occur both in one tumor, sunitinib, affecting its own delivery to remaining tumor tissue in a positive or negative way [26]. In an attempt to evaluate the possible interaction of sunitinib with P-glycoprotein (P-gp, ABCB1), multidrug resistance protein 1 (MRP1, ABCC1), breast cancer resistance protein (BCRP, ABCG2) and lungresistance protein (LRP) in vitro. Dai CL et al. (2009) showed that sunitinib completely reverse drug resistance mediated by ABCG2 at a non-toxic concentration of 2.5 µM and has no significant reversal effect on ABCB1- ABCC1- and LRP-mediated drug resistance [27] . Although a small synergetic effect was observed in combining sunitinib and conventional chemotherapeutic agents in ABCB1 overexpressing MCF-7/adr and parental sensitive MCF-7 cells, ABCC1 overexpressing CA120 and parental sensitive KB-3-1 cells. However, sunitinib neither affect the expression of ABCG2 at mRNA or protein levels nor block the phosphorylation of Akt and Erk1/2 in ABCG2-overexpressing or parental sensitive cells.
6. Dosing Schedules of Sunitinib and Its Combination with Different Chemotherapy Regimens
In an open-label, phase I, dose-escalation study assessed the maximum-tolerated dose (MTD), safety, pharmacokinetics, and antitumor activity of sunitinib in combination with capecitabine in patients with advanced solid tumors [28]. In this study, sunitinib (25, 37.5, or 50 mg) was administered orally once daily on three dosing schedules: 4 weeks on treatment, 2 weeks off treatment (Schedule 4/2); 2 weeks on treatment, 1 week off treatment (Schedule 2/1); and continuous daily dosing (CDD schedule). Capecitabine (825, 1,000, or 1250 mg/m (2)) was administered orally twice daily on days 1 to 14 every 3 weeks for all patients. Sunitinib and capecitabine doses were escalated in serial patient cohorts. The MTD for Schedule 4/2 and the CDD schedule was sunitinib 37.5 mg/d plus capecitabine 1000 mg/m (2) twice per day; the MTD for Schedule 2/1 was sunitinib 50 mg/d plus capecitabine 1000 mg/m (2) twice per day. The study concluded that, in patients with advanced solid tumors, the combination of sunitinib and capecitabine resulted in an acceptable safety profile. Since, there were no clinically significant pharmacokinetic drug-drug interactions. In contrast, in another phase I study [29] , sunitinib was initially administered once daily at 37.5 mg per day on days 1 - 14 of a 21-day cycle, in which irinotecan 250 mg/m (2) was given on day 1. In a second cohort, the sunitinib dose was reduced to 25 mg per day. In the sunitinib 37.5 mg per day cohort, 3 out of 10 evaluable patients had objective responses, but dose-limiting toxicities (DLTs) of neutropenia, pneumococcal sepsis, and fatigue were observed. Therefore, in this study, the maximum tolerated dose was defined as sunitinib 25 mg per day (days 1 - 14) with irinotecan 250 mg/m (2).
7. Vitamins
Vitamins C and E supplementation have been used an attempt to decrease the harmful effects of commonly used anticancer drugs in breast-cancer patients [30]. Coadministration of VCE restored antioxidant status, lowered by the presence of breast-cancer and chemotherapy. DNA damage was also reduced by vitamins C and E. The results suggest that vitamins C and E should be useful in protecting against chemotherapy-related side-effects and a randomized control trial to evaluate the effectiveness of vitamins C and E in breast-cancer patients using clinical outcomes would be appropriate. On the other hand tannic acid, a PARG/PARP inhibitor and an antioxidant, on doxorubicin-induced cardiotoxicity in H9c2 embryonic rat heart myoblasts and its anti-cancer activity in MDAMB-231 human breast cancer cells as well as in DMBAinduced mammary tumor animals [31]. The results show that tannic acid prevents activation of PARP-1, reduces Bax and increases Bcl-2 expression in H9c2 cells, thus, preventing doxorubicin-induced cell death. Further, it reduces the cell viability of MDA-MB-231 breast cancer cells, increases p53 expression in mammary tumors and shows maximum tumor volume reduction, suggesting that tannic acid potentiates the anti-cancer activity of doxorubicin. To the best of our knowledge, this is the first report which shows that tannic acid ameliorates doxorubicin-induced cardiotoxicity and potentiates its anti-cancer activity both in vitro (H9c2 and MDA-MB- 231 cells) as well as in in vivo model of DMBA-induced mammary tumor animals. However, there is no precise information are available about the benefit of the combination of sunitinib and vitamins.
