Health
Vol. 4 No. 9 (2012) , Article ID: 23063 , 4 pages DOI:10.4236/health.2012.49099
Atypical presentations of propionic acidemia
![]()
Department of Pediatrics, Children’s Hospital, King Fahad Medical City, KSA; *Corresponding Author: al_makadma@hotmail.com
Received 8 June 2012; revised 10 July 2012; accepted 27 July 2012
Keywords: Acidemia; Atypical Presentation; Hyperammonemia; Organic; Propionic; Propionyle-CoA Carboxylase
ABSTRACT
One of the most common recessively inherited organic acidemias is the Propionic Acidosis (PA) which results from Propionyle-CoA Carboxylase (PCC) enzyme deficiency that is necessary for the catabolism of the branched chain Amino Acids and other metabolites. Classically this disease presented with high anion gap metabolic acidosis with its clinical consequences. We report 4 patients who presented to our facility with sepsis like picture and no metabolic acidosis. All of them were found to have high ammonia level. Diagnosis was confirmed by tandem MS/MS and urine Gas Chromatography/Mass Spectrometry (GC/MS). All of them were treated supportively and by supplementation of adequate calories and PA formula. The different presentations may be very well attributed to the PCC molecular defects heterogeneity. Mutations in both genes PCCA and PCCB can cause PA with more frequent heterogeneity of PCCA gene. In spite of the fact that PCCB gene is responsible for the most of the oriental cases, our first patient condition was attributed to PCCA gene with a rare mutation which was not described in the literatures.
1. INTRODUCTION
One of the most common recessively inherited organic acidemias is the Propionic Acidosis (PA) [1]. Normally the isoleucine, valine, methionine, and threonine aminoacids as well as odd-chain fatty acids and cholesterol are degraded to propionyl-CoA where propionyl-CoA carboxylase would catalyze further conversion to methylmalonyl-CoA. In propionic acidemia, deficient activity of propionyl-CoA carboxylase prevents this conversion The pathogenesis is due to deficiency of the mitochondrial enzymes Propionyle-CoA Carboxylase (PCC) that leads to accumulation of Propionic acid leading to biochemical features including profound metabolic acidosis, hypoglycemia, and hyperammonemia [2-4]. The PCC is a heteropolymeric enzyme which is composed of 2 sub units α and β in a proposed α6β6 configurative [5-9]. Affected patients fall in 3 groups; PCCA, PCCB and PCCBC (PCCB & PCCC) [8]. The pccBC group possibly reflects mutations of the α-subunit while mutation of β-subunits is reflected by group pccA [9,10]. While the incidence of PA estimated internationally to be 1:50000 at [5], that in the Kingdom of Saudi Arabia (KSA) is estimated to be 1:2000 to 1:5000 depending on the region [11,12].
Most of the affected babies, if not treated, die in the neonatal period or during the first 10 years of life [13,14]. The clinical presentations may start during the first 3 months of life (early onset). About 60% of cases present during the neonatal period, while few of them present as late onset.
The presentation may include lethargy, vomiting, hypotonia, convulsions, decreased activity or loss of consciousness with evidence of high anion gap metabolic acidosis, which is one of the most constant biochemical findings [3,15-17].
Both early and late onset PA can cause severe and permanent neurological damage [17,18]. The degree of neurological insult is related to the degree of Hyperammonemia [17]. Urine Gas Chromatography/Mass Spectrometry (GC/MS) & Mass Spectrometry (MS/MS) and/ or fibroblast culture for enzymatic assay may be used for definite diagnosis [2,12].
2. PATIENTS AND METHODS
We retrospectively reviewed the charts of pediatric patients who were admitted with the diagnosis Propionic Acidosis. Twenty three of those patients were identified. 4 patients were excluded because there was no enough documentation about the initial presentation.
Data like the Age at presentation, sex, clinical picture, the pH, serum bicarbonate, Serum ammonia, white blood cell, Red blood cell and platelets counts were collected.
The results for the Tandem MS and Urine GS/MS were collected as well.
