Advances in Bioscience and Biotechnology
Vol. 3 No. 7 (2012) , Article ID: 24865 , 8 pages DOI:10.4236/abb.2012.37114
Improved methods for cloning and detection in the yeast two hybrid assay
![]()
College of Life Sciences, Key Laboratory of Agricultural Environmental Microbiology of MOA, Nanjing Agricultural University, Nanjing, China
Email: *liangyh@njau.edu.cn
Received 22 August 2012; revised 27 September 2012; accepted 13 October 2012
Keywords: Conventional Method; Improved Method; Cloning; Yeast Two Hybrid; Protein-Protein Interactions
ABSTRACT
The yeast two-hybrid (Y2H) mating assay is a powerful method for detecting protein-protein interactions. Firstly, the gene of interest is cloned into specific Y2H vectors. Although multiple innovations in cloning methods were made in the past two decades, the conventional cloning method of restriction-enzyme (RE) digestion followed by ligation is still widely used. Unfortunately, many researchers, especially new-comers, often encounter difficulties in cloning a gene into a desired vector. Secondly, interaction between two proteins is commonly detected by growth of the diploids in specific media. This step takes about two weeks. Here, we describe improved cloning and detection procedures for the Y2H assay that accelerate the research progress. The changes in procedures involve running an agarose gel after the doubly digested vector and insert are ligated in the cloning step to determine the efficiency of RE digestion and ligation, and performing an additional replica-plating on plates for earlier assessment of interaction in the detection step. We show an example of Y2H interaction between Trs23 and Trs120 (respective subunits of TRAPP I and TRAPP II), as a proof of concept. By following the improved methods described here, the chances of successful cloning increased and the time for the whole Y2H experimental process is significantly shorter.
1. INTRODUCTION
The Y2H assay is a genetic system developed for detecting protein-protein interactions by taking advantage of the properties of the GAL4 protein in Saccharomyces ceravisiae [1]. The native GAL4 protein contains two separable and functionally essential domains: An N-terminal DNA-binding domain (BD) and a C-terminal activation domain (AD). When a protein “X” fused to the GAL4 BD interacts with a protein “Y” fused to the GAL4 AD, it will reconstitute the proximity of the GAL4 domains to function as a transcriptional activator for the expression of genes encoding enzymes of galactose utilization only when X and Y interact in yeast cells [1]. In the past two decades, this assay was greatly modified and developed to suit the needs of the characteristics of the detection proteins as the split-ubiquitin based membrane Y2H [2] or employed as a means of screening cDNA and genomic fusion libraries for protein interaction partners called Y2H library screening [3,4]. Besides GAL4, other complementation proteins including ubiquitin, β-galactosidase and green fluorescent protein or its variants are applied to reconstitute protein activity of two nonfunctional protein portions via their re-association through interacting proteins [2,5-7]. Currently, the Y2H assays are widely used to identify novel protein-protein interactions, confirm suspected interactions, and define interacting domains, although additional biochemical methods are advised because of false positive results in Y2H assay.
In order to perform a Y2H assay, tested proteins need to be fused into a pair of Y2H vectors carrying either the DNA binding domain or the activation domain of the selected complementation protein. The protein fusion step can be done through polymerase chain reaction (PCR) based traditional cloning [8], which was performed by conventional procedures including RE digestion and ligation. The recombined plasmids should be sequenced; and examined for protein expression with western blot before proceeding to the Y2H detection. Now there are a lot of variants of cloning methods including those rely on the commercial kits, such as the GatewayTM system [9] and the In-FusionTM system [10,11], but these high throughout systems are not applicable or affordable to every laboratory, so the traditional cloning method is still prevailing in laboratories worldwide. The traditional cloning method is a common technique which is accessible to all people with basic biology background, but the cloning result is not clear till the last step of cloning and the new learners may have more chances to fail because reasons of failures are often unknown during the process unless proper controls were set up.
We are going to present improved procedures for fusing proteins to Y2H vectors and for performing Y2H assay to determine protein-protein interactions in this study. The methods introduced here will guide people do cloning by confidence on the way instead of by luck at the end, also will greatly shorten the time to obtain Y2H results.
