Open Journal of Pediatrics
Vol. 2 No. 1 (2012) , Article ID: 18009 , 5 pages DOI:10.4236/ojped.2012.21013
Anomalous left coronary artery from pulmonary artery: Case series and brief review
![]()
1Hamad Medical Corporation, Doha, Qatar
2Southampton University Hospital, Southampton, UK
Email: *dilawarmd@yahoo.com, drzaheer@yahoo.com
Received 8 August 2011; revised 10 October 2011; accepted 30 November 2011
Keywords: Anomalous Origin of Left Coronary Artery from Pulmonary Artery
ABSTRACT
Anomalous origin of left coronary artery from the pulmonary artery (ALCAPA) is a rare congenital coronary anomaly. In this study, we present all the ALCAPA patients which were admitted at our institution during April 2007-December 2010. Retrospective review of these patients regarding their clinical presentation and the use of diagnostic modalities will be presented in this series. There were total of five patients, three male and 2 female, with age range of 2 - 12 months. The most common symptoms at presentation were tachypnea (4/5) and poor feeding with irritability (3/5). Electrocardiogram was abnormal in 2/5 cases and chest X ray revealed cardiomegaly with pulmonary congestion in 4/5 patients. Echocardiogram showed mitral valve regurgitation in 5/5 cases (3 with moderate and 2 with mild to moderate), Left ventricular dilatation/dysfunction in 4/5 patients, echogenic left ventricular papillary muscles in 4/5 patients and prominent right coronary with strong suspecision of ALCAPA in 4/5 patients. Coronary angiography was performed in 4/5 cases to confirm the diagnosis. We conclude that by thorough clinical assessment along with ECG and CXR, the diagnosis of ALCAPA can be strongly suspected. Echocardiogram can almost always make the diagnosis of ALCAPA and coronary angiography can confirm the diagnosis in rare atypical cases.
1. INTRODUCTION
Anomalous origin of the left coronary artery from the pulmonary artery is a rare congenital cardiovascular defect that occurs in approximately 1/300,000 live births or 0.5% of children with congenital heart diseases [1]. This anomaly causes coronary blood steal to pulmonary artery leading to myocardial ischemia and if left untreated can lead to dilated cardiomyopathy and death. The mortality of untreated ALCAPA has been estimated to be more than 90% during first year of life [2]. However, patients can survive past infancy and even into adulthood without being symptomatic until later in life. In teenagers and adults, ALCAPA may be an important cause of malignant ventricular arrhythmias and sudden cardiac arrest [3].
Classically, ALCAPA patients present at 2 - 6 weeks of age with history of irritability and inconsolable crying during feeding and with symptoms/signs of heart failure i.e. tachypnea, tachycardia, sweating, poor weight gain, cool extremities, weak pulses, hepatomegaly, crepitations and cardiomegaly with plethoric lung fields. Electrocardiogram reveals deep Q waves and ST-T wave changes in infero-lateral leads and echocardiogram shows poor left ventricular function with possible septal-wall motion abnormalities, mitral valve regurgitation and collaterals flowing in to ALCAPA which ultimately drains in to pulmonary artery. In rare instances, the clinical picture may be atypical and ECG or echocardiogram may not be classical for the diagnosis of ALCAPA. In such atypical cases, cardiac catheterization or computerized axial tomographic scan (CT scan)/magnetic resonance angiography (MRA) may be indicated to confirm the diagnosis. With variable presentation of ALCAPA, it can mimic with non-cardiac causes such as infantile colic, food intolerance and gastroesophageal reflux [4].
2. MATERIALS AND METHODS
From April 2007-December 2010, we retrospectively reviewed all the ALCAPA patients diagnosed here at Hamad General Hospital, Doha, Qatar. Table 1 shows the demographic data and the diagnostic modalities used to confirm the diagnosis of these ALCAPA patients.