8. Strategies of Cancer Cell Resistance for Drugs
Despite the tremendous success in the clinical application of sunitinib for preventing tumor progression, in some patients, treatment with an angiogenesis inhibitor results in an initial response, followed by tumor progression (acquired resistance). For instance, in patients with advanced clear cell renal cell carcinoma (ccRCC), most patients develop sunitinib resistance and progressive disease after about 1 year of treatment [32]. That is actually due to the vascular resistance to the anti-angiogenic effect of sunitinib, through activation of alternative proangiogenic pathways [33]. Therefore, the previous study revealed this resistance to the increased secretion of interleukin-8 (IL-8) from tumors into the plasma. Thus, this study held that IL-8 is an important contributor to sunitinib resistance in ccRCC and a candidate therapeutic target to reverse acquired or intrinsic resistance to sunitinib in this malignancy.
Several potential mechanisms of resistance to anti-angiogenic drugs like sunitinib have been proposed, however, their are two main types of resistance can be distinguished: first, resistance of the tumor vasculature to the inhibition of VEGF and PDGF signaling (vascular resistance); secondly, resistance of cancer cells to the hypoxic and nutrient-depleted microenvironment induced by antiangiogenic effects (hypoxia resistance—resistance to the effector mechanism of anti-angiogenic treatment) [34]. In another vivo study, analysis of tumor protein lysates indicated a greater concentration of hepatocyte growth factor (HGF) in resistant tumors compared with sensitive ones. Furthermore, systemic injection of HGF in the sensitive tumor models conferred resistance to sunitinib through maintaining of tumor angiogenesis [35]. A xenograft study have been emphasized that reversible epithelial to mesenchymal transition may be associated with acquired tumor resistance to TKIs in patients with clear cell renal carcinoma [36].
9. Drug Efflux
P-glycoprotein (P-gp) is a 170-kDa glycosylated transmembrane efflux pump, which was first characterized as the ATP-dependent transporter responsible for efflux of chemotherapeutic agents from multidrug resistant cancer cells [37] . Under normal physiological conditions P-gp is widely expressed in many tissues, such as the membrane of endothelial cells in the intestine [38,39] , liver [38], placenta [40], blood-brain barrier [41]. Hence, P-gp may play a significant role in drug absorption, disposition, and excretion, as well as in drug-drug and drug-food interactions [42,43] . Additional ATP-binding cassette efflux membrane transporters known to play a role in drug pharmacokinetics are the 190-kDa multidrug resistantassociated protein 2 (MRP2) and the breast cancer resistance protein (BCRP) [44]. P-gp may play a significant role in drug absorption, disposition, and excretion, as well as in drug-drug and drug-food interactions, while other H2RIs, i.e., ranitidine, famotidine, and cimetidine, were reported to be P-gp substrates [37] . As mentioned above, in an attempt to evaluate the possible interaction of sunitinib with P-glycoprotein (P-gp, ABCB1), multidrug resistance protein 1 (MRP1, ABCC1), breast cancer resistance protein (BCRP, ABCG2) and lung-resistance protein (LRP) in vitro. C. L. Dai et al. (2009) showed that sunitinib completely reverse drug resistance mediated by ABCG2 at a non-toxic concentration of 2.5 µM and has no significant reversal effect on ABCB1- ABCC1- and LRP-mediated drug resistance ADDIN EN.CITE ADDIN EN.CITE.DATA [27] .
10. Future Directions
Further studies in vitro displaying both cytokines and growth factors mediated forms of multidrug resistance will help us to shed light on the mechanisms of resistance and will guide clinicians to offer the optimal sequence/ combination of targeted agents to patients with advanced tumors. That may also provide a rational basis for the use of chemotherapeutic agents early in the development of this induced impairment. Furthermore, the combination of sunitinib with molecular carriers and or with vitamins may deliver the drug to the particular organ and cells, which could allow the optimization of its use.
11. Acknowledgements
Data for Sunitinib were obtained from http://www.clinicaltrial.gov/ct2/results?term=sunitinib, and analyzed by using SigmaPlot 10 software (Systat Software Inc.).
