Health
Vol.5 No.5(2013), Article ID:31948,8 pages DOI:10.4236/health.2013.55121
The acceleration of aging and Alzheimer’s disease through the biological mechanisms behind obesity and type II diabetes
![]()
1Centre of Excellence in Alzheimer’s Disease Research and Care, School of Medical Sciences, Edith Cowan University, Joondalup, Australia; *Corresponding Author: i.martins@ecu.edu.au
2School of Psychiatry and Clinical Neurosciences, The University of Western Australia, Perth, Australia
3McCusker Alzheimer’s Research Foundation, Hollywood Medical Centre, Perth, Australia
Copyright © 2013 Ian James Martins et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 14 March 2013; revised 20 April 2013; accepted 8 May 2013
Keywords: Telomere; Sirtuin 1; Lifestyle; Nutrition; Diabetes; Obesity; Alzheimer’s Disease
ABSTRACT
The incidence of diabetes is predicted to increase to 21% by 2050. Currently, one third of US adults are obese and over 11% of these individuals have diabetes. Due to the growing need for therapeutic intervention to control and/or stabilize this increase in the incidence of diabetes in Western communities, gaining a comprehensive understanding of the association between obesity and Type 2 diabetes has become increasingly important to diabetes research. The increased cell senescence associated with diabetes has been associated with the limited ability of cells to divide, with indication of telomere shortening and genomic instability of the cells. Obese individuals have shorter telomeres suggesting an inverse relationship between adiposity and telomere length. The implication that Type 2 diabetes has on biological aging is of particular interest since telomere shortening in obesity and diabetes has been associated with an early risk for dementia and even progression to Alzheimer’s disease (AD). Lifestyle, nutrition and longevity are closely related and cellular senescence has been associated with telomere shortening and connected to longevity. Diet, cholesterol lowering drugs and exercise that control food intake and glucose tolerance in aging and diabetic individuals, via connections between liver circadian clocks and the suprachiasmatic nucleus in the brain, also have been shown to alter telomere lengths. Lifestyle interventions, such as diets low in fat and exercise, target the rise in obesity and associated telomere shortening by delaying or preventing the onset of Type 2 diabetes. The implementation of these anti-aging therapies early in life may prevent calorie overload and activation of calorie sensitive genes such as Sirtuin 1 (Sirt1). This may maintain telomere length and the control of obesity, which is linked to cardiovascular disease, diabetes and accelerates aging and AD.
1. BACKGROUND
Age-related diseases are becoming a major concern as the world’s population grows older due to advances in medical technology, health and nutrition. Dementia accounts for a large proportion of age-related diseases and is characterised clinically by deterioration in cognitive processing, including memory. Alzheimer’s disease (AD) is the most common form of age-related dementia. The insulin-resistance syndrome has in many studies been associated with diabetes and AD. The social and economic consequences of this disease present a significant challenge to society, and it is imperative that strategies to prevent or delay the onset of diabetes are developed to prevent the proportion of the age related group with cardiovascular dysfunction progressing towards AD. AD is a neurodegenerative disease which presents clinically with key clinical symptoms including progressive decline in memory. Risk factors for AD include old age, family history of dementia, APOE e4 genotype, Down’s syndrome, obesity and type 2 diabetes.
Epidemiological studies clearly suggest that human obesity is associated with the increased risk for atherosclerosis, contributing to the early onset of coronary artery disease and diabetes. The susceptibility of humans to obesity is far higher compared with other species [1]. Amongst mammals, humans have been reported to have the highest level of fat than any other species as genes and environmental factors predispose humans to obesity [2]. In an editorial by Testa and Ceriello published in 2007, the increased cell senescence associated with diabetes was associated with the limited ability of cells to divide with indication of cellular alteration in genes and genomic instability of the cells [3]. Human obesity may complete the pathogenetic loop between cell senescence and diabetes through teleomere shortening and an association with an increased risk for atherosclerosis. This in turn contributes to the early onset of coronary artery disease and diabetes.
Visceral obesity in particular increases the risk of atherosclerosis owing to both insulin resistance and dyslipoproteinemia and this global rise in obesity is possibly linked to telomere disease. Obesity has been closely linked to diabetes [4,5] and the association of obesity and telomere shortening has now been reported [6]. In particular, telomere length, high density lipoprotein (HDL) cholesterol [7] and other risk factors for atherosclerosis that could be exacerbated by obesity-associated telomere dysfunction include hypertension and hyperlipidemia (particularly hypertriglyceridemia). These are also risk factors for AD. Circadian desynchrony and hyperphagia are also related to obesity and anti-aging, circadian proteins, such as sirtuins are known to regulate several cell functions by deacetylating both histone and nonhisotone targets. Sirtuins are NAD(+)dependent class III histone deacetylase (HDAC) proteins that target transcription factors to adapt gene expression to metabolic activity. Sirtuin 1 (Sirt1) is linked to life span, obesity and cardiovascular disease with effects on liver steatosis, inflammation, food intake, energy metabolism, cognition, mitochondrial biogenesis, neurogenesis, glucose/cholesterol metabolism and amyloidosis [8-22]. Regulation of Sirt1 by calorie restriction in extending life span has been recognized. Novel dietary acitvators of Sirt1 are required and designing compounds that have therapeutic potential for the control of telomere shortening, clock circuitry and generation of Aβ for the treatment of AD is the focus of several research groups [23-26].