2. MATERIALS AND METHODS
2.1. Strains and Plasmids
Yeast strains: PJ69-4A (MATa trp1-901 leu2-3, 112 ura3-52 his3-200 gal4Δ gal80Δ gal2-ade2 lys2::gal1- his3 met2::gal7-lacZ) and PJ69-4α (MATα trp1-901 leu2-3, 112 ura3-52 his3-200 gal4Δ gal80Δ gal2-ade2 lys2::gal1-his3 met2::gal7-lacZ) [12] were used for hosting activation domain-containing and DNA-binding domain-containing plasmids, respectively. The basal vectors used are pACT2 (LEU2, 2u) (Clontech, CA) and pGBDU-C2 (URA3, 2u) [12]. Other used plasmids are: pACT2-Trs130, pACT2-Trs65 and pACT2-Trs120.
2.2. Cloning Trs23 into pACT2 and pGBDU-C2 Vectors
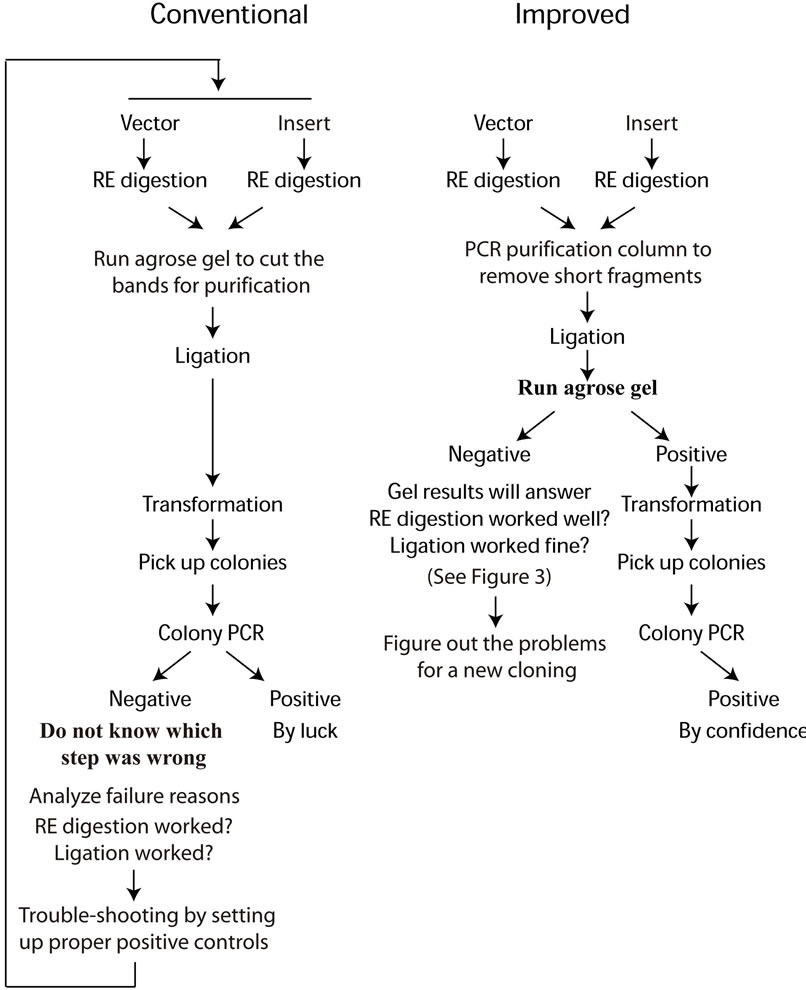
In order to test whether Saccharomyces cerevisiae proteins Trs23 can interact with TRAPP (transport protein particle) II-specific subunits Trs130, Trs65 and Trs120 [13], we cloned TRS23 into both pACT2 and pGBDU-C2 vectors. The primers used for cloning are: primer A (Sma I-TRS23 For) TCCCCCG GGCATGGCCATCGAAACAATACTTGTAATAAAC; primer B (TRS23-Sal I Rev) ACGCGTCGACCCTATTGTA GGTTTTCTACCATTTTTTTGAC. Digestion sites of restriction-enzymes were underlined. TRS23 was amplified at two annealing temperatures (53˚C and 55˚C) as two independent experiments with Pfx polymerase (Invitrogen, CA) according to the manufacturer’s instruction to obtain two independent PCR products for cloning. Vectors and PCR products were digested with Sma I and Sal I (Xho I, which generates the same cohensive end as Sal I, was used to digest pACT2) following the procedures nowadays often omitting the controls in practice as described in Figure 1(a). The improved parts are: 1) Using PCR purification column to clean the short fragments after RE digestion instead of running an agarose gel to cut the desired bands to purify DNA; 2) An agarose gel was run with 4 μL of total 20 μL reaction mixture after ligation for determining the efficiency of RE digestion and ligation, as described in Figure 1(b). Other genes TRS31, BET3 and BET5 for encoding TRAPP I subunits were cloned into both pACT2 and pGBDU-C2 vectors with their corresponding primers in the same way as TRS23 was cloned as described in Figure 1(b).
