Advances in Bioscience and Biotechnology
Vol.3 No.4A(2012), Article ID:21889,13 pages DOI:10.4236/abb.2012.324071
Sickle cell disease: Only one road, but different pathways for inflammation
![]()
1Centro de Pesquisas Gonçalo Moniz, Fundação Oswaldo Cruz (FIOCRUZ), Salvador, Bahia, Brasil
2Instituto Nacional de Ciência e Tecnologia do Sangue, São Paulo, Brasil
3Universidade Estadual de Santa Cruz (UESC), Ilhéus, Bahia, Brasil
4Faculdade de Farmacia da Universidade Federal da Bahia (UFBA), Salvador, Bahia, Brasil
Email: *mari@bahia.fiocruz.br
Received 1 July 2012; revised 2 August 2012; accepted 16 August 2012
Keywords: Sickle Cell Disease; Inflammation; Oxidative Stress; Cells Activation
ABSTRACT
Sickle cell disease (SCD) is a genetic disorder characterized by a chronic inflammatory process, and new biomarkers have been studied as promising molecules for understanding the inflammation in its pathophysiology. The hemolysis and the release of molecules associated to the hemoglobin (Hb) catabolism, such as free Hb, iron, and heme, generate an oxidant environment with production of reactive oxygen and nitrogen species. The immune system plays a very important role in the inflammation, with cells secreting pro-inflammatory cytokines and chemokines. There is also a nitric oxide (NO) resistance state, with an impaired NO bioactivity, leading to a vascular dysfunction; activation of platelet, leukocytes, erythrocytes, and endothelial cells, with expression of adhesion molecules and its ligands, and several receptors, that altogether participate at inflammatory process. During inflammation, there is an increase of dendritic cells (DCs) expressing toll like receptors (TLR), but the role of DCs and TLR in SCD pathogenesis is unclear. Also, there are molecules contributing for enhance the endothelium dysfunction, such as homocysteine that has been associated with vascular complications in the pathology of other diseases and it may contribute to the vascular complications presented by SCD patients. Circulating microparticules (MPs) levels are augmented in several diseases and have been described in SCD, where cells membrane compounds are associated to cell’s thrombotic and coagulation state, such as tissue factor and phosphatidylserine (PS), which may contribute to endothelial dysfunction. The knowledge of all these biomarkers may contribute to new therapeutic approach discover, improving SCD patient life quality.
1. INTRODUCTION
Sickle cell disease (SCD) is a genetic disorder, and the sickle cell anemia (HbSS) is the more severe genotype. The disease is characterized by the presence of the hemoglobin S (HbS), where valine replace glutamic acid (bS 6 Glu→Val) at the beta globin chain, that has a single point mutation (GAG →GTG) at the sixth codon of the b-globin (HBB) gene [1].
Sickle cell disease clinical outcome vary widely from mild to severe and has been associated with multi-organ damage and risk of early mortality [1], with acute and chronic clinical manifestations, including vaso-occlusive episodes (VOE), painful crisis, tissue ischemia/reperfusion injury, hemolysis, impaired blood flow as a result of intravascular sickling in capillary and vessels, inflammation processes and high susceptibility to infection, encephalic vascular accident (EVA), dactylitis, leg ulceration, pulmonary hypertension, acute chest syndrome, and priapism [1,2].
Moreover, the disease pathogenesis comprehends a complex network of mechanisms, involving the vasoocclusive phenomenon and tissue ischemia, with surface and ligands molecules activation from stressed reticulocytes, sickled erythrocytes, leukocytes, platelets and endothelial cells [1-3]; there is also an increase of oxidative stress, secondary to the hemolysis episodes and heme cytotoxicity, electron donation from the iron atom when yet inside the protoporphyrin IX ring, with the generation via Fenton reaction of reactive oxygen and nitrogen species (ROS; RNS), that has a very strong pro-oxidant capacity. Also, there is an increase of nitric oxide (NO) scavenger molecules, a vasodilator that play important role as regulators of vascular homeostasis in SCD pathogenesis [1,4].
Acute and chronic inflammatory phenomenon’s can contribute to activate several cells types and may play important role in the steadyand crisis-states of SCD patients. The immune system pathway, has several mechanisms and needs to be better understood, including the participation of inflammation and cells markers, activation of molecules related to hemolysis, nitric oxide resistance, and some very important inflammatory mediators involved in the arachidonic acid pathway, including the synthesis of molecules such as, prostaglandin E2 (PGE2), thromboxane, and leukotrienes B4 (LTB4) [1].
There are many chemistry and genetic markers, which can modulate symptoms presented by SCD patients, such as alpha-thalassemia presence, reticulocytes count, lactate dehydrogenase (LDH) and bilirubin serum levels [5]. Currently, new biomarkers have been studied as promising molecules for understanding inflammation process in SCD, and have been highlighted the role of lipids metabolism and its participation in vascular injury, C-reactive protein (CRP) and inflammation, and the myeloperoxidase (MPO), a enzyme that had been related with patients susceptibility to infection [6-8].
Bilirubins are resulted of protoporphyrin IX metabolism, which in turn is a heme component. Sickle cell disease patients, particularly those HbSS, are at risk for bile pigment cholelithiasis due to the association of this disease with hemolysis, which produces an unconjugated hyperbilirubinemia [9]. Cholecystitis presents with abdominal pain, nausea and vomiting, fever, and/or jaundice, a constellation of symptoms that has multiple possible etiologies in SCD [10].
C-reactive protein (CRP), an acute-phase protein, increases significantly in inflammatory disorders and nowadays CRP has been used for evaluation of cardiac risk. CRP is produced not only by the liver but also in atherosclerotic lesions by vascular smooth muscle cells and macrophages in response to stimulation by the pro-inflammatory cytokine interleukin-6 (IL-6) [11]. SCD is associated with elevated cardiac output and cardiomegaly to partly compensate for the reduced oxygen-carrying capacity associated to hemolysis and oxidative stress. The combination of these events has been associated with increased levels of CRP in SCD since the childhood [12-14].
Myeloperoxidase is a lysosomal enzyme and plays an important role in the host defense system. MPO deficiency was associated with a higher occurrence of severe and chronic inflammatory processes in SCD patients, and the −463G > A MPO gene polymorphism may be a significant genetic modulator that makes HbSS patients more susceptible to infection [13].
An association between increased low-density lipoprotein cholesterol (LDL-C) and low plasma levels of highdensity lipoprotein cholesterol (HDL-C) is an important risk factor for coronary disease. Actually it has been showed an association between coronary heart disease, high levels of LDL-C and MPO, since MPO catalyses the conversion of chloride and hydrogen peroxide (H2O2) to hypochlorous acid (HOCl), resulting in LDL-C oxidation and conversion into high-uptake forms, such as oxLDL for macrophages, leading to cholesterol deposition and foam cell formation in vivo [14]. Data from our research group showed that some SCD patients can have a specific dyslipidemic subphenotype, characterized by low HDL-C with hypertriglyceridemia and high very low density lipoprotein cholesterol (VLDL-C) in association with other biomarkers, including those related to inflammation like ferritin and CRP [15]. These biomarkers may help in understanding the inflammatory mechanism associated with SCD as well as be used as predictor tests for severe events.
2. VASCULAR DYSFUNCTION AND INFLAMMATION: NITRIC OXIDE SCAVENGING AND ARGININE METABOLISM
In regard to the vascular complication of the HbSS, the decrease of nitric oxide (NO) bioavailability is now associated with the intravascular hemolysis [16], that participate in several important complication of HbSS patients, including pulmonary hypertension, leg ulcers, priapism and different types of stroke [16-18].
The nitric oxide is a diatomic gas produced by vascular endothelial cells that act as a potent vasodilator on smooth muscle cells. The NO synthesis is from the amino acid L-arginine, via an oxidation reaction catalyzed by the enzyme nitric oxide synthase (NOS) [19]. Nitric oxide also tonically inhibits platelet activation and the expression of endothelial adhesion molecules, thus participating in health endothelial function and in the maintenance of blood flow [20,21]. The reaction involveing vascular NO can have a beneficial antioxidant effect. Moreover, NO has been demonstrated to have a cytoprotective effect by scavenging reactive oxygen species (ROS) [22]. Solovey et al. [23] examined the hypothesis that enhanced endothelial tissue factor (TF) expression is modulated by endogenous NO produced by endothelial nitric oxide synthase (eNOS) in animal models. The mechanism by which NO exerts its inhibiting effect on TF have not been completely defined, although it is accompanied by parallel changes in amount of TF mRNA. Because NO exerts the same regulatory influence on vascular cellular adhesion molecule-1 (VCAM-1) expression, current results also have implications beyond inflammation and coagulation system, but also with VCAM-1 inflammatory expression.