Obesity in humans is seen to be associated with decreased life span in man and is an excellent model for the assessment of cell senescence, inflammation, diabetes and AD [27-31]. The understanding of alterations in genes/genomic stability and the environment in the aging brain has been the subject of various anti-aging programs as well as the mechanisms involved in anti-aging strategies related to low fat diets and reversing brain aging. The association between diabetes and neurodegenerative diseases (AD and Parkinson’s disease) have related the majority of the genes to cholesterol metabolism, cytokines and inflammation. Environmental factors such as diet and circadian desynchrony have become important in Western countries since interests in the global increase in obesity is possbily linked to diabetes, timing of food consumption, metabolic activity and alterations in adipose tissue leading to a release of adipokines (leptin and adiponectin). These adipokines increase with age and are associated with age related pathological alterations in cytokines associated with neuroinflammation [32,33].
2. CONVERGENCE OF MECHANISMS LINKING OBESITY TO DIABETES AND ACCELERATION OF AGING AND ALZHEIMER’S DISEASE
The mechanism linking obesity with diabetes is unclear but the global epidemic indicates that most patients with type 2 diabetes are obese; over a third (34%) of US adults are obese and about 11% of these individuals have diabetes [4]. The incidence of diabetes is predicated to increase to 21% by 2050 [5]. The understanding of mechanisms which connects these two diseases has become important to diabetes research since therapeutics to control and stabilize the increase in the incidence of diabetes in Western communities is required. As lipids and proteins accumulate in cells, the cells become impaired and can no longer divide, decreasing the lifespan of the cell resulting in cell telomeres that have eroded or shortened with cell senescence. The telomere is a repeating sequence of DNA at the end of a chromosome (TTAGGG)n [34,35]. Aging results in a progressive loss of telomere repeats and with telomere shortening the cell ceases to replicate with alterations in lipid and protein metabolism with eventual cell death [34,35]. Obese individuals have shorter telomeres and an inverse relationship has been found with adiposity and telomere length [6,36,37]. The pathogenetic loop between obesity and diabetes has recently been related to Sirt1 which is involved in cellular senescence. The genomic instability and telomere dysfunction observed in obesity is now closely associated with the pathogenesis of diabetes, dyslipidemia and cardiovascular disease. Diabetes, genetic and environmental factors have been reported to stress telomeres [38] through telomere shortening (Figure 1).
Sirt1 and cell senescence has been closely linked to telomere biology and global DNA repair which provides mechanistic explanations for SIRT1 functions, in protection from DNA damage, and thus genomic stability [39,40]. Sirt1 has been closely linked to telomerase which is a ribonucleoprotein (RNP) complex responsible
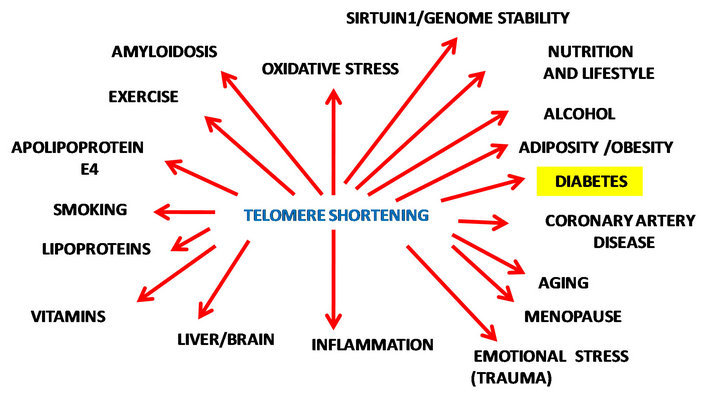
Figure 1. Nutrition, lifestyle and aging determine telomerelength and genomic stability.
for the elongation of telomeres to maintain genomic integrity. Telomerase is composed of the telomerase reverse transcriptase (TERT) and telomerase RNA components (TERC). These factors regulate the catalytic activity of telomerase [41]. Telomerase protects cells from apoptosis via the maintenance of genomic integrity by stabilizing telomeres and adding DNA, in mediating cell survival and anti-apoptotic functions against various cytotoxic stresses [42,43]. Telomerase is closely connected to telomere length maintenance and control of genes involved in growth and cell proliferation [44].
