Open Journal of Veterinary Medicine
Vol.3 No.1(2013), Article ID:29128,7 pages DOI:10.4236/ojvm.2013.31013
A Rapid Bioassay for Classical and L-Type Bovine Spongiform Encephalopathies
1Prion Disease Research Center, National Institute of Animal Health, Tsukuba, Japan
2The Roslin Institute and R(D)SVS, University of Edinburgh, Midlothian, UK
3Department of Biochemistry and Cell Biology, National Institute of Infectious Diseases, Tokyo, Japan
4Toyama Institute of Health, Imizu, Japan
5Department of Prion Research, Tohoku University Graduate School of Medicine, Sendai, Japan
Email: *shirou@affrc.go.jp
Received December 29, 2012; revised February 1, 2013; accepted March 3, 2013
Keywords: Bovine Spongiform Encephalopathy; Follicular Dendritic Cell; Transgenic Mice
ABSTRACT
The rapid detection of infectivity of several agents that cause Creutzfeldt-Jakob disease has previously been achieved by assaying for deposits of abnormal prion protein (PrPSc) in follicular dendritic cells in the spleens of transgenic mice carrying the human prion protein gene. In this study, transgenic mice expressing the bovine prion protein were inoculated intraperitoneally with classical (C-type) or atypical L-type bovine spongiform encephalopathies (BSE). Proteinase-resistant PrPSc were detected in the spleens of all transgenic mice at 75 days after inoculation with both types of BSE. Infectivity in PrPSc-positive spleens of the transgenic mice revealed that prions of Cand L-type BSE replicated. These results suggest that bioassay system by the transgenic mice could be useful for the rapid detection of BSE infectivity with discriminating between Cand L-type BSEs.
1. Introduction
Transmissible spongiform encephalopathies (TSEs) are fatal, infectious neurodegenerative diseases; TSEs include bovine spongiform encephalopathy (BSE) in cattle, scrapie in sheep and goats, chronic wasting disease mainly in deer and elk, and Creutzfeldt-Jakob disease (CJD) in humans. BSE was first defined in 1986 in the United Kingdom [1]. There was little variation in the distribution of histopathological changes and the deposition of disease-associated prion protein (PrPSc), which is converted from the host-encoded normal form (PrPC), and also in the glycoform pattern and molecular mass of proteinase K (PK) resistant fragments of PrPSc, named as PrPcore, among BSE cases [2-5]. In addition, transmissibility of BSE agents to inbred mice has revealed defined incubation period from inoculation to development of clinical signs and distinct histopathological characteristics in the brain, as compared to the other TSE agents [6,7].
It was thought that all BSE cases shared the same origin and a single set of characteristics. However, atypical BSEs possessing different prion characteristics have been reported since 2003 in Japan, France, Italy, the United States, Germany, Poland, the Netherlands, Sweden, and the United Kingdom [8-17]. As determined by Western blotting, atypical BSEs are classified into the two types, which show higher (H-type) or lower (L-type) molecular mass of PrPcore on the basis of that in classical (C-type) BSE [8,9,12]. In 2006, a Japanese black cow was diagnosed as an atypical BSE [11]. Transmission of this agent to cattle or transgenic (Tg) mice over-expressing the bovine PrP gene realized that characteristics of histopathological changes and PrPcore were similar to L-type BSE agents found in Italy, Germany, France and Canada [18-21]. Incubation period of the Tg mice affected with L-type BSE was approximately 180 days post intracerebral (IC) inoculation and was shorter period than that with C-type BSE which was approximately 230 days [20, 22-24].
Transmission of human CJD agents to humanized knock-in (Ki) mice demonstrated that PrPSc could be detected in the follicular dendritic cells (FDCs) in the lymphoreticular tissues within 14 days post intraperitoneal (IP) inoculation [25]. Accordingly, the assessment of PrPSc accumulation in splenic FDCs can be used as a rapid method (FDC assay) to ascertain the infectivity of human CJD at 75 days post IP inoculation [25].
We report here that the FDC assay can be used to demonstrate Cand L-type BSE infection in a Ki+Tg#40 mouse that we generated by crossing Ki mouse with Tg mouse. Transmission of BSE into the Ki+Tg mouse was shown rapidly by detection of PrPSc in splenic FDC after IP inoculation compared to the period when the mice develop clinical signs after IC.