REFERENCES
- M. E. Cockman, N. Masson, D. R. Mole, P. Jaakkola, G. W. Chang, S. C. Clifford, E. R. Maher, C. W. Pugh, P. J. Ratcliffe and P. H. Maxwell, “Hypoxia Inducible Factor-Alpha Binding and Ubiquitylation by the Von Hippel-Lindau Tumor Suppressor Protein,” Journal of Biological Chemistry, Vol. 275, No. 33, 2000, pp. 25733- 25741. doi:10.1074/jbc.M002740200
- D. W. Kim, Y. S. Jo, H. S. Jung, H. K. Chung, J. H. Song, K. C. Park, S. H. Park, J. H. Hwang, S. Y. Rha, G. R. Kweon, S. J. Lee, K. W. Jo and M. Shong, “An Orally Administered Multitarget Tyrosine Kinase Inhibitor, SU11248, Is a Novel Potent Inhibitor of Thyroid Oncogenic RET/Papillary Thyroid Cancer Kinases,” Journal of Clinical Endocrinology & Metabolism, Vol. 91, No. 10, 2006, pp. 4070-4076. doi:10.1210/jc.2005-2845
- W. Y. Kim and W. G. Kaelin, “Role of VHL Gene Mutation in Human Cancer,” Journal of Clinical Oncology, Vol. 22, No, 24, 2004, pp. 4991-5004. doi:10.1200/JCO.2004.05.061
- K. J. Gotink and H. M. W. Verheul, “Anti-Angiogenic Tyrosine Kinase Inhibitors: What Is Their Mechanism of Action?” Angiogenesis, Vol. 13, No. 1, 2010, pp. 1-14. doi:10.1007/s10456-009-9160-6
- R. H. Gunby, E. Sala, C. J. Tartari, M. Puttini, C. Gambacorti-Passerini and L. Mologni, “Oncogenic Fusion Tyrosine Kinases as Molecular Targets for Anti-Cancer Therapy,” Anti-Cancer Agents in Medicinal Chemistry, Vol. 7, No. 6, 2007, pp. 594-611. doi:10.2174/187152007784111340
- T. J. Abrams, L. J. Murray, E. Pesenti, V. W. Holway, T. Colombo, L. B. Lee, J. M. Cherrington and N. K. Pryer, “Preclinical Evaluation of the Tyrosine Kinase Inhibitor SU11248 as a Single Agent and in Combination with “Standard of Care” Therapeutic Agents for the Treatment of Breast Cancer,” Molecular Cancer Therapeutics, Vol. 2, No. 10, 2003, pp. 1011-1021.
- D. W. Kim, Y. S. Jo, H. S. Jung, H. K. Chung, J. H. Song, K. C. Park, S. H. Park, J. H. Hwang, S. Y. Rha, G. R. Kweon, S. J. Lee, K. W. Jo and M. Shong, “An Orally Administered Multitarget Tyrosine Kinase Inhibitor, SU- 11248, Is a Novel Potent Inhibitor of Thyroid Oncogenic RET/Papillary Thyroid Cancer Kinases,” Clinical Endocrinology & Metabolism, Vol. 91, No. 10, 2006, pp. 4070-4076. doi:10.1210/jc.2005-2845
- D. B. Mendel, A. D. Laird, X. Xin, S. G. Louie, J. G. Christensen, G. Li, R. E. Schreck, T. J. Abrams, T. J. Ngai, L. B. Lee, L. J. Murray, J. Carver, E. Chan, K. G. Moss, J. O. Haznedar, J. Sukbuntherng, R. A. Blake, L. Sun, C. Tang, T. Miller, S. Shirazian, G. McMahon and J. M. Cherrington, “In Vivo Antitumor Activity of SU11248, a Novel Tyrosine Kinase Inhibitor Targeting Vascular Endothelial Growth Factor and Platelet-Derived Growth Factor Receptors: Determination of a Pharmacokinetic/ Pharmacodynamic Relationship,” Clinical Cancer Research, Vol. 9, No.1, 2003, pp. 327-337.
- D. J. Hicklin and L. M. Ellis, “Role of the Vascular Endothelial Growth Factor Pathway in Tumor Growth and Angiogenesis,” Journal of Clinical Oncology, Vol. 23, No. 5, 2005, pp. 1011-1027.