Chronic elevated level of cell-free hemoglobin in SCD patients with intravascular hemolysis range from 2 to 20 uM per heme during steady-state and increase to approximately 20 to 40 uM during vaso-occlusive crises [24]. HbSS patients present a NO resistance state, associated with an impaired NO bioactivity that can be due by cell-free hemoglobin accumulation in plasma, intensifying the NO endogenous consumption that contribute for several cellular dysfunction, such as vasoconstriction and inflammation by leukocytes, platelets and endothelial cells activation [25]. Also, ROS are generated by hemolysis, and can react with NO, limiting its bioavailability and contributing to its state of resistance in HbSS patients [18]. The heme that is released from hemolysis also induces the expression of adhesion molecules from leukocytes, such as intercellular adhesion molecules-1 (ICAM-1), and from endothelium, such as VCAM-1.
Another mechanism of NO depletion during the hemolysis is the release of arginase-1 from lysed RBC that converts arginine to ornithine, which competes with the substrate, the eNOS, for L-arginine synthesis [26]. In recent research by our team, we confirm an increased levels of serum arginase-1 in HbSS patients when compared to health controls, as well as an association of serum arginase-1 with biochemical hemolysis markers and cytokines involved in Th17 response, levels of soluble intercellular adhesion molecule-1 (sICAM-1) and soluble vascular cell adhesion molecule-1 (sVCAM-1) [27]. A very new insight in this metabolism lays on a shift in arginine catabolism, where transforming grow factor beta (TGF-beta) may induces the arginase pathway instead of the NO pathway, with a possible involvement of the vascular activation and the increase of serum arginase in chronic hemolysis among HbSS patients.
3. HOMOCYSTEINE AND SICKLE CELL DISEASE
Homocysteine (Hcy), a sulfur-containing amino acid, is found at low concentration in blood and cells and is an important intermediate molecule involved in the biosynthesis of methionine and cysteine [28]. The high plasma concentration of Hcy is a well-established risk factor for several disorders, including cardiovascular disease (CVD) and stroke [29], venous thrombosis and arteriosclerosis [30].
Hyperhomocysteinemia play an important role in vascular disorders and may act through increase cytotoxic activity, especially for endothelial cells; elevating H2O2 levels and decreasing NO synthesis, with pro-inflammatory cytokines synthesis, pro-coagulant factors activetion, and lipid metabolism dysregulation, characterized by LDL-C oxidative modification, enhancing of atherogenesis [31]. High levels of Hcy have also been implicated in changes in the rheological properties of blood, such as decreasing antithrombin III and tissue plasminogen activator (TPA), and increasing factor VII and CRP [32]. Additionally, Hcy is reported to enhance endothelial leukocyte interactions [33].
A chronic inflammatory state in vascular tissue is recognized to contribute to thrombotic and vaso-occlusive events in HbSS patients [34]. Since Hcy has been associated with vascular complications in the pathophysiology of other disease, it may contribute to vascular complications presented by HbSS patients.
The possibility that Hcy may contribute to the ischemic phenomena present in HbSS has attracted some interest in plasma total Hcy. Lowenthal et al. [31] showed that the median plasma concentration of Hcy among HbSS subjects was approximately 1.5-fold higher than that of healthy controls. Additionally, SCD patients have higher plasma Hcy concentration in spite of elevated plasma folate levels and vitamin B12 concentration similar to those observed in controls. In a recent study (2012) from our team, we found significant associations between Hcy levels and increased expression of proinflammatory cytokines and adhesion molecules (data not published) in HbSS patients, supporting the hypothesis that Hcy levels contribute to the vascular activation and with the inflammatory state presented by HbSS patients, and probably has an important role in vaso-occlusive mechanism.
4. INNATE IMMUNITY AND INFLAMMATION IN SCD
Individuals with SCD have transient and periodic painful vaso-occlusive episodes with exposure of organ to ischemia and reperfusion, which may activate inflammatory response in other organs, leading to multiple organ failure [35,36]. Despite ischemia and reperfusion occur in a sterile environment, and activation of innate and adaptive immune responses contribute to injury, including activetion of pattern-recognition receptors, such as Toll like receptors (TLRs) and inflammatory cell trafficking into the damaged organ [37]. Moreover, is well known that the presence of immunodeficiency should be associated with SCD, but no directly deficiency has been related to the immune system was observed to explain the amount of recurrent infections presented by these individuals [34]. On the basis of these, the immune response system seems to be related with health and inflammation in SCD.
The innate immune system is the first line of protecttion against invading microbial pathogens and of responses to inflammatory stimuli that are mediated by phagocytes, polymorphonuclear leukocytes (PMN), monocytes, macrophages, dendritic cells (DCs), and inflammatory T cell subsets (Th1 cells and Natural Killer T cells). This immune response relies on recognition of evolutionarily conserved structures on pathogens, named pathogen associated molecular patterns (PAMPs), through a limited number of germ line-encoded pattern recognition receptors (PRRs), of which the family of TLRs has been studied most extensively [38].
TLRs are integral glycoprotein characterized by an extracellular or luminal ligand binding domain containing leucine rich repeat (LRR) motifs and a cytoplasm signaling Toll/interleukin-1 (IL-1) receptor homology (TIR) domain [38]. Ligand binding to TLRs through PAMP-TLR interaction induces receptor oligomerization, which subsequently triggers intracellular signal transduction. To date, 10 TLRs have been identified in humans, and they can recognize distinct PAMPs derived from various microbial pathogens, including viruses, bacteria, fungi, and protozoa. TLRs can be divided into subfamilies primarily recognizing related PAMPs; TLR1, TLR2, TLR4, and TLR6 recognize lipids, whereas TLR3, TLR7, TLR8, and TLR9 recognize nucleic acids [39].
Cell types expressing TLRs are APCs, including macrophages, DCs, and B lymphocytes. TLRs have been identified in most cell types, expressed either constitutively or in an inducible manner in the course of infection [39]. During inflammation, increased numbers of DCs are rapidly recruited and efficiently capture antigens due to their high phagocytic ability. Subsequent to antigen capture DCs become activated and the mature DCs by pathogens can migrate into draining lymph nodes or the spleen and transforming to powerful antigen-presenting cells that are capable of activating naive T cells. The transition from immature to mature state is followed by production of several cytokines and chemokines by the DCs that regulate their ability to interact with naïve T cells to direct T cell differentiation [40].
Two major DCs subsets can be detected in the peripheral blood, with distinct, but overlapping functions. Myeloid DCs (mDCs) express human leukocyte antigens (HLA) DR, CD11c, and CD1c and are the main producers of interleukin-12 (IL-12), while plasmacytoid DCs (pDCs) express HLA DR, CD123, and blood dendritic cell antigen 2 (BDCA2), and are the main producers of interferon-α (IFN-α) [41]. However, HbSS patients in steady-state seems to have a particular CD1-positive phenotype expression of CD1a, b, and c molecules at DCs, while the classical phenotype found among the general population, in which only 15% of the individuals express the CD1 molecules at the surface of their monocytes [42]. According to Sloma et al. [43], the elevated concentration of cytokines associated with monocyte activation in HbSS patients can contribute for their activated status and it may be hypothesized that CD1 expression on DCs from HbSS patients is a consequence of the elevated plasma levels of endothelin.
The role of DCs in malaria affected population and its association with hemoglobinopathies have been described and show that the activation of mDCs and pDCs during acute malaria may be faster or deeper in children with α+thalassemia than in children with normal hemoglobin profile [44]. Other studies shows the Plasmodium falciparum glycosylphosphatidylinositol (GPI) anchors can bind to TLR2 and TLR4 expressed on mDCs and monocytes [45], whereas a component of schizont lysate as well as hemozoin can bind to TLR9 and activate pDCs [46].
Single-nucleotide polymorphisms (SNPs) have been described for TLR-4 and TLR-9 genes. For TLR-4, the polymorphism (Asp299Gly) has been related to Gramnegative infections susceptibility and septic shock [47]. More recently, the Asp299Gly has been involved as a protective allele against malaria, explaining its high prevalence in sub Saharan Africa [41].
The immune response in SCD is poorly understood. It is knows that the immune system has a close relationship with health and morbidity in SCD, although the complex network involved in the mechanisms of pathogenesis present in this disease, is difficult to understand, once it is a chronic inflammatory condition. Additional studies need to be warranted to elucidate the immunologic processes in SCD.
5. OXIDATIVE STRESS AND INFLAMMATION IN SICKLE CELL ANEMIA
Oxidative stress is a physiological condition that occurs when there is imbalance between the amount of free radicals (ROS; RNS) generated by physiological processes and antioxidant mechanisms. Free radicals are defined as chemical species that contains a pair of electrons unpaired, and this gives the high-capacity reactive free radical [48]. The reactive oxygen of species includes free radicals and non-free radicals, such as hydroxyl, superoxide ( ) and H2O2. In biological systems, the most common source of free radicals is oxygen, and ROS that can be produced from both endogenous and exogenous cellular products [49,50].
) and H2O2. In biological systems, the most common source of free radicals is oxygen, and ROS that can be produced from both endogenous and exogenous cellular products [49,50].