2. Materials and Methods
2.1. Transgenic Mouse with a Bovine PrP Gene
We generated Ki+Tg#40 mice by crossing a Ki mouse with a Tg mouse, which carries the same gene construct with the open reading frame of the bovine PrP gene with 6 octarepeats [26]. The Ki+Tg#40 mice carry 12 copies of the bovine PrP gene on the hemizygote of Tg insertion and express about 6-fold more PrPC in the brain than found in Ki mouse brains (Figure S1). When inoculated via IC route with a BSE agent, the Ki+Tg#40 mice showed shorter incubation period than Ki mice and PrPSc deposition in the brain and spleen, differently from the Tg#40 mice backcrossed into PrP-knockout background that showed in the brain but not in the spleen (Table S1).
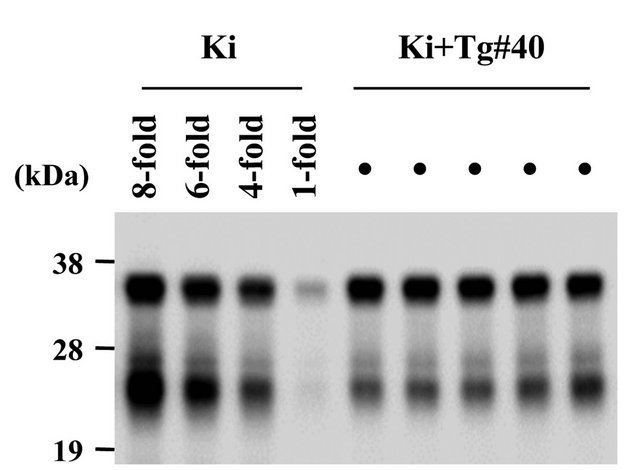
Figure S1. Western blotting of normal brains of Ki+Tg#40 mice and Ki mice. All samples without PK treatment were subjected to Western blotting with 6H4. Five samples of Ki+Tg#40 mice were loaded at 0.1 mg of the brain equivalent to wet weight. For estimating PrPC expression level in the brain of Ki+Tg#40 mice, a sample of Ki mice was loaded at 1-fold, 0.1 mg; 4-fold, 0.4 mg; 6-fold, 0.6 mg; 8-fold, 0.8 mg of the brain equivalent to wet weight. The right bars indicate molecular mass markers, 38 kDa, 28 kDa and 19 kDa.
Table S1. Transmissibility of BSE/UK1 cattle brain to three lines of transgenic mice. Ki mice were generated by genetargeting procedure with the bovine PrP gene. Tg#40 mice were generated by random insertion procedure and by backcrossed into Prnp knockout background. We generated Ki+Tg#40 mice by backcrossing Tg#40 into Ki mouse background.
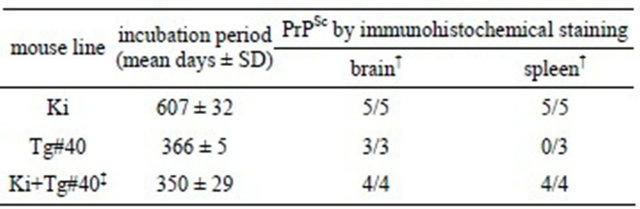
†PrPSc positive number/examined number of mouse; ‡The same data were shown in Table 1.
2.2. BSE Inoculum
Two cattle brains infected with C-type BSE were obtained from the TSE Archive, Veterinary Laboratories Agency, Weybridge, the United Kingdom (reference number: PG2458/98; designated as BSE/UK1) and from the National Institute of Infectious Disease, Tokyo, Japan (the sixth Japanese case of BSE; designated as BSE/JP6). A cattle brain infected with L-type BSE (the 24th Japanese case of BSE; designated as BSE/JP24) was also obtained from the National Institute of Infectious Disease. These 3 cattle brains were found to be positive for BSE PrPSc by Western blotting. As a negative control, a normal brain sample from a healthy cow was obtained from a slaughterhouse and was found to be negative for BSE PrPSc by Western blotting. To prepare the inoculum for transmission and Western blotting, each cattle brain sample was homogenized at 10% (w/v) with phosphate-buffered saline (PBS) using Multi-beads shocker (Yasui Kikai Corp., Osaka, Japan), and debris was removed by centrifuging at 900 × g for 10 min.