2.1. Case #1
This full term male baby was admitted to our neonatal
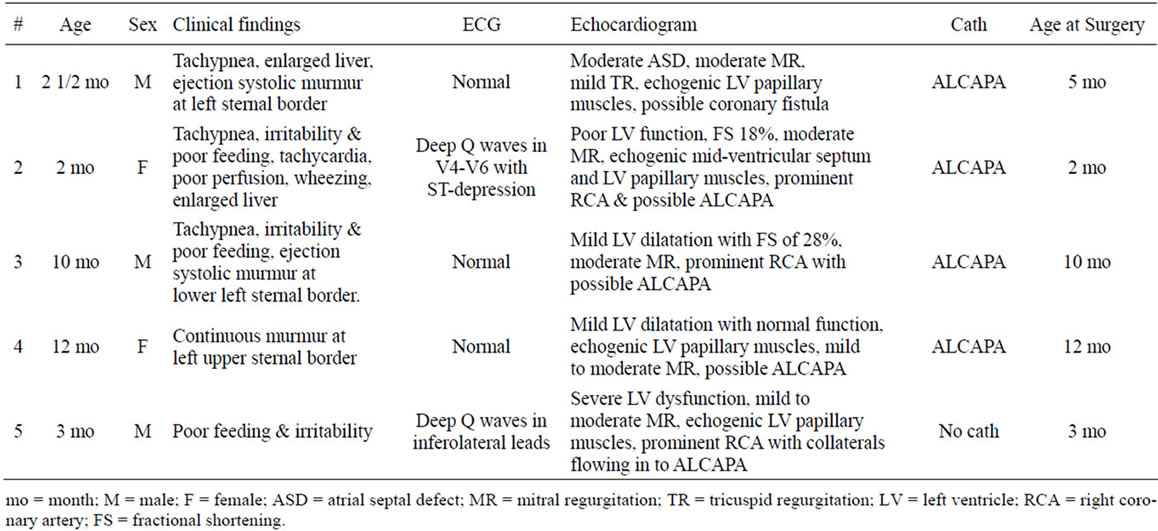
Table 1. Summary of clinical presentation and diagnostic work-up for ALCAPA patients.
intensive care unit immediately after birth because of perinatal asphyxia and cardiology was consulted at four days of age for the evaluation of heart murmur. His cardiovascular examination was unremarkable apart from systolic murmur at left sternal border and electrocardiogram (ECG) and chest x ray (CXR) were normal. Echocardiogram at four days of age showed moderate size secundum atrial septal defect, moderate tricuspid valve regurgitation and normal rest of cardiac anatomy and function. Baby was discharged home with follow up appointment in pediatric cardiology clinic.
Baby was readmitted at two and a half months of age with history of tachypnea and fever. His cardiovascular examination was unremarkable except for II/VI ejection systolic murmur at left sternal border and liver was 3 cm below the right costal margin. Repeat echocardiogram at two and a half months of age revealed moderate size secundum atrial septal defect, moderate mitral valve regurgitation, mild tricuspid valve regurgitation, echogenic left ventricle papillary muscles, coronary fistulae like flow at interventricular septum, normal looking right and left coronary arteries and normal biventricular function. CXR showed mild cardiomegaly with mild pulmonary congestion and ECG was normal.
The patient was offered cardiac catheterization for further evaluation of coronary artery anatomy and possible intervention but family took this patient abroad where he was catheterized, the diagnosis of ALCAPA) was made by coronary angiography and was operated for reimplantation of ALCAPA to aortic root at five months of age.
2.2. Case #2
This two months old female patient was admitted to pediatric intensive care unit with history of tachypnea, poor feeding and irritability especially during feeding for the past two weeks. Her examination revealed tachycardia, poor peripheral perfusion, bilateral wheezing and liver was 4 - 5 cm below the right costal margin. Baby was hemodynamically compromised and needed mechanical ventilation and inotropic support. CXR showed cardiomegaly with plethoric lungs and ECG revealed deep Q waves in leads V4 - V6 with ST depression. Echocardiogram showed dilated left ventricle with poor function, fractional shortening of 18%, moderate mitral valve regurgitation, echogenic mid-ventricular septum with akinesia, echogenic left ventricular papillary muscles, prominent right coronary artery and the diagnosis of ALCAPA was strongly suspected. Baby was catheterized, coronary artery angiogram confirmed the diagnosis of ALCAPA and she was operated for reimplantation of ALCAPA to aortic root.
2.3. Case #3
This ten months old male was admitted to pediatric intensive care unit with tachypnea and poor feeding with irritability for the past few days. Positive physical findings were irritability, mild tachypnea and II/VI ejection systolic murmur at left lower sternal border. CXR showed mild cardiomegaly with plethoric lungs and ECG was normal. Echocardiogram showed mild left ventricular dilatation with fractional shortening of 28%, moderate mitral valve regurgitation, dilated right coronary artery, unable to visualize left coronary artery, prominent coronary flow in ventricular septum and abnormal blood flow was seen in to the main pulmonary artery. ALCAPA was strongly suspected, the diagnosis was confirmed by coronary angiography and reimplantation of ALCAPA to aortic root was done.