- U. McDermott, R. Y. Ames, A. J. Iafrate, S. Maheswaran, H. Stubbs, P. Greninger, K. McCutcheon, R. Milano, S. Y. Rha, A. Tam, D. Y. Lee, L. Lucien, B. W. Brannigan, L. E. Ulkus, X. J. Ma, M. G. Erlander, D. A. Haber, S. V. Sharma and J. Settleman, “Ligand-Dependent PlateletDerived Growth Factor Receptor (PDGFR)-Alpha Activation Sensitizes Rare Lung Cancer and Sarcoma Cells to PDGFR Kinase Inhibitors,” Cancer Research, Vol. 69, No. 9, 2009, pp. 3937-3946. doi:10.1158/0008-5472.CAN-08-4327
- J. O. Haznedar, S. Patyna, C. L. Bello, G. W. Peng, W. Speed, X. Yu, Q. Zhang, J. Sukbuntherng, D. J. Sweeny, L. Antonian and E. Y. Wu, “Singleand Multiple-Dose Disposition Kinetics of Sunitinib Malate, a Multitargeted Receptor Tyrosine Kinase Inhibitor: Comparative Plasma Kinetics in Non-Clinical Species,” Cancer Chemotherapy & Pharmacology, Vol. 64, No. 4, 2009, pp. 691-706. doi:10.1007/s00280-008-0917-1
- H. Xin, C. Zhang, A. Herrmann, Y. Du, R. Figlin and H. Yu, “Sunitinib Inhibition of Stat3 Induces Renal Cell Carcinoma Tumor Cell Apoptosis and Reduces Immunosuppressive Cells,” Cancer Research, Vol. 69, No. 6, 2009, pp. 2506-2513. doi:10.1158/0008-5472.CAN-08-4323
- B. Barré, A. Vigneron, N. Perkins, I. B. Roninson, E. Gamelin and O. Coqueret, “The STAT3 Oncogene as a Predictive Marker of Drug Resistance,” Trends in Molecular Medicine, Vol. 13, No. 1, 2007, pp. 4-11. doi:10.1016/j.molmed.2006.11.001
- N. P. van Erp, K. Eechoute, A. A. van der Veldt, J. B. Haanen, A. K. Reyners, R. H. Mathijssen, E. Boven, T. van der Straaten, R. F. Baak-Pablo, J. A. Wessels, H. J. Guchelaar and H. Gelderblom, “Pharmacogenetic Pathway Analysis for Determination of Sunitinib-Induced Toxicity,” Journal of Clinical Oncology, Vol. 27, No. 26, 2009, pp. 4406-4412. doi:10.1200/JCO.2008.21.7679
- C. L. Bello, L. Sherman, J. Zhou, L. Verkh, J. Smeraglia, J. Mount and K. J. Klamerus, “Effect of Food on the Pharmacokinetics of Sunitinib Malate (SU11248), a Multi-targeted Receptor Tyrosine Kinase Inhibitor: Results from a Phase I Study in Healthy Subjects,” Anticancer Drugs, Vol. 17, No. 3, 2006, pp. 353-358. doi:10.1097/00001813-200603000-00015
- SUTENT (Sunitinib Malate) Prescribing Information, New York, Pfizer Labs.
- V. L. Goodman, E. P. Rock, R. Dagher, R. P. Ramchandani, S. Abraham, J. V. Gobburu, B. P. Booth, S. L. Verbois, D. E. Morse, C. Y. Liang, N. Chidambaram, J. X. Jiang, S. Tang, K. Mahjoob, R. Justice and R. Pazdur, “Approval Summary: Sunitinib for the Treatment of Imatinib Refractory or Intolerant Gastrointestinal Stromal Tumors and Advanced Renal Cell Carcinoma,” Clinical Cancer Research, Vol. 13, No. 5, 2007, pp. 1367-1373. doi:10.1158/1078-0432.CCR-06-2328
- V. R. Adams and M. Leggas, “Sunitinib Malate for the Treatment of Metastatic Renal Cell Carcinoma and Gastrointestinal Stromal Tumors,” Clinical Therapeutics, Vol. 29, No. 7, 2007, pp. 1338-1353. doi:10.1016/j.clinthera.2007.07.022
- Pfizer Labs, Division of Pfizer Inc New York, 2010.
- B. E. Houk, C. L. Bello, D. Kang and M. Amantea, “A Population Pharmacokinetic Meta-Analysis of Sunitinib Malate (SU11248) and Its Primary Metabolite (SU12662) in Healthy Volunteers and Oncology Patients,” Clinical Cancer Research, Vol. 15, No. 7, 2009, pp. 2497-2506. doi:10.1158/1078-0432.CCR-08-1893
- K. M. Sakamoto, “Su-11248 Sugen,” Current Opinion in Investigational Drugs, Vol. 5, No. 12, 2004, pp. 1329- 1339.