The endogenous sources of ROS include mitochondria, cytochrome P450, peroxisomes, and inflammatory cells activated [51]. Mitochondria generates significant quantities of H2O2 and use ~90% of cellular O2. During the process of reducing mitochondrial oxygen for production of water, several short-lived intermediates are produced, including H2O2, 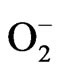 and the hydroxyl radical [OH], which are toxic to the cell. Another molecule is the peroxynitrite (ONOO–), an anion and an unstable isomer of nitrate (
and the hydroxyl radical [OH], which are toxic to the cell. Another molecule is the peroxynitrite (ONOO–), an anion and an unstable isomer of nitrate ( ). The peroxynitrite can be formed in vivo by the reaction of the free
). The peroxynitrite can be formed in vivo by the reaction of the free  with free NO, and is a potentially cytotoxic molecule [52]. Cell destruction also causes further free radical generation [53]. Neutrophils, eosinophils and macrophages are additional endogenous sources of cellular ROS. Activated macrophages initiate increase in oxygen uptake and give rise to a variety of ROS, including O2, NO and H2O2 [54].
with free NO, and is a potentially cytotoxic molecule [52]. Cell destruction also causes further free radical generation [53]. Neutrophils, eosinophils and macrophages are additional endogenous sources of cellular ROS. Activated macrophages initiate increase in oxygen uptake and give rise to a variety of ROS, including O2, NO and H2O2 [54].
In addition, intracellular formation of free radicals can occur by environmental sources including ultraviolet light, ionizing radiation, and pollutants such as paraquat and ozone. All of these sources of free radicals, both enzymatic and non-enzymatic have the potential to inflict oxidative damage on a wide range of biological macromolecules [55]. Membranes are target of free radicals forming due to its lipid composition, lipid peroxide, thus compromising the characteristics of fluidity and elasticity, leading to cell rupture. Other target tissues to ROS are proteins, which may lose their functionality enzyme and cell signaling and DNA; the interaction of ROS with DNA can lead to the DNA strand breaks, point mutations, gene deletions, or gene rearrangement, such changes can be lethal to the cell, with DNA lesion, that accumulate with age, and can be an important etiology of aging processes [56]. The endogenous antioxidant system responsible for neutralize free radicals, include enzymes, such as glutathione peroxidase, superoxide dismutase (SOD) and catalase. The non-enzymatic antioxidants that participate in oxidative stress defense include ascorbic acid (Vitamin C), alpha-tocopherol (Vitamin E), glutathione (GSH), carotenoid, and flavonoids [57-59]. The ROS occur under physiological conditions and in many diseases cause direct or indirect damage in different organs; thus, it is known that oxidative stress (OS) is involved in pathological processes such as obesity, diabetes, cardiovascular disease, and atherogenic processes [60].
Inflammation is an immune system reaction aiming to contain and eliminate pathogens or foreign elements to the body [61,62]. After activation, innate immune system cells secrete pro-inflammatory cytokines and chemokines that induce ROS/RNS production [63]. In the innate immune system, macrophages generate ROS, including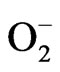 , NO, H2O2, hydroxyl radical, ONOO– and HOCl to play pathogen elimination [62,63]. Chronic inflammation can lead to cellular damage, hyperplasia and, consequently, to the overproduction of ROS by inflammatory cells [61].
, NO, H2O2, hydroxyl radical, ONOO– and HOCl to play pathogen elimination [62,63]. Chronic inflammation can lead to cellular damage, hyperplasia and, consequently, to the overproduction of ROS by inflammatory cells [61].
In the HbSS, the chronic inflammatory state promotes the production of ROS and predicts the disease severity [64-68]. In the HbSS some events contribute to the maintenance of oxidative stress such as the excessive levels of cell-free hemoglobin with its catalytic action on oxidative reactions; the characteristic recurrent ischemiareperfusion injury, a chronic pro-inflammatory state with higher autoxidation of HbS [69-71].
Iron is a chemical element that participates in the reaction of electron transfer between molecules in the process of cellular respiration (redox reactions), and it is deposited in the form of ferritin and hemosiderin. Patients receiving multiple blood transfusions, such as patients with chronic anemia, thalassemia, and HbSS, they can exceed the storage and detoxification capacity of ferritin. Consequently, the free iron begins to accumulate into tissues; this can catalyze the formation of very injurious compounds, such as [OH] by Fenton reaction [71]. Compared with normal red blood cells (RBC) membranes, those from sickle RBC have abnormally increased Fenton reactivity, once that the instability of HbS results in generation of .O– and H2O2, the combination of which potentially forms the .OH [70,71]. The phenomenon of sickling and vaso-occlusive events in HbSS are directly associated with its pathogenesis, and there is evidence that several inflammatory events occur with increased levels of inflammatory and anti-inflammatory cytokines, such as IL1b, IL4, IL6, TNFa, the expression of cell adhesion molecules, such as ICAM-1, VCAM-1, P-selectin and integrins; the adhesion of activated PMN to the endothelium; the participation of activated platelets, and the presence of inflammatory biomarkers, such as CRP and prostaglandins. These factors contribute to the occurrence of vaso-occlusion and chronic organ damage, favoring an increased production of ROS [65,72-77].
6. CYTOKINE IN SICKLE CELL ANEMIA: BREAK THE BALANCE
Pro-inflammatory cytokines mainly expressed by monocytes from stimulation of bacterial components, ROS and growth factors have important role in immune innate response and inflammatory state [78]. These effects are modulated by anti-inflammatory cytokines, such as IL-4 and IL-10, necessary to down regulate leukocytes and the vascular endothelium activation [79]. This delicate balance is broken when inflammatory cytokine increase oxidative stress and overcoming antioxidant barrier by activation of complex transporter of electron in mitochondria [78,80,81], stimulating the transcription of the trans factor NF-kB with degradation of IkB, up regulating the expression of selectins, integrins, ICAM-1, VCAM-1, resulting on pre-activation of leukocytes and with the interaction of these molecules with the activated endothelium [81-83].
Cytokine levels in HbSS patients are elevated not only during crisis-state, but also in different pathophysiologic mechanisms of the disease, like hypoxia and reperfusion rate; beyond clinical inflammatory history generate an increased concentration of these molecules also in steadystate patients. Pro-inflammatory cytokines like IL-1, IL-2, IL-6, IL-8, IL-17 and TNF-alpha are increased on basal state and during vascular occlusion events unaccompanied of an increased level of anti-inflammatory cytokines, such as IL-4 and IL-10 [3,20,79,84-88]. This inflammatory profile is not consensus; maybe it is associated with a high inter-individual variation, mainly due difference of genetic background and environmental aspects that can generate these molecules plasma levels fluctuation.
Soluble vascular cell adhesion molecule-1 (sVCAM-1) amount increase in plasma and change accord to the profile expression of selectins and integrins on leukocytes, such as CD62L and CD11b indirectly reflecting the rate of inflammation, and oxidative stress and have been associated as promising markers of HbSS prognosis [27, 89-91]. Based on these, oxidative stress and inflammatory profile may have complementary and symbiotic effects, mainly in a chronic inflammatory and oxidative disease, such as HbSS.
Inflammatory and oxidative profile, therefore, are extremely connect. This link is also confirmed when it is analyzed the conversion of purines to uric acid by the xanthine oxidase action [92,93]. This reaction generate free radicals, like intermediary products, and is up regulated by pro-inflammatory cytokines due many potential cytokine responsive elements in the xanthine oxidase (XO) gene regulatory region [94].
Therefore, up regulation of XO gene generates more uric acid and contact of this molecule with free sodium driving the monosodium urate that is consider to be a biologically active structure [95]. The monosodium urate is an active form act as a danger signal by activation of inflammasome, resulting on the production of pro-inflammatory cytokines, such as IL-1 and IL-18 [96]. In recent research by our team, inflammasome pathway was observed in HbSS patients, where uric acid, considered as a danger signal was associated with high serum levels of IL-18, further connect to cytokines levels and oxidative stress markers [91]. These effects, accordingly, are amplified on a chronic hemolytic and inflammatory disease, like HbSS, where the intravascular hemolysis contribute to release of cell free hemoglobin, heme and iron, consequently increase of oxygen radicals, limiting NO bioavailability, attracting more leukocytes, activating endothelial cells, contributing to the vascular occlusion [2,15,17,27,91].
7. PLATELETS AND INFLAMMATION IN SICKLE CELL DISEASE
Platelets are small enucleated structures derivatives from megakaryocyte fragmentation and are important to homeostasis process, primary function originally described to platelets. Platelets undergo activation, adhesion and aggregation binding to damage blood vessel, producing a platelet plug and contributing to the generation of thrombin. Furthermore, platelets produce and store a variety of molecules that affect platelet function and modified the vascular tone, the fibrinolysis [97]. Leukocytes and endothelial cells are associated as a critical player in the microvascular alteration induced by inflammation. Actually, have been thought about the role of platelets in inflammatory states through leukocytes interaction, release proteins, chemokines and endothelial disfunction [98].