2.4. Case #4
This twelve months old female was referred to cardiology for evaluation of heart murmur. Apart from rickets, she was asymptomatic and her vital signs were normal. Cardiovascular examination revealed II/VI continuous murmur at left upper sternal border. ECG and CXR were normal and echocardiogram showed mildly dilated left ventricle, echogenic left ventricular papillary muscles, mild to moderate mitral valve regurgitation, normal biventricular function and abnormal blood flow in to main pulmonary artery was seen with strong suspicion of ALCAPA. Coronary angiogram confirmed the diagnosis of ALCAPA (Figure 1) and patient was operated for the reimplantation of ALCAPA to aortic root.
2.5. Case #5
This thirty weeker premature male baby was echoed at five days of age which revealed patent foramen of ovale, small to moderate size patent ductus arteriosus which got treated with indomethacin. He was seen at five weeks of age in pediatric cardiology clinic and was found to be asymptomatic with normal cardiac examination and follow up echocardiogram at three months of age was scheduled. At three months follow up, parents mentioned that baby has been irritable with excessive crying and poor feeding with slow weight gain for the past few weeks. His examination revealed, tachypnea, tachycardia,
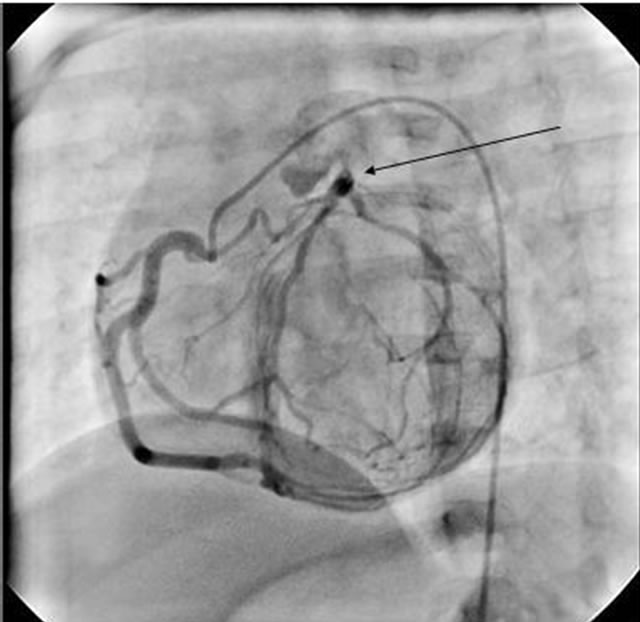
Figure 1. Selective right coronary artery angiogram of case #4, showing right coronary artery with collateral flow in to left coronary artery which ultimately drains in the pulmonary artery.
enlarged liver, cardiomegaly with plethpric lungs on CXR and ECG showed deep Q waves in infero-lateral (lead 1, aVL, V4 - V6) leads.
Echocardiogram (Figures 2(a)-(c)) at three months of age showed severely dilated and dysfunctional left ventricle, mild to moderate mitral valve regurgitation, echogenic left ventricular papillary muscles, prominent right coronary artery with collateral flow in to ALCAPA, small patent foramen of ovale and no ductus arteriosus. The diagnosis of ALCAPA was clear and she was operated for reimplantation to aortic root.
3. RESULTS AND DISCUSSION
The anomalous origin of left coronary artery from the pulmonary trunk is a well known, although rare, congenital anomaly in humans. In most cases, it’s an isolated anomaly but has occasionally been associated with other congenital heart defects such as patent ductus arteriosus, ventricular septal defect, tetralogy of Fallot and coarctation of the aorta [5,6]. In most ALCAPA patients, the left coronary artery originates from the left posterior wall of the pulmonary trunk and occasionally variations of its trajectory may be surgically relevant. An embryological defect during foetal cardiac development leads to the left coronary artery arising from the pulmonary artery instead of the aorta [7].
In fetal and early neonatal life, the origin of the left coronary artery from the pulmonary artery is well tolerated because pulmonary arterial pressure equals systemic pressure which leads to antegrade flow both in left anomalous and the normal right coronary arteries. Soon after birth, pulmonary arterial pressure decreases, so flow in anomalous left coronary artery initially decreases and then gets reversed which leads to myocardial ischemia and infarction [8].
These children usually show clinical findings of severe heart failure during their early weeks or months of life but late presentation or recognition is not that uncommon. Clinical findings resemble those of dilated cardiomyopathy and the diagnosis of ALCAPA must be excluded [9] especially in the presence of ischemic ECG findings. In our patients, tachypnea, irritability and poor feeding were the most common symptoms at the time of presentation.
