Case Reports in Clinical Medicine
Vol.2 No.2(2013), Article ID:31760,2 pages DOI:10.4236/crcm.2013.22038
A novel mutation in NF1 gene in a Chinese patient with Legius syndrome like café au lait spots
![]()
Department of Oral Biology, Clinic of Oral Rare Diseases and Genetic Diseases, School of Stomatology, The Fourth Military Medical University, Xi’an, China; *Corresponding Author: xhduan@fmmu.edu.cn
Copyright © 2013 Dongdong Zheng, Xiaohong Duan. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 10 March 2013; revised 11 April 2013; accepted 20 April 2013
Keywords: Legius Syndrome; Neurofibromatosis Type I
ABSTRACT
Neurofibromatosis type I (NF1) (OMIM, 162200) and Legius syndrome (OMIM, 611431) both have multiple café au lait spots. It is a challenge to differentiate the two diseases. Here we report a Legius syndrome-like patient who finally was identified as NF1 with a novel mutation in NF1 exon1 (c.55G > T). The mutation caused a substitution of a glutamic acid (GAG) with a terminator codon (TAG). For those patients with only multiple café au lait macules, axillary and inguinal freckling, but without any other clinical signs in NF1 and Legius syndrome, a gene testing is necessary to give a final diagnose. We suggested to use “Neurofibromatosis type 1—like syndrome” to describe those patients just like our patient reported here.
1. INTRODUCTION
Legius syndrome (Online Mendelian Inheritance in Man (OMIM), 611431) is an autosomal dominant genetic disorder presenting mainly with café au lait macules (CALMs), axillary freckling, and macrocephaly. The inactivating sprouty-related EVH1 domain containing protein 1 (SPRED1) mutations have recently been described in Legius syndrome [1,2]. Multiple café au lait spots are the identical cutaneous phenotype in both Legius syndrome and neurofibromatosis type I (NF1) (OMIM, 162200) and the differential diagnosis of these two disorders are very challenging. Here we report a Legius syndrome-like patient who finally was identified as NF1.
2. CASE REPORT
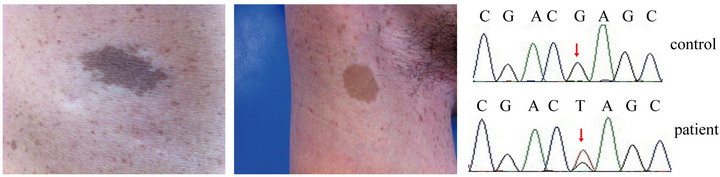
In 2012, a 27-year-old man presented to the Clinic of Oral Rare Diseases and Genetic Diseases, School of Stomatology, the Fourth Military Medical University. He had more than 6 café au lait macules (Figure 1(a)), axillary and inguinal freckling (Figure 1(b)) and did not show any other clinical signs such as skin-fold freckling, macrocephaly, plexiform neurofibromas. MRI and CT image did not show the tumors of the nervous system. Other frequently observed features in NF1 such as certain bone abnormalities, short stature were not observed. The patient did not report a family history. Legius syndrome was diagnosed to the patients according to his identical cutaneous findings. Legius syndrome is caused by the germline loss-of-function SPRED1 mutations, resulting in overactivation of the RAS-MAPK signal transduction cascade [3]. Then the SPRED1 gene of the patient was PCR amplified for sequencing as previously described 1. But sequence analysis revealed no mutations in SPRED1. Since Legius syndrome and NF1 have the similar clinical phenotypes, then neurofibromin 1 (NF1) gene, the candidate gene of NF1, was detected and the final gene mutation analysis revealed that the patient had a novel mutation in NF1 exon1 (c.55G > T). The mutation caused a substitution of a glutamic acid (GAG) with a terminator codon (TAG) (Figure 1(c)). The patient’s parent had no mutation in NF1. In order to remove the possibility of a single nucleotide polymorphism (SNP), we performed NF1 related fragment sequencing on 100 healthy individuals, but the same mutation (NF1 exon1 c.55G > T) was not found in the controls.