Some studies suggest that circulating platelets in HbSS patients are chronically activated, both during steadystate and vaso-occlusive crises, which may result of the overall hypercoagulable state or with the vaso-occlusive process or with the pro-inflammatory characteristics of the microvasculature in HbSS [20,99,100].
Platelets have important organelles to performance its function, such as alpha-granules, lysosomes, peroxisomes, dense bodies and a complex membranous system that contribute to store and rapidly release several factors and proteins [99,101]. Among released products by platelets, there are secretion of adhesion proteins, such as fibrinogen, von Willebrand factor (vWF), thrombospondin, P-selectin, GPIIb/IIIa; there are important chemokines, including RANTES and platelet factor-4; cytokine-like factors as IL1-β, CD40L, β-thromboglobulin or factors essentials for the coagulation process, as plasminogen activator inhibitor (PAI-1), protein S and factors V and XI, and also expression of innate receptors of the Tolllike receptor family, such as TLR2 and TLR4 [102-105].
During inflammatory process, activated endothelial cells and others perivascular cells and leukocytes, release several soluble mediators, such as lipids, IFN-g, IL-2, and CXCL12, which bind to platelet receptors, leading to degranulation of platelets dense and alpha-granules, promoting self adhesion and activation. Furthermore, the OS actives phospholipase A2 and the generation of the arachidonic acid pathway metabolites and platelet activator factor (PAF), and also contribute for platelet activation [103,105]. On the other hand, the platelet activetion can induce several inflammatory responses in monocytes, neutrophils, endothelial cells or endothelial progenitor cells; product released by platelets are potent inflammatory and mitogenics substances, modifying the chemostatic, adhesive and proteolytic properties of cellular microenvironment, mainly of endothelial cells and leukocytes, resulting in an increase of transmigration of leukocytes to the site of inflammation, suggesting that platelets-leukocytes interaction may be a key role in the initiation of inflammation [98,103,104].
Many mechanisms of platelets-leukocytes interactions have been described, but the initial interaction appears to be mediated by P-selectin expressed on the surface activated platelets and P-selectin ligand glycoprotein-1 (PSGL-1) on the surface of neutrophils and monocytes and, subsequently, firmly adhere by binding of Mac-1 to GPIb or other receptors of the platelet membrane [98, 104]. The P-selectin was found in sickle cell transgenic mice with high constitutive levels, which could be attributed to platelet activation, contributing to inflammatory response [98,104]; study suggest the P-selectin-mediated platelet-neutrophil aggregate formation, which activates neutrophils in SCD mouse model and human been carriers [104]. The sCD40L level is increased and biologically active in HbSS patients due to platelet activation, mainly, in patients in crises and positively correlates with an increase of TF and ICAM-1 expression, suggesting that an increase of CD40L may contribute to the chronic inflammation and with the increased pro-coagulant activity in HbSS patients [105].
Thus, several studies suggest that in addition to procoagulant role, platelets contribute directly to constant vascular inflammation state in HbSS patients by activateing neutrophils and monocytes and further research about therapies targeting function and interaction of platelets and endothelial cells and leukocytes may help to control inflammation and vaso-occlusive events among these patients.
8. CIRCULATING MICROPARTICLES: NEW INFLAMMATION BIOMARKERS IN HBSS
Microparticles (MPs) are heterogeneous group of membrane-bound vesicles described as vesicles smaller than 10−15 m in diameter [106]. MPs are shedding from plasma membranes, after cell activation or apoptosis of several cellular types. Essentially any cell type (e.g., leucocytes, and endothelial cells), but also platelets and RBC can release MPs [107,108]. They have been implicated to play a role in inflammation, coagulation and vascular function.
During blebbing, the lipid bilayer forms cytoplasmatic protrusions, culminating in the release of MPs [109]. This process involve an increase in intracellular calcium which affects many important enzymes for the maintaining of the cytoskeletal and membrane structure, such as gelsolin, calpain, flippase, floppase and scramblase [108].
As the lipid and protein composition of the MPs membrane resembles that from the releasing cell, analysis of MPs surface markers by flow cytometry can identify the MPs origin. Internally, MPs contain a variety of cytoplasmatic and nuclear components of their precursor cells [110]. Circulating MPs levels result from the balance between their rates of release from cells and their clearance from the circulation.
MPs levels are augmented in several diseases, such HbSS. Patients have elevated MPs, both in steady-state and crisis, implying that even patients in steady-state are fundamentally different from healthy subjects [111,112]. Total circulating MPs are more elevated in the crisis phase of the disease than in steady-state [111,113]. During crisis, endothelial damage and coagulation activation increase dramatically, and those condition are accompanied by an increases of circulating MPs [113]. Thus, it is worth considering circulating MPs as biomarkers of HbSS.
Sickle cell anemia patients have an increased risk of vascular thrombotic occlusion [114]. In addition, there is strong in vitro evidence that circulating MPs are involved in the coagulation system activation in HbSS [115,112]. The pro-coagulant activity depends of some molecules presence, such as phosphatidylserine (PS) and TF, both exposed on several MPs types outer membranes [110]. Therefore, this pro-coagulant activity may be relevant clinically as MPs concentrations with this phenotype are elevated in HbSS.
The majority of circulating MPs in HbSS has RBC and platelets origin [116]. The report by van Beers et al. [116] showed a strong association between erythrocyte-derived MPs and markers of in vivo coagulation and fibrinolysis activation status as well as endothelial activation [116]. In addition, MPs may support coagulation activation by exposure of PS [115,116], which offers multiple binding sites for the coagulation factors II, Va, and Xa [117]. Sickle cell anemia patients have elevated plasma levels of annexin A5- and PS-exposing MPs [111,112]. Thus, MPs can provide a platform for the assembly of the prothrombinase complex and accelerate the conversion of prothrombin into thrombin.
Importantly to vascular homeostasis has been the discovery of TF, the principal initiator of coagulation, in MPs from HbSS. Sickle blood contains a fraction of MPs originating from platelets [114], endothelial cells and monocytes [113], which are TF positive. Furthermore, once initiated by TF, thrombin generation is greatly accelerated in the presence of PS [67] and co-expression of these molecules can contribute to thrombotic events frequently observed in patients with HbSS [118]. Importantly, MPs could be capable to initiate blood coagulation.
Microparticles are emerging as important biomarker of inflammation, coagulation and thrombosis in HbSS. Linked to crucial steps of HbSS, MPs can now be viewed “partners in disease”, especially in patients in crisis-state. MPs provide a vehicle to couple inflammation and coagulation, contributing to thrombotic tendencies in this disease. This increasingly close relationship between MPs and HbSS demonstrates the need for more studies on this subject. Thus, it is necessary additional research to define the precise role of circulating MPs in HbSS and allow the development of new therapeutic strategies either blocking the release of MPs or modifying their activity.
9. CONCLUSION
It is very well known that SCD is a group of genetic disorders, with 101 years of its first medical relate, but still has several pieces of the puzzle to be solved. The search for pathways and biomarkers involved in the pathophysiology of the disease are still need to be exhausted search, and it will bring the knowledge of molecules that may contribute to increase SCD patients life quality, given opportunity for new therapeutic approaches and clinical management modalities. This review just point several mechanisms associated with SCD, and may contribute to give some ideas about the very complex molecules network involved in the inflammatory process associated with the disease pathogenesis.
10. ACKNOWLEDGEMENTS
This work was supported by grants from the Brazilian National Council of Research (CNPq) (3065427/2007-5 and 484457/2007-1) (M.S.G.); the Foundation of Research and Extension of Bahia (FAPESB) (1431040053063, 9073/2007 and 6234/2010) (M.S.G.); and MCD/ CNPq/MS-SCTIE-DECIT (409800/2006-6), (M.S.G.). The sponsors of this study are public or nonprofit organizations that support science in general. They had no role in gathering, analyzing or interpreting the data.
REFERENCES
- Steinberg, M.H. (2008) Sickle cell anemia, the first molecular disease: Overview of molecular etiology, pathophysiology, and therapeutic approaches. The Scientific World Journal, 8, 1295-1324. doi:10.1100/tsw.2008.157
- Steinberg, M.H. (2005) Predicting clinical severity in sickle cell anaemia. British Journal of Haematology, 129, 465-481. doi:10.1111/j.1365-2141.2005.05411.x
- Lanaro, C., Franco-Penteado, C.F., Albuqueque, D.M., Saad, S.T., Conran, N. and Costa, F.F. (2009) Altered levels of cytokines and inflammatory mediators in plasma and leukocytes of sickle cell anemia patients and effects of hydroxyurea therapy. Journal of Leukocyte Biology, 85, 235-242. doi:10.1189/jlb.0708445
- Bender, M.A. and Hobbs, W. (1993) Sickle cell disease. In: Pagon, R.A., Bird, T.D., Dolan, C.R., Stephens, K. and Adam, M.P., Eds., Genereviews, Seattle, WA.