3. RESULTS
Out of those 19 patients there were only four patients who were diagnosed as PA without Acidosis at the time of presentation. They were 3 females and one male. 3 out of them were the product of consanguineous marriage. All of them presented during the first month of life. The first case was referred only because of the significant family history and decreased activity, the second one presented with seizures and normocytic normochromic anemia, the third had septic like picture and pancytopenia while the fourth one presented with respiratory distress and leukocytosis. All of them had normal pH and high serum ammonia. For all of them, the diagnosis was confirmed by using Tandem MS/MS and urine gas chromatography/mass spectrometry (GC/MS).
A brief report about each case will be discussed below and a summary will be shown in (Table 1).
4. PATIENTS: (TABLE 1)
4.1. Case #1
A 25 days old female a product of non-consanguineous marriage after full term uneventful pregnancy delivered normally with birth weight of 2.9 Kg and normal Apgar score. She was referred to our institute because of a significant family history (FH) suggestive of metabolic disorder; 3 of her brothers died at the age of 40 days and the fourth one at the age of 4 years, one sister died at the age of 8 months and a second one at the age of 5 years, for all of them there was no definite diagnosis.
When first seen she was fairly hydrated, a febrile, growing well and not dysmorphric but was inactive. Otherwise there were unremarkable findings on her sys-
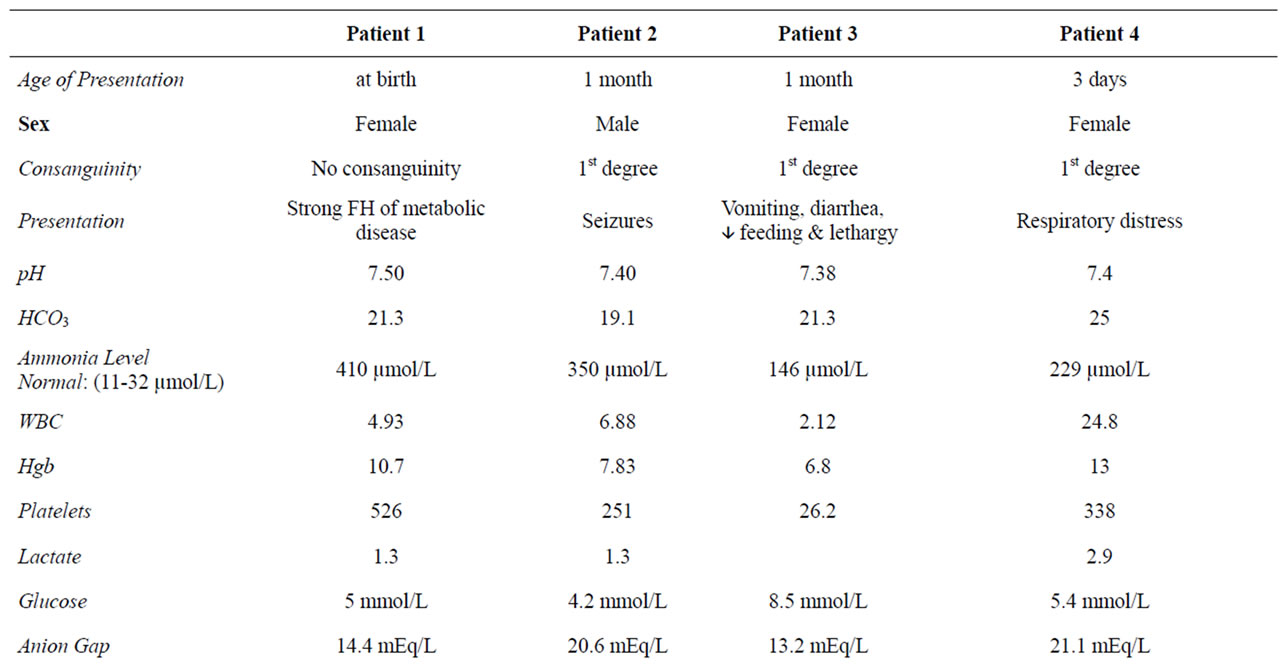

Table 1. Summary for the 4 cases showing the different presentations, investigations and outcome.
temic examination. Her hematological and biochemical results are as shown in Table 1.