- N. P. van Erp, S. D. Baker, A. S. Zandvliet, B. A. Ploeger, M. den Hollander, Z. Chen, J. den Hartigh, J. M. Konig-Quartel, H. J. Guchelaar and H. Gelderblom, “Marginal Increase of Sunitinib Exposure by Grapefruit Juice,” Cancer Chemotherapy and Pharmacology, Vol. 67, No. 3, 2010, pp. 695-703. doi:10.1007/s00280-010-1367-0
- A. Italiano and B. Bui, “Gastrointestinal Stromal Tumors: Molecular Aspects and Therapeutic Implications,” BullCancer, Vol. 95, No. 1, 2008, pp. 107-116.
- T. Nishida, “Imatinib. Sunitinib,” Gan To Kagaku Ryoho, Vol. 34, 2007, pp. 1196-1200.
- J. O. Lee, K. W. Lee, C. J. Kim, Y. J. Kim, H. E. Lee, H. Kim, J. H. Kim, S. M. Bang, J. S. Kim and J. S. Lee, “Metastatic Adrenocortical Carcinoma Treated with Sunitinib: A Case Report” Japanese Journal of Clinical Oncology, Vol. 39,No. 8, 2009, pp. 183-185.
- M. Palmowski, J. Huppert, P. Hauff, M. Reinhardt, K. Schreiner, M. A. Socher, P. Hallscheidt, G. Kauffmann, W. Semmler and F. Kiessling, “Vessel Fractions in Tumor Xenografts Depicted by Flowor Contrast-Sensitive Three-Dimensional High-Frequency Doppler Ultrasound Respond Differently to Antiangiogenic Treatment,” Cancer Research, Vol. 68, No. 17, 2008, pp. 7042-7049. doi:10.1158/0008-5472.CAN-08-0285
- C. L. Dai, Y. J. Liang, Y. S. Wang, A. K. Tiwari, Y. Y. Yan, F. Wang, Z. S. Chen, X. Z. Tong and L. W. Fu, “Sensitization of ABCG2-Overexpressing Cells to Conventional Chemotherapeutic Agent by Sunitinib Was Associated with Inhibiting the Function of ABCG2,” Cancer Letters, Vol. 279, No. 1, 2009, pp. 74-83. doi:10.1016/j.canlet.2009.01.027
- C. J. Sweeney, E. G. Chiorean, C. F. Verschraegen, F. C. Lee, S. Jones, M. Royce, L. Tye, K. F. Liau, A. Bello, R. Chao and H. A. Burris, “A Phase I Study of Sunitinib Plus Capecitabine in Patients with Advanced Solid Tumors,” Journal of Clinical Oncology, Vol. 28, No. 28, 2010, pp. 4513-4520. doi:10.1200/JCO.2009.26.9696
- E. Boven, C. Massard, J. P. Armand, C. Tillier, V. Hartog, N. M. Brega, A. M. Countouriotis, A. Ruiz-Garcia and J. C. Soria, “A Phase I, Dose-Finding Study of Sunitinib In Combination with Irinotecan in Patients with Advanced Solid Tumours,” British Journal of Cancer, Vol. 103, No. 7, 2010, pp. 993-1000. doi:10.1038/sj.bjc.6605852
- N. Suhail, N. Bilal, H. Y. Khan, S. Hasan, S. Sharma, F. Khan, T. Mansoor and N. Banu, “Effect of Vitamins C and E on Antioxidant Status of Breast-Cancer Patients Undergoing Chemotherapy,” Journal of Clinical Pharmacy and Therapeutics, 2011, Online Version of Record Published before Inclusion in an Issue. doi:10.1111/j.1365-2710.2010.01237.x
- K. Tikoo, M. S. Sane and C. Gupta, “Tannic Acid Ameliorates Doxorubicin-Induced Cardiotoxicity and Potentiates Its Anti-Cancer Activity: Potential Role of Tannins in Cancer Chemotherapy,” Toxicology and Applied Pharmacology, Vol. 251, No. 3, 2011, pp. 191-200. doi:10.1016/j.taap.2010.12.012
- D. Huang, Y. Ding, M. Zhou, B. I. Rini, D. Petillo, C. N. Qian, R. Kahnoski, P. A. Futreal, K. A. Furge and B. T. Teh, “Interleukin-8 Mediates Resistance to Antiangiogenic Agent Sunitinib in Renal Cell Carcinoma,” Cancer Research, Vol. 70, No. 3, 2010, pp. 1063-1071. doi:10.1158/0008-5472.CAN-09-3965