- Morris, C.R. (2011) Vascular risk assessment in patients with sickle cell disease. Haematologica, 96, 1-5. doi:10.3324/haematol.2010.035097
- Medina-Urrutia, A., Juarez-Rojas, J.G., Cardoso-Saldana, G., Jorge-Galarza, E., Posadas-Sanchez, R., MartinezAlvarado, R., Caracas-Portilla, N., Mendoza Perez, E. and Posadas-Romero, C. (2011) Abnormal high-density lipoproteins in overweight adolescents with atherogenic dyslipidemia. Pediatrics, 127, e1521-1527.
- Elkind, M.S. (2006) Inflammation, atherosclerosis, and stroke. The Neurologist, 12, 140-148. doi:10.1097/01.nrl.0000215789.70804.b0
- Thanoon, I.A., Abdul-Jabbar, H.A. and Taha, D.A. (2012) Oxidative stress and C-reactive protein in patients with cerebrovascular accident (ischaemic stroke): The role of ginkgo biloba extract. Sultan Qaboos University Medical Journal, 12, 197-205.
- Milton, J.N., Sebastiani, P., Solovieff, N., Hartley, S.W., Bhatnagar, P., Arking, D.E., Dworkis, D.A., Casella, J.F., Barron-Casella, E., Bean, C.J., Hooper, W.C., DeBaun, M.R., Garrett, M.E., Soldano, K., Telen, M.J., AshleyKoch, A., Gladwin, M.T., Baldwin, C.T., Steinberg, M.H. and Klings, E.S. (2012) A genome-wide association study of total bilirubin and cholelithiasis risk in sickle cell anemia. PloS One, 7, e34741. doi:10.1371/journal.pone.0034741
- Ebert, E.C., Nagar, M. and Hagspiel, K.D. (2010) Gastrointestinal and hepatic complications of sickle cell disease. Clinical Gastroenterology and Hepatology: The Official Clinical Practice Journal of the American Gastroenterological Association, 8, 483-489; quiz e470.
- Wakugawa, Y., Kiyohara, Y., Tanizaki, Y., Kubo, M., Ninomiya, T., Hata, J., Doi, Y., Okubo, K., Oishi, Y., Shikata, K., Yonemoto, K., Maebuchi, D., Ibayashi, S. and Iida, M. (2006) C-Reactive protein and risk of firstever ischemic and hemorrhagic stroke in a general japanese population: The hisayama study. Stroke; A journal of Cerebral Circulation, 37, 27-32. doi:10.1161/01.STR.0000194958.88216.87
- Voskaridou, E., Christoulas, D. and Terpos, E. (2012) Sickle-cell disease and the heart: Review of the current literature. British Journal of Haematology, 157, 664-673.
- Costa, R.N., Conran, N., Albuquerque, D.M., Soares, P.H., Saad, S.T. and Costa, F.F. (2005) Association of the G-463a myeloperoxidase polymorphism with infection in sickle cell anemia. Haematologica, 90, 977-979.
- Liu, C., Xie, G., Huang, W., Yang, Y., Li, P. and Tu, Z. (2012) Elevated serum myeloperoxidase activities are significantly associated with the prevalence of ACS and high Ldl-C Levels in Chd patients. Journal of Atherosclerosis and Thrombosis, 19, 435-443. doi:10.1111/j.1365-2141.2012.09143.x
- Seixas, M.O., Rocha, L.C., Carvalho, M.B., Menezes, J.F., Lyra, I.M., Nascimento, V.M., Couto, R.D., Atta, A.M., Reis, M.G. and Goncalves, M.S. (2010) Levels of highdensity lipoprotein cholesterol (Hdl-C) among Children with steady-state sickle cell disease. Lipids in Health and Disease, 9, 91. doi:10.1186/1476-511X-9-91
- Wood, K.C., Hsu, L.L. and Gladwin, M.T. (2008) Sickle cell disease vasculopathy: A state of nitric oxide resistance. Free Radical Biology & Medicine, 44, 1506-1528. doi:10.1016/j.freeradbiomed.2008.01.008
- Reiter, C.D., Wang, X., Tanus-Santos, J.E., Hogg, N., Cannon, R.O., 3rd, Schechter, A.N. and Gladwin, M.T. (2002) Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nature Medicine, 8, 1383- 1389. doi:10.1038/nm1202-799
- Ryter, S.W. and Tyrrell, R.M. (2000) The heme synthesis and degradation pathways: Role in oxidant sensitivity. heme oxygenase has both proand antioxidant properties. Free Radical Biology & Medicine, 28, 289-309. doi:10.1016/S0891-5849(99)00223-3
- Akinsheye, I. and Klings, E.S. (2010) Sickle cell anemia and vascular dysfunction: The nitric oxide connection. Journal of Cellular Physiology, 224, 620-625. doi:10.1002/jcp.22195
- Villagra, J., Shiva, S., Hunter, L.A., Machado, R.F., Gladwin, M.T. and Kato, G.J. (2007) Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cellfree hemoglobin. Blood, 110, 2166-2172. doi:10.1182/blood-2006-12-061697
- Spiecker, M., Darius, H., Kaboth, K., Hubner, F. and Liao, J.K. (1998) Differential regulation of endothelial cell adhesion molecule expression by nitric oxide donors and antioxidants. Journal of Leukocyte Biology, 63, 732-739.
- Aslan, M., Ryan, T.M., Adler, B., Townes, T.M., Parks, D.A., Thompson, J.A., Tousson, A., Gladwin, M.T., Patel, R.P., Tarpey, M.M., Batinic-Haberle, I., White, C.R. and Freeman, B.A. (2001) Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proceedings of the National Academy of Sciences of the United States of America, 98, 15215-15220. doi:10.1073/pnas.221292098
- Solovey, A., Kollander, R., Milbauer, L.C., Abdulla, F., Chen, Y., Kelm, R.J. Jr. and Hebbel, R.P. (2010) Endothelial nitric oxide synthase and nitric oxide regulate endothelial tissue factor expression in vivo in the sickle transgenic mouse. American Journal of Hematology, 85, 41- 45.
- Jain, S. and Gladwin, M.T. (2010) Arginine metabolism and nitric oxide bioavailability in sickle cell disease. Journal of Pediatric Hematology/Oncology, 32, e247-248. doi:10.1097/MPH.0b013e3181ec0b00
- Zhou, Z., Behymer, M. and Guchhait, P. (2011) Role of extracellular hemoglobin in thrombosis and vascular occlusion in patients with sickle cell anemia. ANEMIA, article ID: 918916. doi:10.1155/2011/918916
- Morris, C.R., Kato, G.J., Poljakovic, M., Wang, X., Blackwelder, W.C., Sachdev, V., Hazen, S.L., Vichinsky, E.P., Morris, S.M. Jr. and Gladwin, M.T. (2005) Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA: The Journal of the American Medical Association, 294, 81-90. doi:10.1001/jama.294.1.81
- Vilas-Boas, W., Cerqueira, B.A., Zanette, A.M., Reis, M.G., Barral-Netto, M. and Goncalves, M.S. (2010) Arginase levels and their association with th17-related cytokines, soluble adhesion molecules (Sicam-1 and Svcam-1) and hemolysis markers among steady-state sickle cell anemia patients. Annals of Hematology, 89, 877-882. doi:10.1007/s00277-010-0954-9
- Bialecka, M., Robowski, P., Honczarenko, K., Roszmann, A. and Slawek, J. (2009) Genetic and environmental factors for hyperhomocysteinaemia and its clinical implications in Parkinson’s disease. Neurologia i Neurochirurgia Polska, 43, 272-285.
- Refsum, H., Ueland, P.M., Nygard, O. and Vollset, S.E. (1998) Homocysteine and cardiovascular disease. Annual Review of Medicine, 49, 31-62. doi:10.1146/annurev.med.49.1.31
- Clarke, R., Daly, L., Robinson, K., Naughten, E., Cahalane, S., Fowler, B. and Graham, I. (1991) Hyperhomocysteinemia: An independent risk factor for vascular disease. The New England Journal of Medicine, 324, 1149- 1155. doi:10.1056/NEJM199104253241701
- Lowenthal, E.A., Mayo, M.S., Cornwell, P.E. and Thornley-Brown, D. (2000) Homocysteine elevation in sickle cell disease. Journal of the American College of Nutrition, 19, 608-612.
- Siniscalchi, A., Mancuso, F., Gallelli, L., Ferreri Ibbadu, G., Biagio Mercuri, N. and De Sarro, G. (2005) Increase in plasma homocysteine levels induced by drug treatments in neurologic patients. Pharmacological Research: The Official Journal of the Italian Pharmacological Society, 52, 367-375.