2.3. Transmission Studies
This animal experiment was approved by the Animal Experiment Committee at the National Institute of Animal Health. For the FDC assay, Ki+Tg#40 mice were inoculated via IP route with 50 μl of 10% cattle brain homogenate and were euthanized 75 days after inoculation. The spleens of mice were harvested and divided in two pieces; one was fixed with 4% buffered formalin for immunohistochemical staining and the other was frozen at –80˚C for Western blotting and subsequent transmission. Mice were defined as positive for BSE transmission if PrPSc deposition in splenic germinal center was observed by immunohistochemical staining and/or if the presence of PrPcore in the spleen was detected by Western blotting.
To confirm the BSE infectivity in cattle brain or mouse spleen, Ki+Tg#40 mice were inoculated via IC route with 20 ul of 10% homogenate from cattle brain or PrPSc-positive spleens of three Ki+Tg#40 mice at 75 days after IP inoculation. Incubation periods were defined as the day from inoculation to euthanasia with clinical signs of mouse BSE, including tremors, a high-arched back, incoercible food intake, and significant loss of body weight. BSE transmission to mice was confirmed by histopathological analysis with observation of vacuolation and PrPSc deposition in the brain.
2.4. Immunohistochemical Staining
Hydrolytic autoclaving pretreatment was used to enhance antigen retrieval. An anti-PrP rabbit serum previously raised against the N-terminal of PrP was used as the primary antibody [27]. Universal immunoperoxidase polymer (Histofine MAX PO kit; Nichirei Biosciences Inc., Tokyo, Japan) was used as the secondary antibody for the detection of PrPSc accumulation.
2.5. Western Blotting
The PrPcore was extracted from animal tissues by collagenase and PK treatments, with some modifications to a method described previously [16]. Samples were concentrated after the extractions using the phosphotungstic acid method [28]. PNGase F (New England Biolabs, Beverly, MA) was used for the deglycosylation of PrPcore. Prior to electrophoresis, the samples were resolved in NuPAGE® LDS Sample Buffer (Invitrogen Corp., Carlsbad, CA) containing 1 mM dithiothreitol after treatment at 100˚C for 10 min on a heat block. The samples were subsequently separated by a NuPAGE® 12% Bis-Tris gel (Invitrogen Corp.) in NuPAGE® MOPS SDS Running Buffer (Invitrogen Corp.) and were transferred electrically onto an Immobilon-P transfer membrane (Millipore, Bedford, MA). The antiPrP monoclonal antibody 6H4 (Prionics AG, Zurich, Switzerland) was used as the primary antibody. The PrP signal was generated using Envision (Dako Japan Inc., Tokyo, Japan) and SuperSignal® West Dura Extended Duration Substrate (Pierce Biotechnology Inc., Rockford, IL) and was imaged directly with the Fluochem™ imaging system (Alpha Innotech, San Leandro, CA).
The relative amounts of di-, mono-, and un-glycosylated forms of PrPcore were calculated as the mean percentages of the total signal of the 3 glycoforms from 5 repetitions of the Western blot analysis of the cattle and mouse brains. These calculations were performed using AlphaEaseFC™ software (Alpha Innotech).
3. Results
The spleens of all the Ki+Tg#40 mice at 75 days after IP inoculation with BSE cattle brains were positive for PrPSc deposition (Table 1). Immunohistochemical staining showed that PrPSc accumulations were finely stained in the germinal center of the spleen with no distinction between BSE/UK1 and BSE/JP24 (Figure 1(a)). Western blotting showed PrPcore in the spleen of Ki+Tg#40 mice at 75 days post IP inoculation with BSE cattle brains, but not in the brain (Figure 1(b)). None of the mice inoculated with a normal cattle brain showed any positive results for PrPSc (data not shown).
All Ki+Tg#40 mice inoculated via IC route with the BSE cattle brains developed clinical signs. The mean incubation periods were 350 days with BSE/UK1, 431 days with BSE/JP6, and 313 days with BSE/JP24 (Table 1). Sequential passage with the PrPSc-positive mouse spleens that were harvested at 75 days post IP inoculation showed that all Ki+Tg#40 mice developed clinical signs after IC inoculation (Table 1). The mean incubation periods were 365 days with BSE/UK1 mouse spleen and 338 days with BSE/JP24 mouse spleen.