2.3. Yeast Two Hybrid (Y2H) Assay
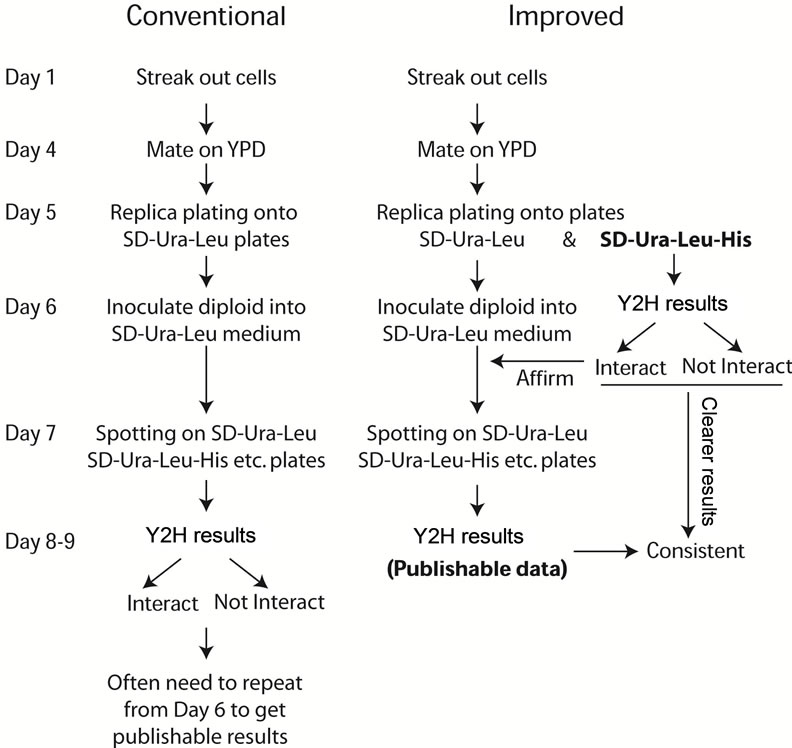
Protein interactions were detected with Y2H assay by mating. Activation-domain containing plasmids were transformed into PJ64-4A strain and DNA-binding-domain containing plasmids were transformed into PJ64-4α strain by the overnight lithium acetate method [14]. The details for the procedures of conventional and improved Y2H assay were described in Figure 2.
3. RESULTS AND DISCUSSION
3.1. Digestion and Ligation Efficiency Can Be Monitored by Running an Agarose Gel after Ligation
Cloning a gene into a vector is a multiple step procedure. Failing in any one of the steps can lead to failure for the whole cloning procedure. In a conventional cloning procedure, after PCR amplification or RE digestion, DNA samples were subjected to agarose gel electrophoresis for isolation and purification of the desired DNA fragment. In order to avoid contamination, it is good practice to wash the chamber and change fresh running buffer every time. When the desired band from the gel was cut under the UV light, there is a chance that the DNA sample may be mutated. In our improved cloning procedure, we use PCR purification kit to remove the unnecessary components and short fragments for both PCR product, and RE digested vector as well as insert DNA samples. Although undigested circular vectors would also be purified by this method, they would not disturb results interpretation of the following steps. This change simplifies the procedure and results in cleaner DNA samples for cloning.