Anti-aging strategies that target telomere shortening in diabetes is of particular interest to biological aging since telomere shortening has been associated as an early risk for dementia [45,46]. Age-related changes in AD lead to neuronal apoptosis and therapy to delay the onset and even progression of Alzheimer’s disease are urgently required. Nutrition related to low fat diets and drugs that reduce intestinal absorption of fat with modulation of adipose tissue Sirt1 activity may improve the adipocyte brain crosstalk that may be assessed as a possible treatment of neuronal diseases that afflict diabetic and AD individuals. Sirt1 is essential for neurogenesis and calorie restriction activates Sirt1 with effects on longevity by modulation of phosphoinositide 3 kinase pathway that determines life span [47-49]. The role of Sirt1 in brain metabolic regulation and synaptic plasticity has been shown and maintenance of Sirt1 expression by calorie restriction regulates lipid metabolism and energy expenditure [19]. Age associated cardiovascular changes have been strongly associated with alterations in Sirt1 [8-11] and genetic as well as experimental evidence of its control of lifespan is of interest to the areas of diabetes, neurodegeneration and AD. Telomere and telomerase activity has now taken an important place in AD therapeutics, using telomere length and telomerase activity in the determination of neuronal populations in these individuals [50,51]. Sirt1 and its role in AD is of interest with reviews that indicate that Sirt1 is closely connected to Abeta production, telomere maintenance and stem cell aging [39,40,52,53]. Interests in Sirt1 control of telomerase is of interest to diabetes and AD with recent publiccations suggesting telomerase inhibition is involved with abeta cell apoptosis. In AD, transgenic mice telomere length has also shown to be associated with amyloid pathology with indications that Sirt1 expression and activity is essential to neuronal population maintenance and prevention of AD.
3. LIFESTYLE, DIET AND DRUG CONNECTIONS TO DIABETES AND ALZHEIMER’S DISEASE
The estimate in global deaths due to diabetes was estimated to be 5.2% of all deaths in 2009 [54]. In poorer countries, mortality related to diabetes was 2% - 3%, in countries such as the USA, Canada and the middle east the mortality was approx. 8% and for individuals between 35 - 64 years between 6% - 27% of the deaths were attributable to diabetes [55]. In 2011, the largest number of people with diabetes were in India, China, United States of America and the Russian Federation and mortality in 2011 reached 8.2% of the global population [55] Nutrition and longevity are closely related and cellular senescence has been associated with telomere shortening as well as the life span of the organism [34,35]. Apart from nutrition, minor alcohol consumption has been shown in midlife to shorten telomeres and accelerate aging in older individuals [56,57]. Diet, cholesterol lowering drugs and exercise may improve telomere lengths. This may provide the classical evolutionary conserved environment to suit anti-inflammatory processes which control of food intake and circadian rhythm and promote normal liver and brain, lipid and protein homeostasis, ultimately preventing glucose intolerance and diabetes.
Sirt1 (nutrient sensitive gene) is closely linked to food intake and the prevention of diabetes, which is linked to amyloidosis and may offer a potential therapy for AD treatment (Figure 2, [58,59]). Interests in high fat intake and the consumption of fat at specific times of the day requires further investigation as a mechanism for diabetes and AD induction. Drugs that reduce intestinal absorption of fat will modulate the tissue anti-aging protein Sirt1, improving the adipocyte brain crosstalk that should be assessed as a possible treatment of neuronal diseases such as AD.
In recent years, the world diabetes epidemic now includes younger individuals and has become of concern because of the risk for telomere shortening and non-alcoholic liver disease (NAFLD) [60]. In Western countries, accelerated aging is associated with NAFLD and has reached epidemic levels, with 40 % of the community with the disease and up to 20% of these individuals developing hepatic fibrosis and cirrhosis [61,62]. In-
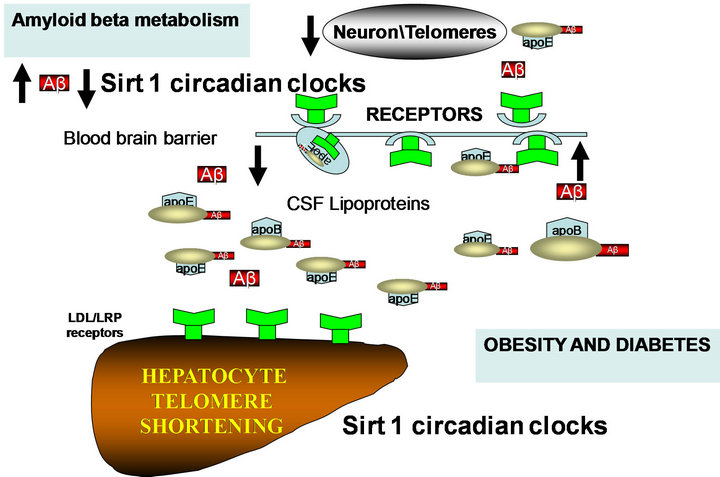
Figure 2. High fat and high cholesterol diets affect Sirt1 control of circadian rhythms with liver steatosis and effects on peripheral and brain amyloid beta metabolism.
dividuals with longer lifespan have been shown to be diabetes free and increased mortality was attributed to increases in obesity [61]. Age related pathologies, such as NAFLD, that have been linked to Type 2 diabetes and AD, indicate association between insulin resistance, oxidative stress and telomere shortening [63]. Hepatocyte senescence, telomere shortening, nuclear size alterations and telomere foci have been closely associated to NAFLD and indicate mitochondrial dysfunction and lack of cell cycle progression beyond the cell cycle G1/S phase [64-68]. Interests in telomere shortening and insulin resistance indicate that the suprachiasmatic nucleus (SCN), which closely regulates peripheral clocks such as the liver (NAFLD) and adipose tissue (increased adiposity), is abnormal with relevance to the pathophysiology of disease [62] progression in obese, diabetic and AD individuals [69-72].