Histopathological analysis using haematoxylin-eosin staining showed severe vacuolation in the hippocampus of the Ki+Tg#40 mice inoculated with the BSE/JP24 cattle brain or mouse spleen (Figures 2(a) and (b)). Moreover, mild vacuolation was observed in the brain cortex and thalamus of these mice. In contrast, less vacuolation
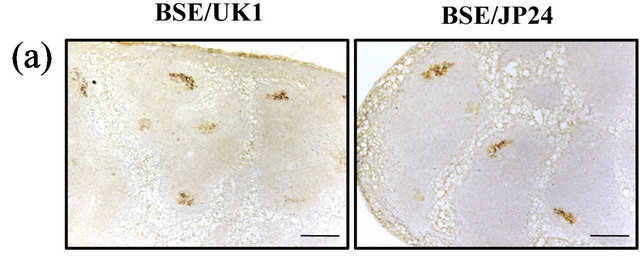
Figure 1. PrPSc accumulation in the Ki+Tg#40 mice at 75 days after IP inoculation. (a) Immunohistochemical staining of the spleen of Ki+Tg#40 mice inoculated with homogenized BSE/UK1 cattle brain (10%) or BSE/JP24 cattle brain (10%). The scale bars indicate a length of 0.2 mm. (b) Western blotting of the spleen and brain of Ki+Tg#40 mice inoculated with BSE cattle brains. All samples of the spleen and brain of Ki+Tg#40 mice were digested with proteinase K (0.1 mg/mL) and were loaded four mg tissue equivalent to the wet weight. The left bars indicate molecular mass markers of 29, 18, and 14 kDa.
Table 1. Transmission of BSE to Ki+Tg#40 mice.
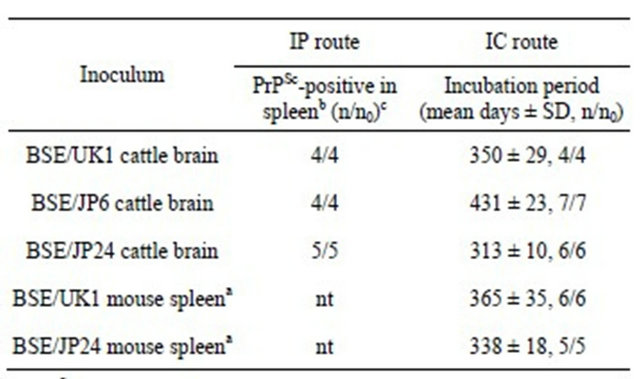
1) PrPSc-positive mouse spleen at 75 days post IP inoculation with BSE cattle brains. 2) 75 days after inoculation. 3) n/n0: number of positive mice per number of inoculated mice, nt: not tested.
Figure 2. Histopathological examination of the clinically affected Ki+Tg#40 mice. (a)-(d) Haematoxylin-eosin staining of the hippocampus and (e)-(h) immunohistochemical staining of the thalamus. Ki+Tg#40 mice were inoculated via IC route with BSE/JP24 cattle brain (a), (e) or mouse spleen (b), (f), and with BSE/UK1 cattle brain (c), (g) or mouse spleen (d), (h). Scale bars indicate a length of 0.1 mm.
was observed in the hippocampus of mice inoculated with the BSE/UK1 cattle brain or mouse spleen (Figures 2(c) and (d)). Immunohistochemical staining revealed a fine granular deposition of PrPSc in the thalamus of mice inoculated with the BSE/JP24 cattle brain or mouse spleen (Figures 2(e) and (f)), while a coarse granular deposition of PrPSc was found in the thalamus of mice inoculated with BSE/UK1 cattle brain or mouse spleen (Figures 2(g) and (h)). A similar pattern of coarse granular deposition was observed in the mice inoculated with BSE/JP6 cattle brain (data not shown).