Two other key steps during cloning which often raise problems are RE digestion and ligation. Since cloning has gotten so easy nowadays, people just do not bother to do the controls in practice. But in a conventional cloning procedure without setting up proper controls, if the cloning does not work in the end, it needs to set up proper positive controls from the beginning to monitor the efficiency in each step of cloning. While in our improved cloning method, these questions would be answered in the middle of cloning by running an additional agarose gel after ligation. If both PCR products and vectors were
 (a) (b)
(a) (b)
Figure 1. Schematic flow chart of both conventional (a) and improved (b) cloning procedures.
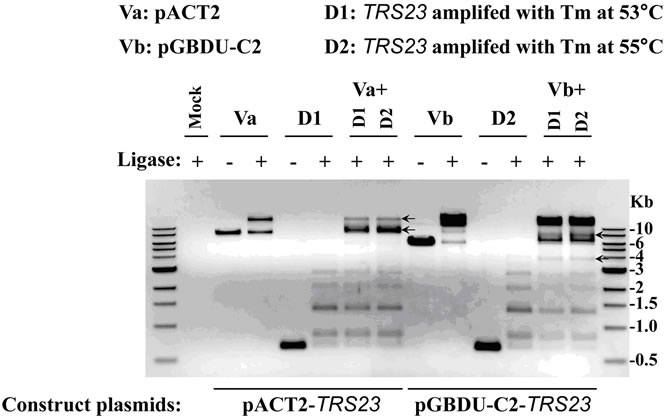
digested with two restriction-enzymes and ligated with DNA ligase properly, theoretically, each type of double digested DNA (vector DNA or insert DNA with the same hangover) will be ligated at both sides to form linear fragments of DNA duplicates at multiple sizes showing as ladders on the agarose gel or to close as circular DNA (with much less chance). In the example demonstrated here, the doubly digested TRS23 DNA (D1 for PCR product amplified at annealing temperature 53˚C and D2 at 55˚C) has a single band at about 600 bp without ligase, but formed ladders with ligase (Figure 3(a)). This is also true for doubly digested vector DNAs, pACT2 (marked as Va) and pGBDU-C2 (marked as Vb), although Va is less efficient in forming bigger concatemers (Figure 3(a)). In case the ligase does not work, ladders cannot be formed on the agarose gel even if the RE digestion works well. DNA will appear as a single band at its original size, as the sample showed in Figure 3(a) without ligase. But if the ligase works, while one of the restriction enzymes does not work, then the DNA may form two bands with the upper one has double size as the lower one which is at its original size. If this situation happened, DNA with single digestion should be included in the ligation step and the ligation mixture should be run on the agarose gel to resolve which RE does not work. These unfavorable situations did not happen in our presented examples. The results in Figure 3(a) clearly show that the two restriction-enzymes we used to digest the inserts and vectors worked well and the ligase added was working. If two types of doubly digested DNAs (vector DNA and insert DNA with the same hangover) were ligated, they may have additional chance to form circular plasmids with one copy of vector DNA and one copy of insert DNA. The recombinant plasmids can be selected by transforming into E. coli. competent cells and growing on bacteria plates supplemented with appropriate antibiotic. The selected plasmids would be verified by colony PCR and sequencing (Figure 1). After the ligation mixture was
 (a) (b)
(a) (b)
Figure 2. Schematic flow chart of both conventional (a) and improved (b) Y2H assay procedures. (a) The traditional Y2H assay procedure includes mating cells on YPD and incubating at 26˚C for 1 day, then replica-plating cells from YPD plates to selective plates SD-Ura-Leu plates to select diploids with incubation at 26˚C for 1 day. The selected diploids were inoculated to liquid SD-Ura-Leu medium overnight at 26˚C and checking interaction on interaction plates (SD-UraLeu-His etc.) at 26˚C for 1 - 2 days. (b) In the improved Y2H assay procedure, when the mated cells were replica-plated onto SD-Ura-Leu to select diploids, meanwhile the cells were replica-plated onto SD-Ura-Leu-His plates to show interactions. At day 6 on the interaction plates (SD-Ura-Leu-His), any strong interaction can be viewed and interaction will be clearer at day 7 - 9. This indicative result will guide the regular frogging step to assure publishable data at day 8 - 9. The results in Figure 4 will demonstrate the benefit of the additional replica-plating to SD-Ura-Leu-His plates helps.