Drug therapies that target the brain have not been successful in preventing amyloid deposition but drugs that target the periphery may be promising by lengthening peripheral cell telomeres, lowering plasma fat and cholesterol and activating adipose tissue, liver and brain metabolic activity. Interest in Sirt1 modulation of various proteins that regulate cellular inflammation, glucose and cholesterol homeostasis are relevant to diabetes and neurodegeneration. Regulation of this calorie-restricted gene, Sirt1, as an important control that targets obesity, diabetes and brain aging by suppression of inflammation as well as maintenance of neuroprotective mechanisms that facilitate normal food intake and vitamin E transport to the central nervous system essential for growth of neurons has been the interest of anti-aging research studies. Designing compounds for the regulation of Sirt1 to extend life span has been recognized and activators of Sirt1 in order for telomere maintenance have been reported. The use of melatonin, which is involved in telomere maintenace and circadian clock control [40], is relevant for the treatment of diabetes and AD as compared with other circadian rhythm drugs such as the benzodiazepines or non-benzodiazepines.
The relationship between telomere erosion and apoE4 is closely associated [23] with the role of apolipoprotein E and its connections with inflammation/cytokines [73, 74] as an important control of telomere attrition in the periphery and the brain. The connections between dyslipidemia and inflammation have been reported in diabetes, where inflammation in the blood plasma and altered cytokines have been associated with changes in lipid metabolism (increased triglyceride and low HDL) and in the brain, alterations in the light/dark cycle [75]. Drugs involved with telomere lengthening are possibly involved in the adaptation of the organism to the environment and require regulation of liver glucose and cholesterol and are controlled by dietary fat and cholesterol intake (Figure 2, [76-78]). The mechanism by which the e4 allele promotes AD risk could be associated with diabetes and telomere shortening as well as with abnormal regulation of food intake, explaining the hyperphagia associated changes in diabetes and AD [79-81].
4. NUTRITIONAL SCIENCE AND DRUGS DELAY OBESITY AND SEVERITY OF DIABETES LINKED TO AGING AND ALZHEIMER’S DISEASE
Biological aging is closely connected to telomere length and control of cellular senescence. Each time a cell replicates and divides the telomere loses some of its length. Eventually the telomere runs out and the cell can no longer divide and rejuvenate, triggering a poor state of cell health that contributes to disease risk and eventual cell death. Telomere length is epigenetically regulated and affected by nutrients, alcohol, drugs, genetics and the environment (Figure 3). It is interesting to note that telomere shortening would be accelerated by synergistic effects of mixing alcohol and fats such as palmitic acid with increased genomic instability and DNA breakdown. The effective function of telomeres require methylation, which uses nutrients like methionine, an essential amino acid that serves as a methyl donor and is involved with the biosynthesis of other nutrients. Improper conversion is associated with production of homocysteine and atherosclerosis, methylsulfonylmethane, sulphur, choline, and trimethylglycine, as building blocks and allow regulation of genes by appropriate telomeres. Vitamins such as vitamin B12, folic acid, and vitamin B6 play multiple roles in genomic stability as well. Foods that are important include protein, eggs, cottage cheese, dairy, red meat, chicken, legumes, duck, nuts, and seeds. Antioxidants and vitamins C, D and E are essential and maintain genomic stability as well as telomeres. A lack of antioxidants leads to increased free radical damage and more risk for damage to telomeres. Minerals, such as mag-
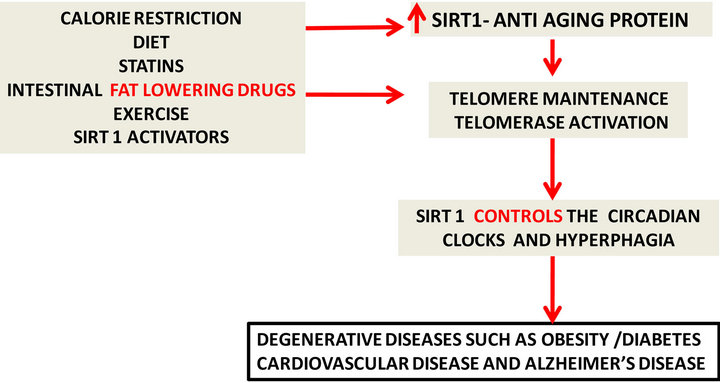
Figure 3. Potential anti-aging therapies (left) delay the aging process by telomere maintenance and control of DNA damage.
nesium, are needed by many enzymes involved with DNA replication and repair and total magnesium intake should be between 400 mg - 800 mg per day. Zinc is intimately involved with DNA as well as DNA repair. The lack of zinc causes an excessive amount of DNA strand breakage and telomere depletion. Inflammation and stress shorten telomeres and omega 3 fatty acids (eicosapentaenoic acid/docosahexaenoic acid) are important as basic nutrients to preserve telomeres. Nutrients such as quercetin, green tea catechins, grape seed extract, curcumin, and resveratrol also help maintain telomeres, with both grape seed extract and curcumin showing the ability to generate longer telomeres. In addition, resveratrol and calorie restriction activate Sirt1 with prevention of telomere attrition.