Her DNA analysis showed two sequence variants (c.111-130del) and (c.136A > G) were detected in both copies of PCCA gene (homozygous). The first variant is a deletion of 20 nucleotides on exon 2 that results in a frame shift of the coding sequence and leads to a premature termination codon. The second variant (c.136A > G) is a replacement of an A nucleotide by a G nucleotide that is located in the frame shifted sequence downstream of the first mutation.
She was treated in a supportive way (IVF, caloric supplementations; PA formula) and L-Carnitine and biotin. She did will and was discharged in good general condition and normal ammonia level. She continued to thrive well and develop normally during her follow up period for 2 years where she didn’t have any relapse and her plasma ammonia and electrolytes levels were maintained normal. But unfortunately she died later on a stormy course of respiratory infection during the last season of H1N1 pandemic (December 2009).
4.2. Case #2
A 7 months old male, who is the first child for the family and is a product of a consanguineous marriage after a full term uneventful pregnancy and normal vaginal delivery with birth weight of 3 Kg and no perinatal complications.
At the age of one month he was admitted in the local hospital because of generalized tonic clonic convulsions. His septic workup, complete blood count, electrolytes, was unremarkable. His serum ammonia was 350 µmol /L. He was treated supportively. One month later he presented to our institute with history of recurrent convulsion with decreased activity and feeding. When examined, he was active, a febrile, and well hydrated with unremarkable systemic findings. Initial investigation revealed that his complete blood count, electrolytes, renal, and liver function tests were within normal limits. The Tandem MS, the urine organic acids profile (GCMS) confirmed the diagnosis of PA.
His DNA analysis showed a sequence variant (c.990_991InsT) in both copies of the PCCB gene (homozygous). Between nucleotides 990 and 991 an insertion of T nucleotide was detected. It leads to the substitution of Glutamic acid at amino acid position 331 by a stop codon (E331X).
Beside the supportive treatment, he was treated by sodium benzoate, L-Carnitine and Biotin. He did well and was discharged home in a good condition on Sodium Benzoate and PA formula.
His followed up for 11 months was satisfactory with reasonable growth and development without any relapses.
4.3. Case#3
A two-month old female baby, a product of a consanguineous marriage after a full term uneventful pregnancy was born by cesarean section because of breech presentation with birth weight of 2.5 Kg and normal Apgar score. After one month, she started to have episodes of fever, vomiting, and diarrhea for which she had no specific management in primary health care clinic to be transferred to our facility at the age of 2 months where she was afebrile, not in distress and apart from being lethargic and not growing well the rest of her clinical examination was normal. She was found to be leukopenic, anemic and thrombocytopenic. Her bone marrow examination ruled out hematological causes. Her septic workup was unremarkable. Her bicarbonate was 22 mmol/L and her serum Ammonia level was 410 µmol/L. The diagnosis of PA was confirmed after the results of Tandem MS/MS and urine gas chromatography/mass spectrometry (GC/MS). She was started on, Na Benzoate, L-Carnitine and Biotin beside the supportive treatment and transfusion of PRBC and platelets. Gradually she was clinically improving and her Ammonia level dropped to 28 (21 - 50 µmol/L) she was discharged home after improvement on Biotin, Na Benzoate, Carnitine, and PA formula. We did not make any molecular study for her.
Unfortunately, two months later we came to hear that she died in her local hospital because of sepsis.
4.4. Case #4
This is a 2 years old female, a product of consanguineous marriage with unremarkable family history. She was delivered after full term uneventful pregnancy by cesarean section because of previous C-sections with birth weight of 3.6 Kg and normal Apgar Scores. She was doing fine till 3rd day of life when she developed respiratory distress and was admitted to the NICU where she was diagnosed to have heart failure due to significant patent ductus arteriosus with bidirectional shunt.
Her initial investigations were unremarkable. Tandem Mass Spectrometry and Urine Organic Acid profiles confirmed the diagnosis of PA. She was stabilized then discharged home in good condition after two weeks. At age of 23 days, she presented to our hospital with history of vomiting, diarrhea, lethargy, and decreased feeding. Her initial workup showed normal complete blood count, electrolytes, and liver function tests. Her pH was 7.4. She had blood ammonia level of 229 µmol.