run on the agarose gel, the circular plasmids will show bands different from the duplicated linear DNA fragments formed by each type of doubly digested DNA. In our example, when Va or Vb was ligated with D1 or D2, they can form bands at size and/or with amount different from each DNA alone does as pointed with black arrows (Figure 3(a)). As the gel picture showed, it was promising that the Va + D1/D2 with ligase and Vb + D1/D2 with ligase would produce circular pACT2-TRS23 and pGBDU-C2-TRS23 plasmids respectively, although the band morphology might be plasmid-dependent. In case the bands just appear at the original size or at two-fold size of each double digested DNA, it indicates the restriction-enzymes and/or ligase do not work. If this situation happened, then the cloning procedure should be paused for trouble-shooting to avoid unnecessary waste of time and labor (Figure 1). The results from an agarose gel after ligation can help to predict the final cloning result or adjust the procedures to get the cloning to work.
After the agarose gel for ligation samples showing both the restriction-enzymes and ligase work well (Figure 3(a)), 4 ul of ligation mixture was transformed into E. coli. competent cells for selecting recombinant plasmids. The growing colonies were subjected for colony PCR with positive candidates as shown in Figure 3(b) for two types of plasmids with insert DNA from two independent DNA resources. For either Va + D1/D2 or Vb + D1/D2, 26 out of 27 colonies are positive with an insert DNA successfully cloned into the vectors. Although the ligation efficiency for Va in Figure 3(b) was less obvious than that for Vb, probably by less efficient double digestion on Va, the restriction enzymes and ligase worked
 (a)
(a)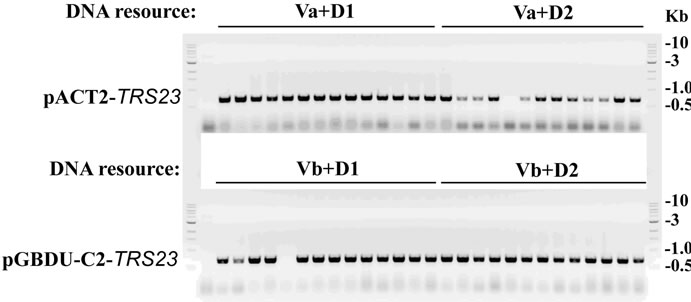 (b)
(b)
Figure 3. Cloning TRS23 into pACT2 and pGBDU-C2 vectors. (a) Ligation efficiency for cloning pACT2-TRS23 and pGBDU-C2-TRS23 plasmids. pACT2 vector was consequentially digested with Sma I and Xho I. pGBDU-C2 vector was consequentially digested with Sma I and Sal I. PCR product of TRS23 amplified from genomic DNA with primer A and primer B as described in materials and methods. Two independent PCR products called D1 and D2 were obtained at two different annealing temperatures 53˚C and 55˚C by using the gradient PCR machine. PCR products were digested with Sma I and Sal I. Then QIAGEN PCR purification kit purified double digestion DNA fragments were ligated with ligase as arranged at the top of panel A. Mixture without ligase was used as control. A control mixture with vector plus insert without ligase gave a combined result of vector plus insert without ligase, which was omitted in the gel. The arrows indicate possible ligation products of vector and insert. (b) Colony PCR to check the colonies containing pACT2-TRS23 or pGBDU-C2-TRS23 plasmids with the same primers as previously used to amplify insert DNA. Thirteen to fourteen colonies were checked for each independent cloning.
well on D1/D2, which was supposed to work on Va and Vb. Under this condition, there is still a chance for a successful cloning (Figure 3(b)). Following the improved cloning methods described in Figure 1, we cloned a few other TRAPP subunits into yeast two hybrid vectors. As summarized in Table 1, the frequencies to get positive colonies are very high. With the improved cloning methods described in Figure 1, we can clone genes to vectors with increased chances for success. At the same time, the modified cloning method can save cloning time from a few days to months over the existing conventional cloning method.
3.2. One Additional Replica-Plating Step Shortens the Y2H Assay
After Y2H plasmids are constructed, they are sequenced to make sure that the genes were cloned in frame. The confirmed plasmids are transformed into corresponding yeast host strains, and frozen at −80˚C.