Drug evaluation and differences in therapeutic effects of various drugs may indicate that the telomere length of tested rodents or diseased individuals may be related to genetic, diet and environmental conditions. Drugs such as statins have been shown to control telomere/telomerase in peripheral leukocytes from individuals with cardiovascular disease [82,83] and have provided potential anti-aging therapy for diabetes and AD. Statins, however, are not Sirt1 activators and Sirt1 activators are primarily in control of telomere length and attrition. The close association of hyperphagia with diabetes and AD indicate that fat consumption (calorie excess) may have marked effects on telomeres/telomerase activation [42,43]. Experimental drugs in mice, such as an acyl coA cholesterol acyltransferase (ACAT) inhibitor Avasimibe, that prevents fat absorption (calorie excess) have shown clear effects on body weight, liver and brain growth in mice consuming high fat diets (Abstract AD/PD Italy, 2013) possibly as a Sirt1/telomerase activator. Interests in telomerase activation have increased with the recent findings of TA-65 (purified from the root of Astragalus membranaceus/legume), a small molecule activator of telomerase involved in glucose regulation [84]. In contrast, various drugs that are Sirt1 inhibitors should be assessed with various diets since telomere shortening and attrition by Sirt1 inhibitors will lead to degenerative diseases such as obesity, diabetes, cardiovascular disease and AD. Lifestyles changes such as diet and exercise early in the life of obese or diabetic individuals may prevent circadian alterations and calorie overload. Continued Sirt1 activation and the maintenance of peripheral (liver) and CNS (brain) cell telomeres with prevention of obesity linked to diabetes may decrease accelerated aging and the risk of AD.
5. CONCLUSION
The biological mechanism behind the global epidemic of obesity and Type 2 diabetes has become important to Alzheimer’s research since therapeutic interventions with the potential to control and stabilize their increased incidence in Western society may also have the potential to delay the acceleration in aging and AD. The increased cell senescence associated with obesity and diabetes has indicated an increase in telomere shortening and genomic instability of the cells from these individuals. Stabilization of biological aging is of particular interest in obesity and diabetes and the delay of telomere shortening in these individuals may result in a decrease in dementia and even a delay in the progression to (AD). Lifestyle, nutrition and longevity are closely connected to life span of obese and Type 2 diabetic individuals and therapeutics such as cholesterol lowering drugs, exercise and diets low in fat, target the rise in obesity and associated telomere shortening by delaying or preventing the onset of Type 2 diabetes. Implementing these anti-aging therapies early in life may prevent calorie overload and activation of calorie sensitive genes, such as (Sirt1), that may maintain telomere length and the control of obesity, which is linked to cardiovascular disease, diabetes and accelerates aging and AD.
REFERENCES
- Wells, J.C. (2006) The evolution of human fatness and susceptibility to obesity: An ethological approach. Biological Reviews of the Cambridge Philosophical Society, 81, 183-205. doi:10.1017/S1464793105006974
- O’Rahilly, S., et al. (2003) Minireview: Human obesitylessons from monogenic disorders. Endocrinology, 144, 3757-3764. doi:10.1210/en.2003-0373
- Testa, R. and Ceriello, A. (2007) Pathogenetic loop between diabetes and cell senescence. Diabetes Care, 30, 2974-2975. doi:10.2337/dc07-1534
- Eckel, R.H., et al. (2011) Obesity and type 2 diabetes: What can be unified and what needs to be individualized? Diabetes Care, 34, 1424-1430. doi:10.2337/dc11-0447
- Steinberger, J. and Daniels, S.R. (2003) Obesity, insulin resistance, diabetes, and cardiovascular risk in children: An American Heart Association scientific statement from the atherosclerosis, hypertension, and obesity in the Young Committee (council on cardiovascular disease in the young) and the Diabetes Committee (council on nutrition, physical activity, and metabolism). Circulation, 107, 1448- 1453. doi:10.1161/01.CIR.0000060923.07573.F2
- Lee, M., et al. (2011) Inverse association between adiposity and telomere length: The Fels Longitudinal Study. American Journal of Human Biology, 23, 100-106. doi:10.1002/ajhb.21109
- Khalaf, D., Ye, L. and Shil, A.B. (2012) Telomere length and high-density lipoprotein cholesterol. Journal of the American Geriatrics Society, 60, 599. doi:10.1111/j.1532-5415.2011.03856.x
- Hsu, C.P., et al. (2008) Sirt1 protects the heart from aging and stress. The Journal of Biological Chemistry, 389, 221-231. doi:10.1515/BC.2008.032
- Borradaile, N.M. and Pickering, J.G. (2009) NAD(+), sirtuins, and cardiovascular disease. Current Pharmaceutical Design, 15, 110-117. doi:10.2174/138161209787185742
- Stein, S. and Matter, C.M. (2011) Protective roles of SIRT1 in atherosclerosis. Cell Cycle, 10, 640-647. doi:10.4161/cc.10.4.14863
- Shi, Y., Camici, G.G. and Luscher, T.F. (2010) Cardiovascular determinants of life span. Pflügers Archiv, 459, 315-324. doi:10.1007/s00424-009-0727-2
- Purushotham, A., et al. (2009) Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metabolism, 9, 327-338. doi:10.1016/j.cmet.2009.02.006
- Elliott, P.J. and Jirousek, M. (2008) Sirtuins: Novel targets for metabolic disease. Current Opinion in Investigational Drugs, 9, 371-378.