- Dudman, N.P., Temple, S.E., Guo, X.W., Fu, W. and Perry, M.A. (1999) Homocysteine enhances neutrophil-endothelial interactions in both cultured human cells and rats in vivo. Circulation Research, 84, 409-416. doi:10.1161/01.RES.84.4.409
- Chies, J.A. and Nardi, N.B. (2001) Sickle cell disease: A chronic inflammatory condition. Medical Hypotheses, 57, 46-50. doi:10.1054/mehy.2000.1310
- Wallace, K.L. and Linden, J. (2010) Adenosine A2a receptors induced on Inkt and Nk cells reduce pulmonary inflammation and injury in mice with sickle cell disease. Blood, 116, 5010-5020. doi:10.1182/blood-2010-06-290643
- Park, S.W., Kim, M., Brown, K.M., D’Agati, V.D. and Lee, H.T. (2011) Paneth cell-derived interleukin-17a causes multiorgan dysfunction after hepatic ischemia and reperfusion injury. Hepatology, 53, 1662-1675. doi:10.1002/hep.24253
- Chen, G.Y. and Nunez, G. (2010) Sterile inflammation: Sensing and reacting to damage. Nature reviews. Immunology, 10, 826-837. doi:10.1038/nri2873
- Akira, S., Uematsu, S. and Takeuchi, O. (2006) Pathogen recognition and innate immunity. Cell, 124, 783-801. doi:10.1016/j.cell.2006.02.015
- Iwasaki, A. and Medzhitov, R. (2004) Toll-like receptor control of the adaptive immune responses. Nature Immunology, 5, 987-995. doi:10.1038/ni1112
- Pulendran, B. (2005) Variegation of the immune response with dendritic cells and pathogen recognition receptors. Journal of Immunology, 174, 2457-2465.
- Ferwerda, B., McCall, M.B., Alonso, S., GiamarellosBourboulis, E.J., Mouktaroudi, M., Izagirre, N., Syafruddin, D., Kibiki, G., Cristea, T., Hijmans, A., Hamann, L., Israel, S., ElGhazali, G., Troye-Blomberg, M., Kumpf, O., Maiga, B., Dolo, A., Doumbo, O., Hermsen, C.C., Stalenhoef, A.F., van Crevel, R., Brunner, H.G., Oh, D.Y., Schumann, R.R., de la Rua, C., Sauerwein, R., Kullberg, B.J., van der Ven, A.J., van der Meer, J.W. and Netea, M.G. (2007) Tlr4 polymorphisms, infectious diseases, and evolutionary pressure during migration of modern humans. Proceedings of the National Academy of Sciences of the United States of America, 104, 16645-16650. doi:10.1073/pnas.0704828104
- Gregory, S., Zilber, M., Charron, D. and Gelin, C. (2000) Human Cd1a molecule expressed on monocytes plays an accessory role in the superantigen-induced activation of T lymphocytes. Human Immunology, 61, 193-201. doi:10.1016/S0198-8859(99)00129-9
- Sloma, I., Zilber, M.T., Charron, D., Girot, R., Tamouza, R. and Gelin, C. (2004) Upregulation and atypical expression of the Cd1 molecules on monocytes in sickle cell disease. Human Immunology, 65, 1370-1376. doi:10.1016/j.humimm.2004.09.009
- Urban, B.C., Shafi, M.J., Cordery, D.V., Macharia, A., Lowe, B., Marsh, K. and Williams, T.N. (2006) Frequencies of peripheral blood myeloid cells in healthy kenyan children with alpha+ thalassemia and the sickle cell trait. The American Journal of Tropical Medicine and Hygiene, 74, 578-584.
- Krishnegowda, G., Hajjar, A.M., Zhu, J., Douglass, E.J., Uematsu, S., Akira, S., Woods, A.S. and Gowda, D.C. (2005) Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of plasmodium falciparum: Cell signaling receptors, glycolsylphosphatidylinositol (Gpi) structural requirement, and regulation of Gpi activity. The Journal of Biological Chemistry, 280, 8606-8616. doi:10.1074/jbc.M413541200
- Pichyangkul, S., Yongvanitchit, K., Kum-arb, U., Hemmi, H., Akira, S., Krieg, A.M., Heppner, D.G., Stewart, V.A., Hasegawa, H., Looareesuwan, S., Shanks, G.D. and Miller, R.S. (2004) Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a toll-like receptor 9-dependent pathway. Journal of Immunology, 172, 4926-4933.
- Lorenz, E., Mira, J.P., Frees, K.L. and Schwartz, D.A. (2002) Relevance of mutations in the Tlr4 receptor in patients with gram-negative septic shock. Archives of Internal Medicine, 162, 1028-1032. doi:10.1001/archinte.162.9.1028
- Khansari, N., Shakiba, Y. and Mahmoudi, M. (2009) Chronic inflammation and oxidative stress as a major cause of age-related diseases and cancer. Recent Patents on Inflammation & Allergy Drug Discovery, 3, 73-80. doi:10.2174/187221309787158371
- Valdivia, P.A., Zenteno-Savin, T., Gardner, S.C. and Aguirre, A.A. (2007) Basic oxidative stress metabolites in eastern pacific green turtles (chelonia mydas agassizii). Comparative biochemistry and physiology. Toxicology & Pharmacology: CBP, 146, 111-117.
- Inoue, M., Sato, E.F., Nishikawa, M., Park, A.M., Kira, Y., Imada, I. and Utsumi, K. (2003) Mitochondrial generation of reactive oxygen species and its role in aerobic life. Current Medicinal Chemistry, 10, 2495-2505. doi:10.2174/0929867033456477
- Akopova, O.V., Kolchinskaya, L.I., Nosar, V.I., Bouryi, V.A., Mankovska, I.N. and Sagach, V.F. (2012) Cytochrome C as an amplifier of ros release in mitochondria. Fiziolohichnyi Zhurnal, 58, 3-12.
- Gutteridge, J.M., Rowley, D.A. and Halliwell, B. (1982) Superoxide-dependent formation of hydroxyl radicals and lipid peroxidation in the presence of iron salts. Detection of “catalytic” iron and anti-oxidant activity in extracellular fluids. The Biochemical Journal, 206, 605-609.
- Wickens, A.P. (2001) Ageing and the free radical theory. Respiration Physiology, 128, 379-391. doi:10.1016/S0034-5687(01)00313-9
- Conner, E.M. and Grisham, M.B. (1996) Inflammation, free radicals, and antioxidants. Nutrition, 12, 274-277. doi:10.1016/S0899-9007(96)00000-8
- Mateos, R. and Bravo, L. (2007) Chromatographic and electrophoretic methods for the analysis of biomarkers of oxidative damage to macromolecules (dna, lipids, and proteins). Journal of Separation Science, 30, 175-191. doi:10.1002/jssc.200600314
- Pastore, A., Federici, G., Bertini, E. and Piemonte, F. (2003) Analysis of glutathione: Implication in redox and detoxification. Clinica Chimica Acta; International Journal of Clinical Chemistry, 333, 19-39.
- Esposito, K., Ciotola, M., Schisano, B., Misso, L., Giannetti, G., Ceriello, A. and Giugliano, D. (2006) Oxidative stress in the metabolic syndrome. Journal of Endocrinological Investigation, 29, 791-795.
- Peterhans, E. (1997) Reactive oxygen species and nitric oxide in viral diseases. Biological Trace Element Research, 56, 107-116. doi:10.1007/BF02778986
- Stehbens, W.E. (2003) Oxidative stress, toxic hepatitis, and antioxidants with particular emphasis on zinc. Experimental and Molecular Pathology, 75, 265-276. doi:10.1016/S0014-4800(03)00097-2
- Emmendoerffer, A., Hecht, M., Boeker, T., Mueller, M. and Heinrich, U. (2000) Role of inflammation in chemical-induced lung cancer. Toxicology Letters, 112-113, 185-191. doi:10.1016/S0378-4274(99)00285-4
- Segal, A.W. (2006) How Superoxide production by neutrophil leukocytes kills microbes. Novartis Foundation Symposium, 279, 92-98; Discussion 98-100, 216-109.
- Costa, A.D. and Garlid, K.D. (2008) Intramitochondrial signaling: Interactions among mitokatp, pkcepsilon, ROS, and MPT. American Journal of Physiology, Heart and Circulatory Physiology, 295, H874-882. doi:10.1152/ajpheart.01189.2007
- Fialkow, L., Wang, Y. and Downey, G.P. (2007) Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radical Biology & Medicine, 42, 153-164. doi:10.1016/j.freeradbiomed.2006.09.030
- Hebbel, R.P. and Vercellotti, G.M. (1997) The endothelial biology of sickle cell disease. The Journal of Laboratory and Clinical Medicine, 129, 288-293. doi:10.1016/S0022-2143(97)90176-1
- Kaul, D.K. and Hebbel, R.P. (2000) Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. The Journal of Clinical Investigation, 106, 411-420. doi:10.1172/JCI9225
- Osarogiagbon, U.R., Choong, S., Belcher, J.D., Vercellotti, G.M., Paller, M.S. and Hebbel, R.P. (2000) Reperfusion injury pathophysiology in sickle transgenic mice. Blood, 96, 314-320.