The Y2H assay can be done through mating by following the procedures as described in Figure 2. We used our improved Y2H assay for determining whether the TRAPP I subunit Trs23 interacts with the three TRAPP II-specific subunits, Trs130, Trs65 and Trs120 [15,16], as an example to demonstrate the improvement of the Y2H
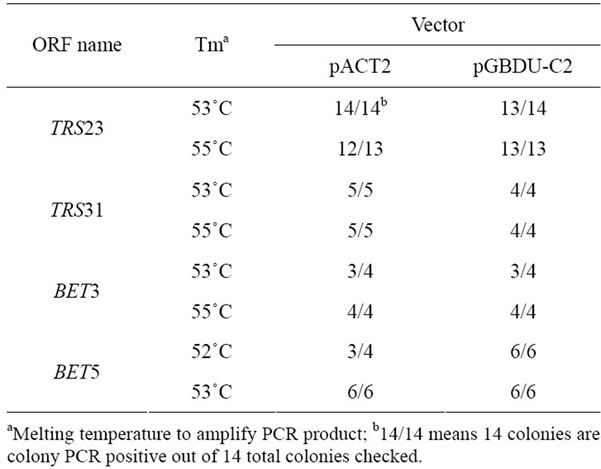
Table 1. Cloning efficiency of some TRAPP I subunits into pACT2 and pGBDU-C2 vectors showed by colony PCR.
procedure and its advantages (Figure 4). By the conventional method, it usually takes about 8 - 9 days to perform a Y2H assay through the mating, replica-plating and frogging process to know the final result at the end of this assay, as shown by the time line at the left side of Figure 2.
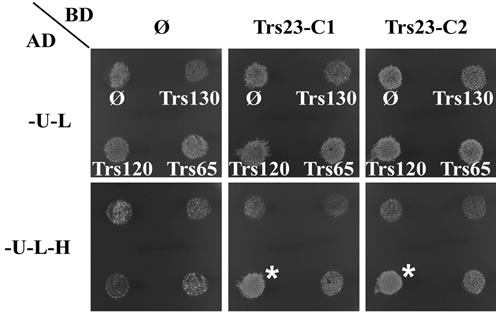
A modification of this conventional procedure will change the general picture of this Y2H assay. Duplicate experiments can be easily set up with the improved method. In the improved Y2H assay, before the mated cells were replica-plated from YPD plates to SD-UraLeu plates, we first replica-plated them to SD-Ura-LeuHis plates for detecting interaction, then to SD-Ura-Leu plates for selecting diploids. This order of operation assures there are enough cells being transferred onto SDUra-Leu-His plates. Usually there are fewer cells transferred onto SD-Ura-Leu plates after the first replicaplating to SD-Ura-Leu-His plates, but they can propagate quickly on SD-Ura-Leu plates. The Y2H interaction results will be available at day 6 through the improved procedure, at least 2 days earlier than that with the conventional method (Figure 2). Moreover, two parallel duplicates can be easily set up as shown in Figure 4(a) for TRS23 in BD domain vectors as marked as TRS23-C1 and TRS23-C2.