- G. Bergers and D. Hanahan, “Modes of Resistance to Anti-Angiogenic Therapy,” Nature Reviews Cancer, Vol. 8, No. 8, 2008, pp. 592-603. doi:10.1038/nrc2442
- S. Charles, M. L. James, M. Gerlinger., C. E. Aron, H. Michael, S. Gordon, D. Julian, M. Gore., F. P. Andrew, E. Bernard, A. Fabrice, A. Laurence, B. Benoit, O. Stephane, H. Jens, G. Balázs, J. T. Chris, A. B. Karen, V. Hansjuergen, T. Luisella, N. Barbara, B. Marlene and S. Zoltan, “Predictive Biomarker Discovery Through the Parallel Integration of Clinical Trial and Functional Genomics Datasets,” Genome Medicine, Vol. 2, No. 8, 2010, p. 53. doi:10.1186/gm174
- F. Shojaei, J. H. Lee, B. H. Simmons, A. Wong, C. O. Esparza, P. A. Plumlee, J. Feng, A. E. Stewart, D. D. HuLowe and J. G. Christensen, “HGF/c-Met Acts as an Alternative Angiogenic Pathway in Sunitinib-Resistant Tumors,” Cancer Research, Vol. 70, No. 24, 2010, pp. 10090-10100. doi:10.1158/0008-5472.CAN-10-0489
- H. J. Hammers, H. M. Verheul, B. Salumbides, R. Sharma, M. Rudek, J. Jaspers, P. Shah, L. Ellis, L. Shen, S. Paesante, K. Dykema, K. Furge, B. T. Teh, G. Netto and R. Pili, “Reversible Epithelial to Mesenchymal Transition and Acquired Resistance to Sunitinib in Patients with Renal Cell Carcinoma: Evidence from a Xenograft Study,” Molecular Cancer Therapeutics, Vol. 9, No. 6, 2010, pp. 1525-1535.
- A. Dahan, H. Sabit and G. L. Amidon, “The H2 Receptor Antagonist Nizatidine Is a P-Glycoprotein Substrate: Characterization of Its Intestinal Epithelial Cell Efflux Transport,” AAPS Journal, Vol. 11, No. 2, 2009, pp. 205- 213. doi:10.1208/s12248-009-9092-5
- L. Z. Benet, T. Izumi, Y. Zhang, J. A. Silverman, V. J. Wacher, “Intestinal MDR Transport Proteins and P-450 Enzymes as Barriers to Oral Drug Delivery,” Journal of Controlled Release, Vol. 62. No. 1, 1999, pp. 25-31. doi:10.1016/S0168-3659(99)00034-6
- V. J. Wacher, J. A. Silverman, Y. Zhang and L. Z. Benet, “Role of P-Glycoprotein and Cytochrome P450 3A in Limiting Oral Absorption of Peptides and Peptidomimetics,” Journal of Pharmaceutical Sciences, Vol. 87, No. 11, 1998, pp. 1322-1330. doi:10.1021/js980082d
- M. Ceckova-Novotna, P. Pavek and F. Staud, “P-Glycoprotein in the Placenta: Expression, Localization, Regulation and Function,” Reproductive Toxicology, Vol. 22, No. 3, 2006, pp. 400-410. doi:10.1016/j.reprotox.2006.01.007
- H. Sugimoto, H. Hirabayashi, Y. Kimura, A. Furuta, N. Amano and T. Moriwaki, “Quantitative Investigation of the Impact of P-Glycoprotein Inhibition on Drug Transport across Blood-Brain Barrier in Rats,” Drug Metabolism and Disposition, Vol. 39, No. 1, 2010, pp. 8-14. doi:10.1124/dmd.110.035774
- A. Dahan and H. Altman, “Food-Drug Interaction: Grapefruit Juice Augments Drug Bioavailability—Mechanism, Extent and Relevance,” European Journal of Clinical Nutrition, Vol. 58, No. 1, 2004, pp. 1-9. doi:10.1038/sj.ejcn.1601736
- L. Z. Benet, C. L. Cummins and C. Y. Wu, “Transporter-Enzyme Interactions: Implications for Predicting Drug-Drug Interactions from in Vitro Data,” Current Drug Metabolism, Vol. 4, No. 5, 2003, pp. 393-398. doi:10.2174/1389200033489389
- M. Takano, R. Yumoto and T. Murakami, “Expression and Function of Efflux Drug Transporters in the Intestine,” Pharmacology & Therapeutics, Vol. 109, No. 1-2, 2006, pp. 137-161. doi:10.1016/j.pharmthera.2005.06.005