Based on survival pattern, two types of coronary circulations, the adult and infantile types have been described. In infantile type, the collateral circulation is poorly developed and there is early presentation with ischemia, ventricular dysfunction and mitral regurgitation [10]. However, in adult type, the collateral circulation is relatively better and the patient presents much later in life. There is an estimated 80% - 90% incidence of sudden death at a mean age of 35 years in the adult type.
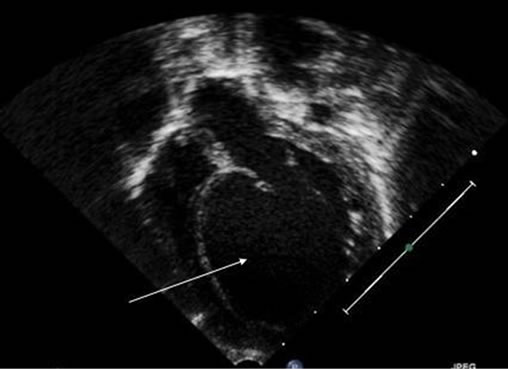 (a)
(a) (b)
(b) (c)
(c)
Figure 2. Echocardiographic pictures of case #5. (a) Apical 4-chamber view showing dilated left ventricle; (b) Parasternal short axis view showing echogenic left ventricle papillary muscles and midventricular septum; (c) Parasternal short axis view with color Doppler revealing blood flow from left coronary artery to pulmonary artery.
Twelve lead ECG can alert the possibility of ALCAPA if ischemic changes are seen especially in young age group. Broad deep Q waves and associated T wave inversion in leads I and aVL has been described as being characteristic for ALCAPA [11]. In our group, ECG was abnormal in 40% of the patients and was absolutely normal in rest of the 60% cases. Echocardiography is an important diagnostic tool for the diagnosis of ALCAPA. Sometime the transverse sinus of pericardium is misjudged as left coronary artery [12]. In infants with suspicion of endocardial fibroelastosis or dilated cardiomyopathy, anomalous origin of coronary artery must be ruled out. ALCAPA patients surviving beyond one year without treatment, coronary collaterals have been found to be obvious septal color flow signals echocardiographically. The identification of these septal collaterals is the initial clue for the diagnosis of ALCAPA in patients over one year of age [13,14]. In our study group, 80% of the patients were strongly suspected as ALCAPA echocardiographically and the most common findings were mild to moderate mitral valve regurgitation, echogenic left ventricular papillary muscles, some degree of left ventricle dilatation/dysfunction and prominent right coronary artery with collaterals like flow in interventricular septum and then to pulmonary artery. Only one (20%) patient was not suspected as ALCAPA and was considered to be a coronary fistula echocardiographically. If the ostium of the left coronary artery cannot be visualized from pulmonary trunk, one should search the proximal right pulmonary artery to exclude anomalous origin of left coronary artery from right pulmonary artery.
Additional imaging techniques such as CT scan/MRA are undertaken only when definitive diagnosis by echocardiography is not possible, or in an effort to exclude other potential diagnoses [15]. Selective right coronary angiography is a simple and quick invasive diagnostic modality in the centers like ours where CT scan/MRA facilities are not fully established. Therefore we performed selective right coronary angiography in 80% of our cases to confirm the diagnosis prior to surgical intervention.
When coronary artery anomalies like ALCAPA are diagnosed, urgent surgery is often indicated in order to prevent myocardial ischemia, malignant ventricular arrhythmias and sudden cardiac death. In cases of ALCAPA in young children, favourable results have been followed after re-establishment of a dual coronary system through re-implantation of the left coronary artery to the aorta or by bypass grafting and ligation of proximal ALCAPA [16]. When dual coronary system cannot be established either due to anatomical limitations or because of significant surgical co-morbidities, closure of the anomalous artery through surgical ligation or by the use of a percutaneous vascular plug may lead to clinical improvement [17].
4. CONCLUSION
Clinical diagnosis of ALCAPA can be challenging but features like episodes of colic or irritability during feeding associated with sweating or pallor should alert the physician to suspect ALCAPA. ECG may be helpful to suspect ALCAPA if inferol-lateral leads show deep Q waves. It’s mandatory to exclude ALCAPA in an infant presenting with congestive heart failure and echocardiographic findings of a dilated and depressed left ventricular function. Two dimensional echocardiogram has its limitations to show the exact origin of ALCAPA and color Doppler is much more sensitive. Awareness of this condition is essential for prompt recognition and referral to a tertiary care centre to enable early surgical intervention and better prognosis for these children.