- Colak, Y., et al. (2011) SIRT1 as a potential therapeutic target for treatment of nonalcoholic fatty liver disease. Medical Science Monitor, 17, HY5-HY9. doi:10.12659/MSM.881749
- Zhang, Z., et al. (2010) Roles of SIRT1 in the acute and restorative phases following induction of inflammation. The Journal of Biological Chemistry, 285, 41391-41401. doi:10.1074/jbc.M110.174482
- Yoshizaki, T., et al. (2009) SIRT1 exerts anti-inflammatory effects and improves insulin sensitivity in adipocytes. Molecular and Cellular Biology, 29, 1363-1374. doi:10.1128/MCB.00705-08
- Sasaki, T. and Kitamura, T. (2010) Roles of FoxO1 and Sirt1 in the central regulation of food intake. Endocrine Journal, 57, 939-946. doi:10.1507/endocrj.K10E-320
- Michan, S., et al. (2010) SIRT1 is essential for normal cognitive function and synaptic plasticity. The Journal of Neuroscience, 30, 9695-9707. doi:10.1523/JNEUROSCI.0027-10.2010
- Gao, J., et al. (2010) A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature, 466, 1105- 1109. doi:10.1038/nature09271
- Guarente, L. (2008) Mitochondria—A nexus for aging, calorie restriction, and sirtuins? Cell, 132, 171-176. doi:10.1016/j.cell.2008.01.007
- Libert, S., Cohen, D. and Guarente, L. (2008) Neurogenesis directed by Sirt1. Nature Cell Biology, 10, 373-374. doi:10.1038/ncb0408-373
- Silva, J.P. and Wahlestedt, C. (2010) Role of Sirtuin 1 in metabolic regulation. Drug Discovery Today, 15, 781-791. doi:10.1016/j.drudis.2010.07.001
- Takata, Y., et al. (2012) Association between ApoE phenotypes and telomere erosion in Alzheimer’s disease. Journals of Gerontology. Series A: Biological Sciences and Medical Sciences, 67, 330-335. doi:10.1093/gerona/glr185
- Moghekar, A. and O’Brien, R.J. (2012) Con: Alzheimer’s disease and circadian dysfunction: Chicken or egg? Alzheimer’s Research & Therapy, 4, 26. doi:10.1186/alzrt129
- Wang, J., et al. (2010) The role of Sirt1: At the crossroad between promotion of longevity and protection against Alzheimer’s disease neuropathology. Biochimica et Biophysica Acta, 1804, 1690-1694. doi:10.1016/j.bbapap.2009.11.015
- Bonda, D.J., et al. (2011) The sirtuin pathway in ageing and Alzheimer disease: Mechanistic and therapeutic considerations. The Lancet Neurology, 10, 275-279. doi:10.1016/S1474-4422(11)70013-8
- Galimberti, D. and Scarpini, E. (2011) Inflammation and oxidative damage in Alzheimer’s disease: Friend or foe? Frontiers in Bioscience (Scholar Edition), 3, 252-266. doi:10.2741/s149
- Fresno, M., Alvarez, R. and Cuesta, N. (2011) Toll-like receptors, inflammation, metabolism and obesity. Archives of Physiology and Biochemistry, 117, 151-164. doi:10.3109/13813455.2011.562514
- Ikeoka, D., Mader, J.K. and Pieber, T.R. (2010) Adipose tissue, inflammation and cardiovascular disease. Revista da Associação Médica Brasileira, 56, 116-121. doi:10.1590/S0104-42302010000100026
- Maccioni, R.B., et al. (2009) The role of neuroimmunomodulation in Alzheimer’s disease. Annals of the New York Academy of Sciences, 1153, 240-246. doi:10.1111/j.1749-6632.2008.03972.x
- Arosio, B., et al. (2004) Interleukin-10 and interleukin-6 gene polymorphisms as risk factors for Alzheimer’s disease. Neurobiology of Aging, 25, 1009-1015. doi:10.1016/j.neurobiolaging.2003.10.009
- Godbout, J.P. and Johnson, R.W. (2004) Interleukin-6 in the aging brain. Journal of Neuroimmunology, 147, 141- 144. doi:10.1016/j.jneuroim.2003.10.031
- Scheff, J.D., et al. (2010) Modeling the influence of circadian rhythms on the acute inflammatory response. Journal of Theoretical Biology, 264, 1068-1076. doi:10.1016/j.jtbi.2010.03.026
- Shammas, M.A. (2011) Telomeres, lifestyle, cancer, and aging. Current Opinion in Clinical Nutrition & Metabolic Care, 14, 28-34. doi:10.1097/MCO.0b013e32834121b1
- Jennings, B.J., Ozanne, S.E. and Hales, C.N. (2000) Nutrition, oxidative damage, telomere shortening, and cellular senescence: individual or connected agents of aging? Molecular Genetics and Metabolism, 71, 32-42. doi:10.1006/mgme.2000.3077
- Farzaneh-Far, R., et al. (2010)Telomere length trajectory and its determinants in persons with coronary artery disease: Longitudinal findings from the heart and soul study. PLoS One, 5, e8612. doi:10.1371/journal.pone.0008612
- Zannolli, R., et al. (2008) Telomere length and obesity. Acta Paediatrica, 97, 952-954. doi:10.1111/j.1651-2227.2008.00783.x
- Ly, H. (2009) Genetic and environmental factors influencing human diseases with telomere dysfunction. International Journal of Clinical and Experimental Medicine, 2, 114-130.