Western blot analysis showed that the PrPcore characteristics of BSE/JP24 in the cattle brain, the spleen and brain of the transmitted Ki+Tg#40 mice were different from those of BSE/UK1 and BSE/JP6 in the corresponding tissues (Figure 3(a)). The relative amounts of the di-, mono-, and un-glycosylated forms of PrPcore in the cattle brain were present in the approximate ratio of 37:40:23 in BSE/JP24, 71:21:7 in BSE/UK1, and 81:17:2 in BSE/ JP6 (Figure 3(b)). Similarly, the different forms of PrPcore in the mouse brains were present in an approximate ratio of 43:35:22 in BSE/JP24, 77:17:7 in BSE/UK1, and 82:14:3 in BSE/JP6. In the mouse spleens, the different forms of PrPcore were present in an approximate ratio of 54:34:12 in BSE/JP24, 77:18:5 in BSE/UK1, and 79:18:3 in BSE/JP6. The relative amount of the di-glycosylated form in the brains and the spleens of mice transmitted
Figure 3. Western blot analysis of BSE cattle and inoculated Ki+Tg#40 mice. All samples were treated with PK (0.1 mg/ml) for 30 min. The left bars indicate molecular mass markers. (a) Western blot analysis of cattle brain (applied at 3 mg of tissue equivalent to the wet weight), mouse spleen (4 mg of tissue equivalent to the wet weight), and mouse brain (0.05 mg of tissue equivalent to the wet weight). Lanes 1 - 4 indicate the cattle brains of the BSE negative control, BSE/UK1, BSE/JP24, and BSE/JP6, respectively. Lanes 5 - 7 indicate mouse spleens of the Ki+Tg#40 at 75 days after IP inoculation with BSE/UK1, BSE/JP24, and BSE/JP6, respectively. Lanes 8 - 10 indicate brains of the Ki+Tg#40 inoculated via IC route with BSE/UK1, BSE/JP24, and BSE/JP6, respectively. Lanes 11 and 12 are the brain and spleen, respectively, of Ki+Tg#40 inoculated with normal cattle brain. (b) Comparison of the glycoform ratio of PrPSc. The proportion, mean (standard deviation), of di-(black bars), mono-(gray bars), and un-glycosylated (white bars) PrPSc were measured. Standard deviation is indicated by the error bar. (c) Deglycosylation experiment. Lanes indicate the same samples as in (a). The following quantities of tissue equivalent to the wet weight were applied, in order of lanes 1 - 12: 0.75 mg; 0.75 mg; 0.75 mg; 0.75 mg; 1 mg; 1 mg; 1 mg; 0.01 mg; 0.01 mg; 0.01 mg; 0.01 mg; 1 mg.
with BSE/JP24 was slightly higher than that in the original cattle brain, but was not predominant (Figure 3(b)).
Western blot analysis also revealed that the unglycosylated molecular masses of PrPcore in the BSE/JP24 cattle brain, mouse spleen and mouse brain were lower than those in BSE/UK1 and BSE/JP6, which was confirmed by deglycosylation with PNGase F (Figure 3(c)).
4. Discussion
The FDC assay in the Ki+Tg#40 mice realized that PrPSc was generated in the mouse spleens within 75 days post IP inoculation with BSE cattle brains (Figure 1(a)). The PrPSc-positive mouse spleen had a well-established infectivity to Ki+Tg#40 mice (Table 1). In the brain of the Ki+Tg#40 mice, no PrPSc was not detected at 75 days post IP inoculation with C-type or L-type BSE (Figure 1(b)) although the PrPSc in the brain was detected when the Ki+Tg#40 mice developed overt clinical signs post IC (Figures 2 and 3) and IP inoculation (data not shown). It is reported in other TSEs following peripheral routes of infection that PrPSc accumulation occurs in FDC of lymphoid tissues prior to in the neural tissues [29,30]. These results support that Ki+Tg#40 mice showing PrPSc accumulation in FDC should develop BSE at preclinical stage after IP inoculation.
Immunohistochemical staining of the mouse spleen at 75 days post IP inoculation showed appearance of PrPSc accumulation in L-type BSE was indistinguishable from that in C-type (Figure 1(a)). Subsequent passage of the PrPSc-positive mouse spleen, however, realized in the affected brain that histopathological properties of L-type and C-type BSEs were distinct from each other and similar to those after transmitted with the original cattle brain (Figure 2). These results suggest that prions of Ltype and C-type BSEs should replicate in the mouse spleen at 75 days post IP inoculation.
It was easy to discriminate L-type BSE from C-type by Western blotting of the mouse spleen at 75 days post IP inoculation. The molecular mass of deglycosylated PrPcore in L-type BSE was apparently lower than that from C-type BSE (Figure 3(c)). The relative amount of the diglycosylated PrPcore in L-type BSE was less than that in C-type BSE, but slightly increased comparing to that in the original cattle brain (Figure 3). It has reported in other TSEs that the glycoform pattern and molecular mass of PrPcore was controlled not only by the prionspecific conformation but also by the PrPSc distribution in the tissue and brain regions [31]. The slight increasing of di-glycosylated PrPcore in the L-type BSE was shown in the mouse brain as like as in the spleen (Figure 3), suggesting that glycoform pattern of PrPcore could be controlled slightly by unknown factors in mouse during the replication of prion.