One day after the mated cells were replica-plated, the four spots on SD-Ura-Leu plates are similar, but the spot marked with white star on the SD-Ura-Leu-His plates are obviously thicker than the other three spots. There are more cells grow up on SD-Ura-Leu plates for all four spots and on SD-Ura-Leu-His plates for the marked spot two days after replica-plating (Figure 4(a)). These results indicated that diploid cells were successfully selected on SD-Ura-Leu plates and Trs23 interacts with Trs120. Since the duplicate samples TRS23-C1 and TRS23-C2 produce the same result, we can just record these
 (a)
(a)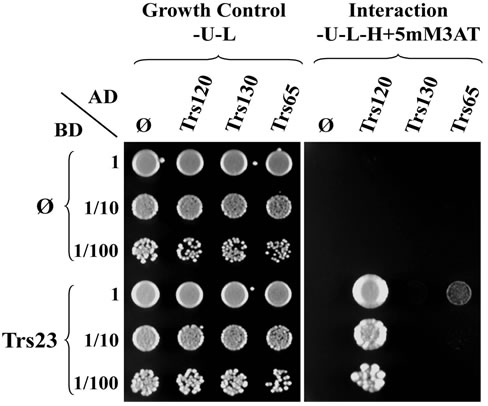 (b)
(b)
Figure 4. Trs23 interacts with Trs120. (a) Growth of diploid cells replica-plated from YPD to -U-L (SD-Ura-Leu) and -U-L-H (SD-Ura-Leu-His) plates. TRS23-C1 and TRS23-C2 represent two cloned independent pGBUD-C2-TRS23 colonies from Figure 3. Mat cells carrying AD plasmids were mated with Mat α cells carrying BD plasmids on YPD and replica-plated onto -U-L and -U-L-H plates. Diploid cells were selected on -U-L plates and grew well there. Only the diploids with interactions between the fusion proteins on the two plasmids were able to grow on -U-L-H plates. In this example, Trs23 interacted with Trs120 so that those diploids could grow on -U-L-H plate as marked with white star. Other spots are just background due to the sticky cells during the replica-plating. (b) Diploid cells selected from the -U-L plated were inoculated into -U-L liquid medium and grown overnight at 26˚C. Then diploid cells were spotted onto growth control plates (-U-L) and interaction plates (only present -U-L-H + 5 mM3AT plates here) with 1:10 serial dilutions from top to bottom. Trs23 strongly interacts with Trs120, weakly interacts with Trs65. These results are generally consistent with the results shown in panel A.
replica-plating results and continue with one set of cells for the following inoculation and spotting steps. From day 6 to day 8, the diploid cells selected from SD-Ura-Leu plates will be preceded exactly as the conventional steps. At the end of this assay, one set of Y2H assay result will be available (Figure 4(b)), which is consistent with the preliminary replica-plating results on SD-Ura-Leu-His plates (Figure 4(a)). Once the Y2H results were further confirmed at the biochemical level, the Y2H is publishable. Our Y2H result of Trs23 interacting with Trs120 is consistent with the earlier result by affinity capture-mass spectrometry [17]. The SD-Ura-Leu plates and SD-UraLeu-His plates can be replaced with other double dropout plates for selecting diploids and other triple dropout plates or variations for detecting interaction, respectively, depending on which Y2H system was used. The biggest advantage of the improved Y2H method is that when the experiment is on the way, the final result is already known so that the experiment can be done more precisely. In contrast, using the conventional method the Y2H assay result is not known until the last step, and it takes longer time if the experiment has to be redone. This modification helps to see the repeatable results earlier and guide the rest of the experiment to save time and resources.
3.3. Summary
In summary, we made two main modifications in the construction of Y2H plasmids and the detection of interactions by Y2H assay. In the cloning step, an additional agarose gel was run after the double digested samples were ligated to examine the efficiency of RE digestion and ligation, so that the success of cloning can be predicted or cloning procedures should be adjusted. In the Y2H assay, an additional replica-plating step on the interaction plates (e.g., SD-Ura-Leu-His) show the results about 2 days earlier, allowing better arrangement for Y2H assay to obtain finalized results 2 - 5 days earlier. This study provides an example to modify the experiment procedures so that the work can be done in shorter time and with increased chances for success.
4. ACKNOWLEDGEMENTS
We thank Drs. N. Segev and Z. Lipotova for reagents and helpful discussions. We thank Dr. S. Yu for critical reading of this manuscript. This research was supported by the Natural Science Foundation of China (31271520), the Research Fund for the Doctoral Program of Higher Education of China (20090097120039), the Project-sponsored by SRF for ROCS, SEM ([2011]508) to Y. Liang, Q. Wang, X. Kang and Y. Liao are recipients of SRT program from both Nanjing Agricultural University (1110A030) and Jiangsu Province (JSS1103).