- Palacios, J.A., et al. (2010) SIRT1 contributes to telomere maintenance and augments global homologous recombination. The Journal of Cell Biology, 191, 1299-1313. doi:10.1083/jcb.201005160
- Kagawa, Y. (2012) From clock genes to telomeres in the regulation of the healthspan. Nutrition Reviews, 70, 459- 471. doi:10.1111/j.1753-4887.2012.00504.x
- Sung, Y.H., et al. (2005) The pleiotropy of telomerase against cell death. Molecules and Cells, 19, 303-309.
- Chan, S.W. and Blackburn, E.H. (2002) New ways not to make ends meet: Telomerase, DNA damage proteins and heterochromatin. Oncogene, 21, 553-563. doi:10.1038/sj.onc.1205082
- Saretzki, G. (2009) Telomerase, mitochondria and oxidative stress. Experimental Gerontology, 44, 485-492. doi:10.1016/j.exger.2009.05.004
- Smith, L.L., Coller, H.A. and Roberts, J.M. (2003) Telomerase modulates expression of growth-controlling genes and enhances cell proliferation. Nature cell biology, 5, 474-479.
- Jiang, H., Ju, Z. and Rudolph, K.L. (2007) Telomere shortening and ageing. Zeitschrift für Gerontologie und Geriatrie, 40, 314-324. doi:10.1007/s00391-007-0480-0
- Grodstein, F., et al. (2008) Shorter telomeres may mark early risk of dementia: Preliminary analysis of 62 participants from the nurses’ health study. PLoS One, 3, e1590.
- Frojdo, S., et al. (2011) Phosphoinositide 3-kinase as a novel functional target for the regulation of the insulin signaling pathway by SIRT1. Molecular and Cellular Endocrinology, 335, 166-176. doi:10.1016/j.mce.2011.01.008
- Zhang, J. (2006) Resveratrol inhibits insulin responses in a SIRT1-independent pathway. Biochemical Journal, 397, 519-527.doi:10.1042/BJ20050977
- Qin, W., et al. (2006) Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. The Journal of Biological Chemistry, 281, 21745-21754. doi:10.1074/jbc.M602909200
- Sonneborn, J.J. (2012) Alternative strategy for Alzhei- mer’s disease: Stress response triggers. International Journal of Alzheimer’s Disease, 2012, Article ID: 684283.
- Franco, S., et al. (2006) Telomeres and telomerase in Alzheimer’s disease: Epiphenomena or a new focus for therapeutic strategy? Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association, 2, 164-168. doi:10.1016/j.jalz.2006.03.001
- Donmez, G., et al. (2010) SIRT1 suppresses beta-amyloid production by activating the alpha-secretase gene ADAM10. Cell, 142, 320-332. doi:10.1016/j.cell.2010.06.020
- Mantel, C. and Broxmeyer, H.E. (2008) Sirtuin 1, stem cells, aging, and stem cell aging. Current Opinion in Hematology, 15, 326-331. doi:10.1097/MOH.0b013e3283043819
- Roglic, G., et al. (2005) The burden of mortality attributeable to diabetes: Realistic estimates for the year 2000. Diabetes Care, 28, 2130-2135. doi:10.2337/diacare.28.9.2130
- Colagiuri, S., et al. (2005) There really is an epidemic of type 2 diabetes. Diabetologia, 48, 1459-1463. doi:10.1007/s00125-005-1843-y
- Pavanello, S., et al. (2011) Shortened telomeres in individuals with abuse in alcohol consumption. International Journal of Cancer, 129, 983-992. doi:10.1002/ijc.25999
- Strandberg, T., et al. (2012) Association between alcohol consumption in healthy midlife and telomere length in older men. The Helsinki businessmen study. European Journal of Epidemiology, 27, 815-822. doi:10.1007/s10654-012-9728-0
- Pfluger, P.T., et al. (2008) SIRT1 protects against high-fat diet-induced metabolic damage. Proceedings of the National Academy of Sciences of the United States of America, 105, 9793-9798. doi:10.1073/pnas.0802917105
- Deng, X.Q., Chen, L.L. and Li, N.X. (2007) The expression of SIRT1 in nonalcoholic fatty liver disease induced by high-fat diet in rats. Liver International, 27, 708-715. doi:10.1111/j.1478-3231.2007.01497.x
- [61] Mulder, H. (2010) Is shortening of telomeres the missing link between aging and the type 2 diabetes epidemic? Aging (Albany NY), 2, 634-636.