3. DISCUSSION
Legius syndrome is often mistaken for NF1 [2]. NF1 diagnostic criteria were established by the National Institutes
 (a) (b) (c)
(a) (b) (c)
Figure 1. Clinical cutaneous phenotypes such as café au lait macules (a) and axillary freckling (b) of the patient. Sequence analysis results of NF1 gene (c). A mutation with G > T transition at cDNA nucleotide 55 in exon 1 was found in the patient.
of Health (NIH) and are widely used to make the diagnosis using information obtained from physical examination, family history, and radiologic studies. NF1 is characterized by multiple CALMs, skin-fold freckling, iris Lisch nodules, and tumors of the nervous system such as optic pathway gliomas, astrocytomas, neurofibromas, and malignant peripheral nerve sheath tumors. NF1 is caused by the mutations in NF1 gene (a regulator of the GTPase activating enzyme (GAP)). A further genetically testing in NF1 gene was used to distinguish NF1 from Legius syndrome in our patient.
The symptoms of Legius syndrome and NF1 are very similar. This is the reason why they are easily confused. An important difference is the absence of tumor growths (Lisch nodules and neurofibromas which are common in NF1) in Legius syndrome [4], so Legius syndrome is often regarded to be less severe of the two syndromes. A genetic testing is often the only way to make sure whether a patient has Legius syndrome or NF1. The similarity of symptoms stem from the fact that the different genes affected in the two syndromes code for proteins that carry out a similar task in the same reaction pathway.
The previous studies evaluated cohorts of individuals with clinical features of NF1 who did not have identifiable NF1 mutations but had SPRED1 mutations, and many patients who met the NIH clinical diagnostic criteria for NF1 did not have an identifiable NF1 mutation by exhaustive mutation analysis [5].
4. CONCLUSION
Due to the hard clinical differential diagnosis between NF1 and Legius syndrome, we suggested to use “Neurofibromatosis Type 1-like Syndrome” to describe those patients with only multiple café au lait macules, axillary and inguinal freckling, but without any other clinical signs in NF1 and Legius syndrome, just like our patient reported here. A gene testing is necessary to give a final diagnose. There are only 7 exons in SPRED1 gene, which is shorter than NF1 gene with 58 exons. The gene structure of SPRED1 is not so complicated as NF1 gene, thus the gene testing is easier in SPRED1 gene than in NF1 gene. As for a patient with the similar clinical symptoms in our report, we suggest to test mutations in SPRED1 gene first in order to save man power, material and financial resources if a mutation in SPRED1 is finally found.
REFERENCES
- Pasmant, E., Sabbagh, A., Hanna, N., Masliah-Planchon, J., Jolly, E., Goussard, P., Ballerini, P., Cartault, F., Barbarot, S., Landman-Parker, J., Soufir, N., Parfait, B., Vidaud, M., Wolkenstein, P., Vidaud, D. and France, R.N. (2009) SPRED1 germline mutations caused a neurofibromatosis type 1 overlapping phenotype. Journal of Medical Genetics, 46, 425-430. doi:10.1136/jmg.2008.065243
- Brems, H., Chmara, M., Sahbatou, M., Denayer, E., Taniguchi, K., Kato, R., Somers, R., Messiaen, L., De Schepper, S., Fryns, JP., Cools, J., Marynen, P., Thomas, G., Yoshimura, A. and Legius, E. (2007) Germline lossof-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nature Genetics, 39, 1120-1126. doi:10.1038/ng2113
- Muram-Zborovski, T.M., Vaughn, C.P., Viskochil, D.H., Hanson, H., Mao, R. and Stevenson, D.A. (2010) NF1 exon 22 analysis of individuals with the clinical diagnosis of neurofibromatosis type 1. Journal of Medical Genetics, 152, 1973-1978.
- Muram-Zborovski, T.M., Stevenson, D.A., Viskochil, D.H., Dries, D.C., Wilson, A.R. and Rong, M. (2010) SPRED 1 mutations in a neurofibromatosis clinic. Journal of Child Neurology, 25, 1203-1209. doi:10.1177/0883073809359540
- Messiaen, L., Yao, S., Brems, H., Callens, T., Sathienkijkanchai, A., Denayer, E., Spencer, E., Arn, P., BabovicVuksanovic, D., Bay, C., Bobele, G., Cohen, B.H., Escobar, L., Eunpu, D., Grebe, T., Greenstein, R., Hachen, R., Irons, M., Kronn, D., Lemire, E., Leppig, K., Lim, C., McDonald, M., Narayanan, V., Pearn, A., Pedersen, R., Powell, B., Shapiro, L.R., Skidmore, D., Tegay, D., Thiese, H., Zackai, E.H., Vijzelaar, R., Taniguchi, K., Ayada, T., Okamoto, F., Yoshimura, A., Parret, A., Korf, B. and Legius, E. (2009) Clinical and mutational spectrum of neurofibromatosis type1-like syndrome. JAMA, 302, 2111- 2118. doi:10.1001/jama.2009.1663