- Stuart, M.J. and Setty, B.N. (2001) Acute chest syndrome of sickle cell disease: New light on an old problem. Current Opinion in Hematology, 8, 111-122. doi:10.1097/00062752-200103000-00009
- Nath, K.A., Grande, J.P., Croatt, A.J., Frank, E., Caplice, N.M., Hebbel, R.P. and Katusic, Z.S. (2005) Transgenic sickle mice are markedly sensitive to renal ischemiareperfusion injury. The American Journal of Pathology, 166, 963-972. doi:10.1016/S0002-9440(10)62318-8
- Nagababu, E., Fabry, M.E., Nagel, R.L. and Rifkind, J.M. (2008) Heme degradation and oxidative stress in murine models for hemoglobinopathies: Thalassemia, sickle cell disease and hemoglobin C disease. Blood Cells, Molecules & Diseases, 41, 60-66. doi:10.1016/j.bcmd.2007.12.003
- Nur, E., Biemond, B.J., Otten, H.M., Brandjes, D.P., Schnog, J.J. and Group, C.S. (2011) Oxidative stress in sickle cell disease; Pathophysiology and potential implications for disease management. American Journal of Hematology, 86, 484-489. doi:10.1002/ajh.22012
- Repka, T. and Hebbel, R.P. (1991) Hydroxyl radical formation by sickle erythrocyte membranes: Role of pathologic iron deposits and cytoplasmic reducing agents. Blood, 78, 2753-2758.
- Setty, B.N., Rao, A.K. and Stuart, M.J. (2001) Thrombophilia in sickle cell disease: The red cell connection. Blood, 98, 3228-3233. doi:10.1182/blood.V98.12.3228
- Francis, R.B. Jr. and Haywood, L.J. (1992) Elevated immunoreactive tumor necrosis factor and interleukin-1 in sickle cell disease. Journal of the National Medical Association, 84, 611-615.
- Aslan, M., Thornley-Brown, D. and Freeman, B.A. (2000) Reactive species in sickle cell disease. Annals of the New York Academy of Sciences, 899, 375-391. doi:10.1111/j.1749-6632.2000.tb06201.x
- Gladwin, M.T., Schechter, A.N., Ognibene, F.P., Coles, W.A., Reiter, C.D., Schenke, W.H., Csako, G., Waclawiw, M.A., Panza, J.A. and Cannon, R.O. 3rd (2003) Divergent nitric oxide bioavailability in men and women with sickle cell disease. Circulation, 107, 271-278. doi:10.1161/01.CIR.0000044943.12533.A8
- Selvaraj, S.K., Giri, R.K., Perelman, N., Johnson, C., Malik, P. and Kalra, V.K. (2003) Mechanism of monocyte activation and expression of proinflammatory cytochemokines by placenta growth factor. Blood, 102, 1515- 1524. doi:10.1182/blood-2002-11-3423
- Musa, B.O., Onyemelukwe, G.C., Hambolu, J.O., Mamman, A.I. and Isa, A.H. (2010) Pattern of serum cytokine expression and t-cell subsets in sickle cell disease patients in vaso-occlusive crisis. Clinical and Vaccine Immunology: CVI, 17, 602-608. doi:10.1128/CVI.00145-09
- Goossens, V., Grooten, J., De Vos, K. and Fiers, W. (1995) Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proceedings of the National Academy of Sciences of the United States of America, 92, 8115-8119. doi:10.1073/pnas.92.18.8115
- Muhl, D., Woth, G., Drenkovics, L., Varga, A., Ghosh, S., Csontos, C., Bogar, L., Weber, G. and Lantos, J. (2011) Comparison of oxidative stress & leukocyte activation in patients with severe sepsis & burn injury. The Indian Journal of Medical Research, 134, 69-78.
- Read, M.A., Whitley, M.Z., Williams, A.J. and Collins, T. (1994) Nf-kappa B and I kappa B alpha: An inducible regulatory system in endothelial activation. The Journal of Experimental Medicine, 179, 503-512. doi:10.1084/jem.179.2.503
- Patel, S.J., Jindal, R., King, K.R., Tilles, A.W. and Yarmush, M.L. (2011) The inflammatory response to double stranded DNA in endothelial cells is mediated by Nfkappab and Tnfalpha. PloS One, 6, e19910. doi:10.1371/journal.pone.0019910
- Croizat, H. (1994) Circulating cytokines in sickle cell patients during steady state. British Journal of Haematology, 87, 592-597. doi:10.1111/j.1365-2141.1994.tb08318.x
- Raghupathy, R., Haider, M.Z., Azizieh, F., Abdelsalam, R., D'Souza, T.M. and Adekile, A.D. (2000) Th1 and Th2 cytokine profiles in sickle cell disease. Acta Haematologica, 103, 197-202. doi:10.1159/000041049
- Goncalves, M.S., Queiroz, I.L., Cardoso, S.A., Zanetti, A., Strapazoni, A.C., Adorno, E., Albuquerque, A., Sant’Ana, A., dos Reis, M.G., Barral, A. and Barral Netto, M. (2001) Interleukin 8 as a vaso-occlusive marker in Brazilian patients with sickle cell disease. Brazilian Journal of Medical and Biological Research, 34, 1309-1313.
- Pathare, A., Al Kindi, S., Alnaqdy, A.A., Daar, S., Knox-Macaulay, H. and Dennison, D. (2004) Cytokine profile of sickle cell disease in Oman. American Journal of Hematology, 77, 323-328. doi:10.1002/ajh.20196
- Akohoue, S.A., Shankar, S., Milne, G.L., Morrow, J., Chen, K.Y., Ajayi, W.U. and Buchowski, M.S. (2007) Energy expenditure, inflammation, and oxidative stress in steady-state adolescents with sickle cell anemia. Pediatric Research, 61, 233-238. doi:10.1203/pdr.0b013e31802d7754
- Lard, L.R., Mul, F.P., de Haas, M., Roos, D. and Duits, A.J. (1999) Neutrophil activation in sickle cell disease. Journal of Leukocyte Biology, 66, 411-415.
- Okpala, I. (2002) Steady-state platelet count and complications of sickle cell disease. The Hematology Journal: The Official Journal of the European Haematology Association/EHA, 3, 214-215.
- Schnog, J.B., Rojer, R.A., Mac Gillavry, M.R., Ten Cate, H., Brandjes, D.P. and Duits, A.J. (2003) Steady-state svcam-1 serum levels in adults with sickle cell disease. Annals of Hematology, 82, 109-113.
- Dworkis, D.A., Klings, E.S., Solovieff, N., Li, G., Milton, J.N., Hartley, S.W., Melista, E., Parente, J., Sebastiani, P., Steinberg, M.H. and Baldwin, C.T. (2011) Severe sickle cell anemia is associated with increased plasma levels of Tnf-R1 and Vcam-1. American Journal of Hematology, 86, 220-223. doi:10.1002/ajh.21928
- Cerqueira, B.A., Boas, W.V., Zanette, A.D., Reis, M.G. and Goncalves, M.S. (2011) Increased concentrations of Il-18 and uric acid in sickle cell anemia: Contribution of hemolysis, endothelial activation and the inflammasome. Cytokine, 56, 471-476. doi:10.1016/j.cyto.2011.08.013
- Berry, C.E. and Hare, J.M. (2004) Xanthine oxidoreductase and cardiovascular disease: Molecular mechanisms and pathophysiological implications. The Journal of Physiology, 555, 589-606. doi:10.1113/jphysiol.2003.055913
- Xu, P., Huecksteadt, T.P. and Hoidal, J.R. (1996) Molecular cloning and characterization of the human xanthine dehydrogenase gene (Xdh). Genomics, 34, 173-180. doi:10.1006/geno.1996.0262
- Zhang, C., Hein, T.W., Wang, W., Ren, Y., Shipley, R.D. and Kuo, L. (2006) Activation of Jnk and xanthine oxidase by Tnf-alpha impairs nitric oxide-mediated dilation of coronary arterioles. Journal of Molecular and Cellular Cardiology, 40, 247-257. doi:10.1016/j.yjmcc.2005.11.010
- Martinon, F., Mayor, A. and Tschopp, J. (2009) The inflammasomes: Guardians of the body. Annual Review of Immunology, 27, 229-265. doi:10.1146/annurev.immunol.021908.132715
- Martinon, F., Petrilli, V., Mayor, A., Tardivel, A. and Tschopp, J. (2006) Gout-associated uric acid crystals activate the Nalp3 inflammasome. Nature, 440, 237-241. doi:10.1038/nature04516
- Vieira-de-Abreu, A., Campbell, R.A., Weyrich, A.S. and Zimmerman, G.A. (2012) Platelets: Versatile effector cells in hemostasis, inflammation, and the immune continuum. Seminars in Immunopathology, 34, 5-30. doi:10.1007/s00281-011-0286-4
- Maugeri, N., Baldini, M., Ramirez, G.A., Rovere-Querini, P. and Manfredi, A.A. (2012) Platelet-leukocyte deregulated interactions foster sterile inflammation and tissue damage in immune-mediated vessel diseases. Thrombosis Research, 129, 267-273. doi:10.1016/j.thromres.2011.12.001
- Stokes, K.Y. and Granger, D.N. (2012) Platelets: A critical link between inflammation and microvascular dysfunction. The Journal of Physiology, 590, 1023-1034.