- [62] Cunningham, S.A., et al. (2011) Decreases in diabetes-free life expectancy in the US and the role of obesity. Diabetes Care, 34, 2225-2230. doi:10.2337/dc11-0462
- [63] Farrell, G.C., et al. (2012) NASH is an inflammatory disorder: Pathogenic, prognostic and therapeutic implications. Gut Liver, 6, 149-171. doi:10.5009/gnl.2012.6.2.149
- [64] Katic, M. and Kahn, C.R. (2005) The role of insulin and IGF-1 signaling in longevity. Cellular and Molecular Life Sciences, 62, 320-343. doi:10.1007/s00018-004-4297-y
- [65] Aravinthan, A., et al. (2012) Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. Journal of Hepatology, 58, 549-556.
- [66] Hewitt, G., et al. (2012) Telomeres are favoured targets of a persistent DNA damage response in ageing and stressinduced senescence. Nature Communications, 3, 708. doi:10.1038/ncomms1708
- [67] Monickaraj, F., et al. (2012) Accelerated aging as evidenced by increased telomere shortening and mitochondrial DNA depletion in patients with type 2 diabetes. Molecular and Cellular Biochemistry, 365, 343-350. doi:10.1007/s11010-012-1276-0
- [68] Nakajima, T., et al. (2010) Nuclear size measurement is a simple method for the assessment of hepatocellular aging in non-alcoholic fatty liver disease: Comparison with telomere-specific quantitative FISH and p21 immunohistochemistry. Pathology International, 60, 175-183. doi:10.1111/j.1440-1827.2009.02504.x
- [69] Nakajima, T., et al. (2006) Premature telomere shortening and impaired regenerative response in hepatocytes of individuals with NAFLD. Liver International, 26, 23-31. doi:10.1111/j.1478-3231.2005.01178.x
- [70] Weldemichael, D.A. and Grossberg, G.T. (2010) Circadian rhythm disturbances in patients with Alzheimer’s disease: A review. International Journal of Alzheimer’s Disease, 2010, Article ID: 716453.
- [71] Okamura, H. (2007) Suprachiasmatic nucleus clock time in the mammalian circadian system. Cold Spring Harbor Symposia on Quantitative Biology, 72, 551-556. doi:10.1101/sqb.2007.72.033
- [72] Coomans, C.P., et al. (2012) The suprachiasmatic nucleus controls circadian energy metabolism and hepatic insulin sensitivity. Diabetes, 62, 1102-1108.
- [73] Garaulet, M. and Madrid, J.A. (2010) Chronobiological aspects of nutrition, metabolic syndrome and obesity. Advanced Drug Delivery Reviews, 62, 967-978. doi:10.1016/j.addr.2010.05.005
- [74] Baitsch, D., et al. (2011) Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arteriosclerosis, Thrombosis, and Vascular Biology, 31, 1160-1168. doi:10.1161/ATVBAHA.111.222745
- [75] Ali, K., et al. (2005) Apolipoprotein E suppresses the type I inflammatory response in vivo. Circulation Research, 97, 922-927. doi:10.1161/01.RES.0000187467.67684.43
- [76] Esteve, E., Ricart, W. and Fernandez-Real, J.M. (2005) Dyslipidemia and inflammation: An evolutionary conserved mechanism. Clinical Nutrition, 24, 16-31. doi:10.1016/j.clnu.2004.08.004
- [77] Romon, M., et al. (1997) Circadian variation of postprandial lipemia. The American Journal of Clinical Nutrition, 65, 934-940.
- [78] Mondola, P., et al. (1995) Circadian rhythms of lipid and apolipoprotein pattern in adult fasted rats. Physiology & Behavior, 58, 175-180. doi:10.1016/0031-9384(95)00016-C
- [79] Kang, J.E., et al. (2009) Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science, 326, 1005-1007. doi:10.1126/science.1180962
- [80] Bhagavati, S. (2008) Marked hyperphagia associated with total loss of satiety in Alzheimer’s disease. The Journal of Neuropsychiatry and Clinical Neurosciences, 20, 248- 249.
- [81] Martins, I.J., et al. (1994) Lipid and apolipoprotein B48 transport in mesenteric lymph and the effect of hyperphagia on the clearance of chylomicron-like emulsions in insulin-deficient rats. Diabetologia, 37, 238-246. doi:10.1007/BF00398049
- [82] Tsang, S.W., et al. (2010) A serotoninergic basis for hyperphagic eating changes in Alzheimer’s disease. Journal of the Neurological Sciences, 288, 151-155. doi:10.1016/j.jns.2009.08.066
- [83] Olivieri, F., et al. (2012) Telomere/telomerase system: A new target of statins pleiotropic effect? Current Vascular Pharmacology, 10, 216-224. doi:10.2174/157016112799305076
- [84] Brouilette, S.W., et al. (2007) Telomere length, risk of coronary heart disease, and statin treatment in the west of Scotland primary prevention study: A nested case-control study. The Lancet, 369, 107-114. doi:10.1016/S0140-6736(07)60071-3
- [85] Bernardes de Jesus, B., et al. (2011) The telomerase acti- vator TA-65 elongates short telomeres and increases health span of adult/old mice without increasing cancer incidence. Aging Cell, 10, 604-621. doi:10.1111/j.1474-9726.2011.00700.x