Her molecular study showed a sequence variant (c.425G > A) was detected in both copies of PCCA gene (homozygous). It results in the substitution of Glycine at amino acid position 142 by Aspartic acid (G142D).
She was treated by Na Benzoate, L-Carnitine, biotin and the usual supportive treatment. Her condition improved gradually and her blood ammonia dropped to57 µmol/L. She was discharged home after three days on L-Carnitine, Biotin and PA formula. Her condition remained clinically stable without relapse or elevation of her ammonia level for the last three years.
5. DISCUSSION
These unusual presentations involving different systems highlight the importance of high index of suspicion when dealing with general, neurological, hematological and/or gastrointestinal presenta-tions which cannot be explained by definite disease.
Although Propionic acidosis typically presents with high anion gap metabolic acidosis, we encoun-tered 4 patients who presented without acidosis. Yet the diagnosis of P.A. was confirmed.
We can notice in the 4 cases described above the variability of the initial presentation, while the first case was referred because of the very significant family history, the second was admitted because of the clonic seizures and the third case was admitted mainly with gastrointestinal presentation to be found after admission to have pancytopenia. The reason for the fourth patient to be admitted was severe respiratory distress.
None of the four cases had a pH less than 7.38 while all of them had high serum ammonia level.
In all, the diagnosis of P.A. was established by the results of tandem MS/MS and urine gas chromatography/mass spectrometry (GC/MS). All of the cases responded very well to the supportive treatment, special formulas, Biotin and Carnitine. All of them did well initially without relapses but unfortunately two of them died later on; the first one died at the age of 2 years due to respiratory distress on December 2009 during the outbreak of H1N1 pandemic, the third one died because of sepsis at the age of 4 months.
Mutation in PCCA gene on chromosome 13q32 and PCCB on chromosome 3q21-22 encoding both subunits of PCC results in PA.
We performed molecular studies for our patients for two reasons, the first being to confirm our diagnosis and to correlate it with the phenotype, and the second is to provide family counseling.
Although the (homozygous) PCCA gene is more common in the Arab population, the second case in our series was homozygous for the PCCB, and the first case had a homozygous PCCA gene, yet with a mutation that was not reported in literature. There were two sequence variants as described in Table 1. The first variant is deletion of 20 nucleotides on exon 2 that resulted in frame shift of the coding sequence leading to a premature termination codon. The second variant (c.136A > G) is a replacement of an A nucleotide by a G nucleotide that is located in the shifted frame sequence downstream of the first mutation.
These mutations were not reported in literature. However, the fact that the first one results in a frame shift and a premature termination codon makes it highly suggestive of a pathogenic mutation. We did not perform the molecular study on the third case due to the early death and the non-availability of the parental consent upon first encounter.
6. CONCLUSIONS
Absence of acidosis should not be considered as an exclusion criterion for Propionic acidosis especially in highly suspected or unclear cases presented with acute symptoms. The presence of hyperammonemia should increase the suspicion of PA diagnosis in such patients Diagnosis can be confirmed by using tandem MS/MS and urine gas chromatography/mass spectrometry (GC/ MS). Early recognition and proper management can decrease the morbidity and mortality.
Although the homozygous PCCA is known to be more common in the Arabic population, our series harbors different mutations on different genes. We describe new mutations for the PCCA gene for the first time in the Arab patient population (as seen in the first case).
![]()
![]()
REFERENCES
- Ogier, H., Charpentier, C. and Saudubray, J.M. (1990) Organic acidemias. In: Fernandes, J., Saudubray, J.M. and Tada, K., Eds., Inborn Metabolic Diseases, Springer-Verlag, Berlin, 271-299.
- Rosenbery, L.E. and Fenton, W.A. (1989) Disorders of propionate and methylmalonate metabolism. In: Scriver, C.R., Beaudet, A.L. and Valle, D., Eds., The Metabolic Basis of Inherited Disease, 6th Edition, McGraw-Hill, New York, 821-844.