In conclusion, the FDC assay in Ki+Tg#40 mouse enables to verify the infectivity of Cand L-type BSEs rapidly and could also discriminate L-type from C-type BSE. For establishing the FDC assay as diagnosis tool of BSE, we have preceded the further studies of sensitivity and specification. This FDC assay system may archive a more rapid risk-evaluation of BSE infection to humans and cattle.
5. Acknowledgements
The authors thank the researchers and technical assistants at the Prion Disease Research Center, National Institute of Animal Health, and H. Kudo of the Tohoku University Graduate School of Medicine for providing technical assistance. This study was supported in part by a Grantin-Aid from the BSE and other Prion Disease Control Project of the Ministry of Agriculture, Forestry, and Fisheries of Japan; a grant for BSE research from the Ministry of Health, Labour and Welfare of Japan; and Research and Development Program for New Bioindustry Initiative from the Ministry of Agriculture, Forestry, and Fisheries of Japan.
REFERENCES
- G. A. Wells, A. C. Scott, C. T. Johnson, R. F. Gunning, R. D. Hancock, M. Jeffrey, M. Dawson and R. Bradley, “A Novel Progressive Spongiform Encephalopathy in Cattle,” Veterinary Record, Vol. 121, No. 18, 1987, pp. 419- 420. doi:10.1136/vr.121.18.419
- G. A. Wells and J. W. Wilesmith, “The Neuropathology and Epidemiology of Bovine Spongiform Encephalopathy,” Brain Pathology, Vol. 5, No. 1, 1995, pp. 91-103. doi:10.1111/j.1750-3639.1995.tb00580.x
- J. Collinge, K. C. Sidle, J. Meads, J. Ironside and A. F. Hill, “Molecular Analysis of Prion Strain Variation and the Aetiology of ‘New Variant’ CJD,” Nature, Vol. 383, No. 6602, 1996, pp. 685-690. doi:10.1038/383685a0
- A. F. Hill, M. Desbruslais, S. Joiner, K. C. Sidle, I. Gowland, J. Collinge, L. J. Doey and P. Lantos, “The Same Prion Strain Causes vCJD and BSE,” Nature, Vol. 389, No. 6650, 1997, pp. 448-450, 526. doi:10.1038/38925
- T. Kuczius, I. Haist and M. H. Groschup, “Molecular Analysis of Bovine Spongiform Encephalopathy and Scrapie Strain Variation,” Journal of Infectious Diseases, Vol. 178, No. 3, 1998, pp. 693-699. doi:10.1086/515337
- M. E. Bruce, R. G. Will, J. W. Ironside, I. McConnell, D. Drummond, A. Suttie, L. McCardle, A. Chree, J. Hope, C. Birkett, S. Cousens, H. Fraser and C. J. Bostock, “Transmissions to Mice Indicate That ‘New Variant’ CJD Is Caused by the BSE Agent,” Nature, Vol. 389, No. 6650, 1997, pp. 498-501. doi:10.1038/39057
- M. E. Bruce, A. Boyle, S. Cousens, I. McConnell, J. Foster, W. Goldmann and H. Fraser, “Strain Characterization of Natural Sheep Scrapie and Comparison with BSE,” Journal of General Virology, Vol. 83, No. 3, 2002, pp. 695-704.
- A. G. Biacabe, J. L. Laplanche, S. Ryder and T. Baron, “Distinct Molecular Phenotypes in Bovine Prion Diseases,” EMBO Reports, Vol. 5, No. 1, 2004, pp. 110-115. doi:10.1038/sj.embor.7400054
- A. Buschmann, A. Gretzschel, A. G. Biacabe, K. Schiebel, C. Corona, C. Hoffmann, M. Eiden, T. Baron, C. Casalone and M. H. Groschup, “Atypical BSE in Germany— Proof of Transmissibility and Biochemical Characterization,” Veterinary Microbiology, Vol. 117, No. 2-4, 2006, pp. 103-116. doi:10.1016/j.vetmic.2006.06.016
- C. Casalone, G. Zanusso, P. Acutis, S. Ferrari, L. Capucci, F. Tagliavini, S. Monaco and M. Caramelli, “Identification of a Second Bovine Amyloidotic Spongiform Encephalopathy: Molecular Similarities with Sporadic Creutzfeldt-Jakob Disease,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 101, No. 9, 2004, pp. 3065-3070. doi:10.1073/pnas.0305777101
- K. Hagiwara, Y. Yamakawa, Y. Sato, Y. Nakamura, M. Tobiume, M. Shinagawa and T. Sata, “Accumulation of Mono-Glycosylated Form-Rich, Plaque-Forming PrP(Sc) in the Second Atypical Bovine Spongiform Encephalopathy Case in Japan,” Japanese Journal of Infectious Diseases, Vol. 60, No. 5, 2007, pp. 305-308.