- Chiang, E.Y. and Frenette, P.S. (2005) Sickle cell vasoocclusion. Hematology/Oncology Clinics of North America, 19, 771-784. doi:10.1016/j.hoc.2005.08.002
- Charneski, L. and Congdon, H.B. (2010) Effects of antiplatelet and anticoagulant medications on the vasoocclusive and thrombotic complications of sickle cell disease: A review of the literature. American Journal of HealthSystem Pharmacy: AJHP: Official Journal of the American Society of Health-System Pharmacists, 67, 895-900.
- May, A.E., Seizer, P. and Gawaz, M. (2008) Platelets: Inflammatory firebugs of vascular walls. Arteriosclerosis, Thrombosis, and Vascular Biology, 28, s5-10. doi:10.1161/ATVBAHA.107.158915
- Gawaz, M., Langer, H. and May, A.E. (2005) Platelets in inflammation and atherogenesis. The Journal of Clinical Investigation, 115, 3378-3384. doi:10.1172/JCI27196
- Polanowska-Grabowska, R., Wallace, K., Field, J.J., Chen, L., Marshall, M.A., Figler, R., Gear, A.R. and Linden, J. (2010) P-selectin-mediated platelet-neutrophil aggregate formation activates neutrophils in mouse and human sickle cell disease. Arteriosclerosis, Thrombosis, and Vascular Biology, 30, 2392-2399. doi:10.1161/ATVBAHA.110.211615
- Lee, S.P., Ataga, K.I., Orringer, E.P., Phillips, D.R. and Parise, L.V. (2006) Biologically active Cd40 ligand is elevated in sickle cell anemia: Potential role for plateletmediated inflammation. Arteriosclerosis, Thrombosis, and Vascular Biology, 26, 1626-1631. doi:10.1161/01.ATV.0000220374.00602.a2
- Wolf, P. (1967) The nature and significance of platelet products in human plasma. British Journal of Haematology, 13, 269-288. doi:10.1111/j.1365-2141.1967.tb08741.x
- Amabile, N., Guerin, A.P., Leroyer, A., Mallat, Z., Nguyen, C., Boddaert, J., London, G.M., Tedgui, A. and Boulanger, C.M. (2005) Circulating endothelial microparticles are associated with vascular dysfunction in patients with end-stage renal failure. Journal of the American Society of Nephrology: JASN, 16, 3381-3388. doi:10.1681/ASN.2005050535
- Piccin, A., Murphy, W.G. and Smith, O.P. (2007) Circulating microparticles: Pathophysiology and clinical implications. Blood Reviews, 21, 157-171. doi:10.1016/j.blre.2006.09.001
- Cocucci, E., Racchetti, G. and Meldolesi, J. (2009) Shedding microvesicles: Artefacts no more. Trends in Cell Biology, 19, 43-51. doi:10.1016/j.tcb.2008.11.003
- Ardoin, S.P., Shanahan, J.C. and Pisetsky, D.S. (2007) The role of microparticles in inflammation and thrombosis. Scandinavian Journal of Immunology, 66, 159-165. doi:10.1111/j.1365-3083.2007.01984.x
- van Tits, L.J., van Heerde, W.L., Landburg, P.P., Boderie, M.J., Muskiet, F.A., Jacobs, N., Duits, A.J. and Schnog, J.B. (2009) Plasma annexin A5 and microparticle phosphatidylserine levels are elevated in sickle cell disease and increase further during painful crisis. Biochemical and Biophysical Research Communications, 390, 161- 164. doi:10.1016/j.bbrc.2009.09.102
- Noubouossie, D.C., Le, P.Q., Rozen, L., Debaugnies, F., Ferster, A. and Demulder, A. (2011) Evaluation of the procoagulant activity of endogenous phospholipids in the platelet-free plasma of children with sickle cell disease using functional assays. Thrombosis Research. doi:10.1016/j.thromres.2011.10.016
- Shet, A.S., Aras, O., Gupta, K., Hass, M.J., Rausch, D.J., Saba, N., Koopmeiners, L., Key, N.S. and Hebbel, R.P. (2003) Sickle blood contains tissue factor-positive microparticles derived from endothelial cells and monocytes. Blood, 102, 2678-2683. doi:10.1182/blood-2003-03-0693
- Ataga, K.I., Brittain, J.E., Desai, P., May, R., Jones, S., Delaney, J., Strayhorn, D., Hinderliter, A. and Key, N.S. (2012) Association of coagulation activation with clinical complications in sickle cell disease. PloS One, 7, e29786. doi:10.1371/journal.pone.0029786
- Mahfoudhi, E., Lecluse, Y., Driss, F., Abbes, S., Flaujac, C. and Garcon, L. (2012) Red cells exchanges in sickle cells disease lead to a selective reduction of erythrocytes-derived blood microparticles. British Journal of Haematology, 156, 545-547. doi:10.1111/j.1365-2141.2011.08897.x
- Van Beers, E.J., Schaap, M.C., Berckmans, R.J., Nieuwland, R., Sturk, A., van Doormaal, F.F., Meijers, J.C., Biemond, B.J. and Group, C.S. (2009) Circulating erythrocyte-derived microparticles are associated with coagulation activation in sickle cell disease. Haematologica, 94, 1513-1519. doi:10.3324/haematol.2009.008938
- Michelson, A.D., Rajasekhar, D., Bednarek, F.J. and Barnard, M.R. (2000) Platelet and platelet-derived microparticle surface factor V/Va binding in whole blood: Differences between neonates and adults. Thrombosis and Haemostasis, 84, 689-694.
- Adedeji, M.O., Cespedes, J., Allen, K., Subramony, C. and Hughson, M.D. (2001) Pulmonary thrombotic arteriopathy in patients with sickle cell disease. Archives of Pathology & Laboratory Medicine, 125, 1436-1441.
LIST OF ABBREVIATIONS
BDCA 2: blood dendritic cell antigen 2;
CRP: C-reactive protein;
CVD: cardiovascular disease;
DC: dendritic cells;
EVA: encephalic vascular accident;
GPI: glycosylphosphatidylinositol;
GSH: glutathione;
Hb: hemoglobin;
HBB: β-globin gene;
HBSS: sickle cell anemia;
Hcy: Homocysteine;
HDL-C: high-density lipoprotein cholesterol;
HLA: human leukocyte antigen;
H2O2: hydrogen peroxide;
HOCl: hypochlorous acid;
ICAM-1: intercellular adhesion molecule-1;
IL-1: interleukin-1;
IL-12: interleukin-12;
IL-6: interleukin-6;
INF-α: interferon-α;
LDH: lactate dehydrogenase;
LDL-C: low-density lipoprotein cholesterol;
LRR: leucine rich repeat;
LTB4: leukotriene B4;
mDC: myeloid dendritic cells;
MPO: myeloperoxidase;
MPs: Microparticles;
NO: nitric oxide;
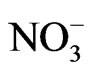 : nitrate;
: nitrate;
NOS: nitric oxide synthase;
[OH]: hydroxyl radical;
ONOO–: peroxynitrite;
OS: oxidative stress;
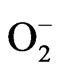 : superoxide;
: superoxide;
PAF: platelet activator factor;
PAI: plasminogen activator inhibitor;
PAMP: pathogen associated molecular patterns;
pDC: plasmacytoid dendritic cells;
PGE2: prostaglandin E2;
PMN: polymorphonuclear leukocytes;
PRR: pattern recognition receptors;
PS: phosphatidylserine;
PSGL-1: P-selectin ligand glycoprotein-1;
RBC: red blood cells;
RNS: reactive nitrogen species;
ROS: reactive oxygen species;
SCD: sickle cell disease;
SNP: Single-nucleotide polymorphisms;
SOD: superoxide dismutase;
sICAM-1: soluble intercellular adhesion molecule-1;
sVCAM-1: soluble vascular cellular adhesion molecule-1;
TF: tissue factor;
TGF-beta: transforming grow factor beta;
TLR: toll like receptor;
TPA: tissue plasminogen activator;
VCAM-1: vascular cellular adhesion molecule-1;
VLDL-C: very low density lipoprotein cholesterol;
VOE: vaso-occlusive episodes;
vWF: von Willebrand factor;
XO: xanthine oxidase.
NOTES
*Corresponding author.