- Sass, J.O., Hofmann, M., Skladal, D., Mayatepek, E., Schwahn, B. and Sperl, W. (2004) Propionic acidemia revisited: A workshop report. Clinical Pediatrics, 43, 837- 843. doi:10.1177/000992280404300908
- Wolf, B., Hsia, Y.E., Sweetman, L., et al. (1981) Propionic acedimia: A clinical update. Journal of Pediatrics, 99, 835-846. doi:10.1016/S0022-3476(81)80004-2
- Gravel, R.A., Lam, K.F., Mahuran, D. and Kronis, A. (1980) Purification of human liver propionyl-CoA carboxylase by carbon tetrachloride extraction and monomeric avidin affinity chromatography. Archives of Biochemistry and Biophysics, 201, 669-673. doi:10.1016/0003-9861(80)90557-3
- Kalousek, F., Darigo, M.D. and Rosenberg, L.E. (1980) Isolation and characterization of propionyl-CoA carboxylase from normal human liver: Evidence for a protomeric tetramer of non-identical subunits. Journal of Biological Chemistry, 255, 60-65.
- Lamhonwah, A.M., Troxel, C.E., Schuster, S. and Gravel, R.A. (1990) Two distinct mutations at the same site in the PCCB gene in propionic acidemia. Genomics, 8, 249-254. doi:10.1016/0888-7543(90)90279-4
- Urgarte, M., Perez-Cerda, C., Rodriquez-Pombo, P., Desviat, L.R., Perez, B., Richard, E., et al. (1999) Overview of mutations in the PCCA and PCCB genes causing propionic acidemia. Human Mutation, 14, 275-282. doi:10.1002/(SICI)1098-1004(199910)14:4<275::AID-HUMU1>3.0.CO;2-N
- Ohura, T., Miyabashi, S., Narisawa, K. and Tada, K. (1991) Genetic heterogeneity of propionic acidemia: Analysis of Japanese patients. Human Genetics, 87, 41- 44. doi:10.1007/BF01213089
- Ozand, P.T., Rashed, M., Gascon, G.G., Youssef, N.G., Harfi, H., Rahbeeni, Z., Al Garawi, S. and Al Aqeel, A. (1994) Unusual presentations of propionic acidemia. Brain and Development, 16, 46-57. doi:10.1016/0387-7604(94)90096-5
- Al Essa, M., Rahbeeni, Z., Jumaah, S., Joshi, S., Al Jishi, E., Rashed, M.S., Al Amoudi, M. and Ozand, P.T. (1998) Infectious complications of propionic acidemia in Saudia Arabia. Clinical Genetics, 54, 90-94. doi:10.1111/j.1399-0004.1998.tb03702.x
- Rashed, M. (2001) Clinical applications of tandem mass spectrometry: Ten years of diagnosis and screening for inherited metabolic diseases. Journal of Chromatography, 758, 27-48.
- Leonard, J.V., Daish, P., Naughten, E.R. and Bartlett, K. (1984) The management and long term outcome of organic acidaemias. Journal of Inherited Metabolic Disease, 7, 13-17. doi:10.1007/BF03047367
- Surtees, R.A., Matthews, E.E. and Leonard, J.V. (1992) Neurologic outcome of propionic acidemia. Pediatr Neurol, 8, 333-337. doi:10.1016/0887-8994(92)90085-D
- Lucke, T., Perez-Cerda, C., Baumgartner, M., et al. (2004) Propionic acidemia: Unusual course with late onset and fatal outcome. Metabolism, 53, 809-810. doi:10.1016/j.metabol.2003.12.025
- North, K.N., Korson, M.S., Gopal, Y.R., et al. (1995) Neonatal onset propionic acidemia: Neurologic and developmental profiles, and implications of management. Journal of Pediatrics, 126, 916-922.
- Tokatli, A., Coskun, T. and Ozalp, I. (1993) A retrospective evaluation of 78 cases with organic acidemia. Turkish Journal of Medical Sciences, 18, 47-53.
- Desviat, L.R., Pérez, B., Pérez-Cerdá, C., RodríguezPombo, P., Clavero, S. and Ugarte, M. (2004) Propionic acidemia: Mutation update and functional and structural effects of the variant alleles. Molecular Genetics and Metabolism, 83, 28-37. doi:10.1016/j.ymgme.2004.08.001