- J. G. Jacobs, J. P. Langeveld, A. G. Biacabe, P. L. Acutis, M. P. Polak, D. Gavier-Widen, A. Buschmann, M. Caramelli, C. Casalone, M. Mazza, M. Groschup, J. H. Erkens, A. Davidse, F. G. van Zijderveld and T. Baron, “Molecular Discrimination of Atypical Bovine Spongiform Encephalopathy Strains from a Geographical Region Spanning a Wide Area in Europe,” Journal of Clinical Microbiology, Vol. 45, No. 6, 2007, pp. 1821-1829. doi:10.1128/JCM.00160-07
- M. P. Polak, J. F. Zmudzinski, J. G. Jacobs and J. P. Langeveld, “Atypical Status of Bovine Spongiform Encephalopathy in Poland: A Molecular Typing Study,” Archives of Virology, Vol. 153, No. 1, 2008, pp. 69-79. doi:10.1007/s00705-007-1062-6
- J. A. Richt, R. A. Kunkle, D. Alt, E. M. Nicholson, A. N. Hamir, S. Czub, J. Kluge, A. J. Davis and S. M. Hall, “Identification and Characterization of Two Bovine Spongiform Encephalopathy Cases Diagnosed in the United States,” Journal of Veterinary Diagnostic Investigation, Vol. 19, No. 2, 2007, pp. 142-154. doi:10.1177/104063870701900202
- L. A. Terry, R. Jenkins, L. Thorne, S. J. Everest, M. J. Chaplin, L. A. Davis and M. J. Stack, “First Case of HType Bovine Spongiform Encephalopathy Identified in Great Britain,” Veterinary Record, Vol. 160, No. 25, 2007, pp. 873-874. doi:10.1136/vr.160.25.873
- Y. Yamakawa, K. Hagiwara, K. Nohtomi, Y. Nakamura, M. Nishijima, Y. Higuchi, Y. Sato and T. Sata, “Atypical Proteinase K-Resistant Prion Protein (PrPres) Observed in an Apparently Healthy 23-Month-Old Holstein Steer,” Japanese Journal of Infectious Diseases, Vol. 56, No. 5-6, 2003, pp. 221-222.
- D. Gavier-Widen, M. Noremark, J. P. Langeveld, M. Stack, A. G. Biacabe, J. Vulin, M. Chaplin, J. A. Richt, J. Jacobs, C. Acin, E. Monleon, L. Renstrom, B. Klingeborn and T. G. Baron, “Bovine Spongiform Encephalopathy in Sweden: An H-Type Variant,” Journal of Veterinary Diagnostic Investigation, Vol. 20, No. 1, 2008, pp. 2-10. doi:10.1177/104063870802000102
- S. Fukuda, Y. Iwamaru, M. Imamura, K. Masujin, Y. Shimizu, Y. Matsuura, Y. Shu, M. Kurachi, K. Kasai, Y. Murayama, S. Onoe, K. Hagiwara, T. Sata, S. Mohri, T. Yokoyama and H. Okada, “Intraspecies Transmission of L-Type-Like Bovine Spongiform Encephalopathy Detected in Japan,” Microbiology and Immunology, Vol. 53, No. 12, 2009, pp. 704-707. doi:10.1111/j.1348-0421.2009.00169.x
- Y. Iwamaru, M. Imamura, Y. Matsuura, K. Masujin, Y. Shimizu, Y. Shu, M. Kurachi, K. Kasai, Y. Murayama, S. Fukuda, S. Onoe, K. Hagiwara, Y. Yamakawa, T. Sata, S. Mohri, H. Okada and T. Yokoyama, “Accumulation of LType Bovine Prions in Peripheral Nerve Tissues,” Emerging Infectious Diseases, Vol. 16, No. 7, 2010, pp. 1151- 1154. doi:10.3201/eid1607.091882
- K. Masujin, Y. Shu, Y. Yamakawa, K. Hagiwara, T. Sata, Y. Matsuura, Y. Iwamaru, M. Imamura, H. Okada, S. Mohri and T. Yokoyama, “Biological and Biochemical Characterization of L-Type-Like Bovine Spongiform Encephalopathy (BSE) Detected in Japanese Black Beef Cattle,” Prion, Vol. 2, No. 3, 2008, pp. 123-128. doi:10.4161/pri.2.3.7437