![]()
![]()
REFERENCES
- Fields, S. and Song, O. (1989) A novel genetic system to detect protein-protein interactions. Nature, 340, 245-246. doi:10.1038/340245a0
- Johnsson, N. and Varshavsky, A. (1994) Split ubiquitin as a sensor of protein interactions in vivo. Proceedings of the National Academy of Sciences of the United States of America, 91, 10340-10344. doi:10.1073/pnas.91.22.10340
- Young, K., Lin, S., Sun, L., Lee, E., Modi, M., Hellings, S., Husbands, M., Ozenberger, B. and Franco, R. (1998) Identification of a calcium channel modulator using a high throughput yeast two-hybrid screen. Nature Biotechnology, 16, 946-950. doi:10.1038/nbt1098-946
- Young, K.H. (1998) Yeast two-hybrid: So many interactions, (in) so little time. Biology of Reproduction, 58, 302- 311. doi:10.1095/biolreprod58.2.302
- Rossi, F., Charlton, C.A. and Blau, H.M. (1997) Monitoring protein-protein interactions in intact eukaryotic cells by beta-galactosidase complementation. Proceedings of the National Academy of Sciences of the United States of America, 94, 8405-8410. doi:10.1073/pnas.94.16.8405
- Stagljar, I., Korostensky, C., Johnsson, N. and te Heesen, S. (1998) A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo. Proceedings of the National Academy of Sciences of the United States of America, 95, 5187-5192. doi:10.1073/pnas.95.9.5187
- Hu, C.D. and Kerppola, T.K. (2003) Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nature Biotechnology, 21, 539-545. doi:10.1038/nbt816
- Saiki, R.K., Gelfand, D.H., Stoffel, S., Scharf, S.J., Higuchi, R., Horn, G.T., Mullis, K.B. and Erlich, H.A. (1988) Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science, 239, 487- 491. doi:10.1126/science.2448875
- Landy, A. (1989) Dynamic, structural, and regulatory aspects of lambda site-specific recombination. Annual Review of Biochemistry, 58, 913-949. doi:10.1146/annurev.bi.58.070189.004405
- Au, K., Berrow, N.S., Blagova, E., Boucher, I.W., Boyle, M.P., Brannigan, J.A., Carter, L.G., Dierks, T., Folkers, G., Grenha, R., Harlos, K., Kaptein, R., Kalliomaa, A.K., Levdikov, V.M., Meier, C., Milioti, N., Moroz, O., Muller, A., Owens, R.J., Rzechorzek, N., Sainsbury, S., Stuart, D.I., Walter, T.S., Waterman, D.G., Wilkinson, A.J., Wilson, K.S., Zaccai, N., Esnouf, R.M. and Fogg, M.J. (2006) Application of high-throughput technologies to a structural proteomics-type analysis of Bacillus anthracis. Acta Crystallographica Section D: Biological Crystallography, 62, 1267-1275. doi:10.1107/S0907444906033555
- Berrow, N.S., Alderton, D., Sainsbury, S., Nettleship, J., Assenberg, R., Rahman, N., Stuart, D.I. and Owens, R.J. (2007) A versatile ligation-independent cloning method suitable for high-throughput expression screening applications. Nucleic Acids Research, 35, e45. doi:10.1093/nar/gkm047
- James, P., Halladay, J. and Craig, E.A. (1996) Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics, 144, 1425-1436.
- Liang, Y., Morozova, N., Tokarev, A.A., Mulholland, J.W. and Segev, N. (2007) The role of Trs65 in the Ypt/Rab guanine nucleotide exchange factor function of the TRAPP II complex. Molecular Biology of the Cell, 18, 2533-2541. doi:10.1091/mbc.E07-03-0221
- Gietz, D., Stjean, A., Woods, R.A. and Schiestl, R.H. (1992) Improved method for high-efficiency transformation of intact yeast-cells. Nucleic Acids Research, 20, 1425-1425. doi:10.1093/nar/20.6.1425
- Sacher, M., Barrowman, J., Schieltz, D., Yates 3rd, J.R. and Ferro-Novick, S. (2000) Identification and characterization of five new subunits of TRAPP. European Journal of Cell Biology, 79, 71-80. doi:10.1078/S0171-9335(04)70009-6
- Sacher, M., Barrowman, J., Wang, W., Horecka, J., Zhang, Y., Pypaert, M. and Ferro-Novick, S. (2001) TRAPP I implicated in the specificity of tethering in ER-to-Golgi transport. Molecular Cell, 7, 433-442.
- Yip, C.K., Berscheminski, J. and Walz, T. (2010) Molecular architecture of the TRAPPII complex and implications for vesicle tethering. Nature Structural & Molecular Biology, 17, 1298-1304. doi:10.1038/nsmb.1914
NOTES
*Corresponding author.