REFERENCES
- Keith, J.D. (1959) The anomalous origin of the left coronary artery from the pulmonary artery. British Heart Journal, 21, 149-161. doi:10.1136/hrt.21.2.149
- Wesselhoeft, H., Fawcett, J.S. and Johnson, A.L. (1968) Anomalous origin of the left coronary from the pulmonary trunk. Its clinical spectrum, pathology, and pathophysiology, based on a review of 140 cases with seven further cases. Circulation, 38, 403-425.
- Orem, C., Kiriş, A., Korkmaz, L., Oztürk, S., Kahraman, N., Koşucu, P., Karaci, A.R. and Celik, S. (2009) Adulttype anomalous origin of the left coronary artery from the main pulmonary artery: One case report. Echocardiography, 26, 1232-1235. doi:10.1111/j.1540-8175.2009.00977.x
- Mahle, W.T. (1998) A dangerous case of colic: Anomalous left coronary artery presenting with paroxysms of irritability. Pediatric Emergency Care, 14, 24-27. doi:10.1097/00006565-199802000-00007
- Neufeld, H.N. and Schnweeweiss, A. (1983) Coronary artery disease in infants and children. LeeandFebige, Philadelphia.
- Ogden, J.A. (1970) Congenital anomalies of the coronary arteries. American Journal of Cardiolog, 25, 474-479. doi:10.1016/0002-9149(70)90016-0
- Zheng, J., Ding, W., Xiao, Y., Jin, M., Zhang, G., Cheng, P. and Han, L. (2011) Anomalous origin of the left coronary artery from the pulmonary artery in children: 15 years experience. Pediatric Cardiology, 32, 24-31. doi:10.1007/s00246-010-9798-2
- Allen, H., Gutgesell, H., Clark, E. and Driscoll, D. (2001) Moss and Adams’ Heart disease in infants, children, and adolescent including the fetus and young adult. 6th Edition, Lippincott Williams and Wilkins, Philadelphia.
- Han, L., Du, J.H. and Zhang, G.Z. (1999) Left coronary artery arising from pulmonary artery in infants. Chinese Journal of Contemporary Pediatrics, 14, 664-666.
- Peña, E., Nguyen, E.T., Merchant, N. and Dennie, G. (2009) ALCAPA syndrome: Not just a pediatric disease. Radiographics, 29, 553-565. doi:10.1148/rg.292085059
- Chang, R.R. and Allada, V. (2001) Electrocardiographic and echocardiographic features that distinguish anomalous origin of the left coronary artery from pulmonary artery from idiopathic dilated cardiomyopathy. Pediatric Cardiology, 22, 3-10. doi:10.1007/s002460010142
- King, D.H., Danford, D.A., Huhta, J.C. and Gutgesell, H.P. (1985) Noninvasive detection of anomalous origin of the left main coronary from the pulmonary trunk by pulsed doppler echocardiography. American Journal of Cardiolog, 55, 608-609. doi:10.1016/0002-9149(85)90269-3
- Frommelt, M.A., Miller, E., Willianmson, J., et al. (2002) Detection of septal coronary collaterals by color flow Doppler mapping is a marker for anomalous origin of a coronary artery from the pulmonary artery. Journal of the American Society of Echocardiography, 15, 259-263. doi:10.1067/mje.2002.115658
- Hildreth, B., Junkel, P., Allada, V., Sintek, C. and Sapin, S. (2001) An uncommon echocardiographic marker for anomalous origin of the left coronary artery from the pulmonary artery: Visualization of intercoronary collaterals within the ventricular septum. Pediatric Cardiology, 22, 406-408. doi:10.1007/s002460010263
- Pena, E., Nquyen Merchant, N. and Dennie, G. (2009) ALCAPA syndrome: Not just a pediatric disease. Radiographics, 29, 553-565. doi:10.1148/rg.292085059
- Lange, R., Vogt, M., Hörer, J., Cleuziou, J., Menzel, A., Holper, K., Hess, J. and Schreiber, C. (2007) Long-term results of repair of anomalous origin of the left coronary artery from the pulmonary artery. Annals of Thoracic Surgery, 83, 1463-1471. doi:10.1016/j.athoracsur.2006.11.005
- Collins, N., Colman, J., Benson, L., Hansen, M., Merchant, N. and Horlick, E. (2007) Successful percutaneous treatment of anomalous left coronary artery from pulmonary artery. International Journal of Cardiology, 122, e29-e31. doi:10.1016/j.ijcard.2006.11.075
Abbreviations
ALCAPA = anomalous left coronary artery from pulmonary artery; ECG = electrocardiogram; CXR = chest x ray; CT scan = computerized axial tomographic scan; MRA = magnetic resonance angiography.
NOTES
*Corresponding author.