- K. Masujin, R. Miwa, H. Okada, S. Mohri and T. Yokoyama, “Comparative Analysis of Japanese and Foreign LType BSE Prions,” Prion, Vol. 6, No. 1, 2012, pp. 89-93. doi:10.4161/pri.6.1.18429
- A. Buschmann and M. H. Groschup, “Highly Bovine Spongiform Encephalopathy-Sensitive Transgenic Mice Confirm the Essential Restriction of Infectivity to the Nervous System in Clinically Diseased Cattle,” Journal of Infectious Diseases, Vol. 192, No. 5, 2005, pp. 934- 942. doi:10.1086/431602
- V. Béingue, A. Bencsik, A. Le Dur, F. Reine, T. L. Lai, N. Chenais, G. Tilly, A. G. Biacabe, T. Baron, J. L. Vilotte and H. Laude, “Isolation from Cattle of a Prion Strain Distinct from That Causing Bovine Spongiform Encephalopathy,” PLoS Pathogens, Vol. 2, No. 10, 2006, p. e112. doi:10.1371/journal.ppat.0020112
- R. Capobianco, C. Casalone, S. Suardi, M. Mangieri, C. Miccolo, L. Limido, M. Catania, G. Rossi, G. Di Fede, G. Giaccone, M. G. Bruzzone, L. Minati, C. Corona, P. Acutis, D. Gelmetti, G. Lombardi, M. H. Groschup, A. Buschmann, G. Zanusso, S. Monaco, M. Caramelli and F. Tagliavini, “Conversion of the BASE Prion Strain into the BSE Strain: The Origin of BSE?” PLoS Pathogens, Vol. 3, No. 3, 2007, p. e31. doi:10.1371/journal.ppat.0030031
- T. Kitamoto, S. Mohri, J. W. Ironside, I. Miyoshi, T. Tanaka, N. Kitamoto, S. Itohara, N. Kasai, M. Katsuki, J. Higuchi, T. Muramoto and R. W. Shin, “Follicular Dendritic Cell of the Knock-In Mouse Provides a New Bioassay for Human Prions,” Biochemical and Biophysical Research Communications, Vol. 294, No. 2, 2002, pp. 280- 286. doi:10.1016/S0006-291X(02)00476-X
- M. Asano, S. Mohri, J. W. Ironside, M. Ito, N. Tamaoki and T. Kitamoto, “vCJD Prion Acquires Altered Virulence through Trans-Species Infection,” Biochemical and Biophysical Research Communications, Vol. 342, No. 1, 2006, pp. 293-299. doi:10.1016/j.bbrc.2006.01.149
- T. Kitamoto, K. Doh-ura, T. Muramoto, M. Miyazono and J. Tateishi, “The Primary Structure of the Prion Protein Influences the Distribution of Abnormal Prion Protein in the Central Nervous System,” American Journal of Pathology, Vol. 141, No. 2, 1992, pp. 271-277.
- J. D. Wadsworth, S. Joiner, A. F. Hill, T. A. Campbell, M. Desbruslais, P. J. Luthert and J. Collinge, “Tissue Distribution of Protease Resistant Prion Protein in Variant Creutzfeldt-Jakob Disease Using a Highly Sensitive Immunoblotting Assay,” Lancet, Vol. 358, No. 9277, 2001, pp. 171-180. doi:10.1016/S0140-6736(01)05403-4
- T. Muramoto, T. Kitamoto, J. Tateishi and I. Goto, “The Sequential Development of abnormal Prion Protein Accumulation in Mice with Creutzfeldt-Jakob Disease,” American Journal of Pathology, Vol. 140, No. 6, 1992, pp. 1411-1420.
- L. McCulloch, K. L. Brown, B. M. Bradford, J. Hopkins, M. Bailey, K. Rajewsky, J. C. Manson and N. A. Mabbott, “Follicular Dendritic Cell-Specific Prion Protein (PrP) Expression Alone Is Sufficient to Sustain Prion Infection in the Spleen,” PLoS Pathogens, Vol. 7, No. 12, 2011, Article ID: e1002402. doi:10.1371/journal.ppat.1002402
- B. E. Schreuder and R. A. Somerville, “Bovine Spongiform Encephalopathy in Sheep?” Revue Scientifique et Technique, Vol. 22, No. 1, 2003, pp. 103-120.
NOTES
*Corresponding author.