International Journal of Organic Chemistry
Vol. 2 No. 1 (2012) , Article ID: 17864 , 7 pages DOI:10.4236/ijoc.2012.21013
Lipopeptides for the Fragment-Based Pharmaceutics Design
1The University of Queensland, School of Chemistry and Molecular Biosciences, Brisbane, Australia
2The University of Queensland, Centre for Integrated Preclinical Drug Development, TetraQ-Pharmaceutics, Brisbane, Australia
3Proxima Concepts Ltd., The London Bioscience Innovation Centre, London, UK
Email: *i.toth@uq.edu.au
Received November 25, 2011; revised December 27, 2011; accepted January 5, 2012
Keywords: Lipoamino Acids; Lipopeptides; Amphiphiles; Fragment Based Design; Supra-Molecular Assembly
ABSTRACT
This paper describes the synthesis of peptide fragments for use in a new type of combinatorial discovery technology, in which the building blocks are brought together by non-covalent interactions, rather than direct chemical bonding. The building blocks of interest—in this case different amino acids—are converted to amphiphiles by attachment to lipid tails. The amphiphiles, when mixed together in aqueous phase, are designed so that they aggregate spontaneously to form micelles. The building blocks form the headgroups of each of the amphiphiles, and these headgroups cover the surface of the micelle in a dynamic close-packed fluid mosaic array. These building blocks come together so closely that twoor three-dimensional structures are created on the surface of the micelles, and these can be screened in biological assays to find out which combination of building blocks is able to elicit a biological response. Lipopeptides consisting of two residues of lipoamino acid and other amino acids moieties have been designed, synthesized, characterized and the ability of these constructs to form supra-molecular assemblies is demonstrated.
1. Introduction
Conventional methods of drug discovery often employ combinatorial chemistry as part of the methodology to create new structures as potential ligands for binding to biological targets [1-4]. When using amino acids as building blocks, the individual subunits are linked together via peptide bonds. Unfortunately, these techniques suffer from a number of disadvantages [5]. The created peptides are time-consuming and expensive to make and purify, even using parallel synthesis, and the quantities available for testing are severely limited. Furthermore, the need for chemical linkages introduces physical constraints on the molecules under study. They are usually linear, and do not have free rotation about the bonds which joining them together so that even if the right combination of building blocks is selected, it is possible that they will not have the right orientation with respect to each other to interact appropriately with the target.
For these reasons, we have devised a combinatorial screening system in which amino-acid building blocks are brought together without the use of covalent linkages. The amino acids are conjugated to lipid tails so that amphiphiles are formed, in which the amino-acid building blocks made up the headgroups of the amphiphiles. A series of different amphiphiles has been constructed, where each member of the series has a different headgroup, but the lipid tails are identical throughout. The amphiphiles form micelles when mixed in aqueous phase, thus bringing the headgroups together sufficiently closely to form structures capable of binding to biological targets (Figure 1). In order to achieve the right degree of spacing between headgroups on the surface of the micelles, design of the lipid tail is important [6,7]. Here, the entire amphiphile is synthesized in the form of a linear peptide, where the lipid tail is provided by two lipoamino acids in tandem in which the side-chain residues are extended straight-chain hydrocarbons. In addition, amino acids, usually glycine and serine, are used a spacers between the lipid tail and headgroup.
2. Results and Discussion
We designed and synthesized amphiphilic peptides 1 and 2, containing lipophilic amino acidic residues (Figure 2) combined with representatives of coded amino acids. The lipoamino acids are alpha amino acids containing long hydrocarbon side chains [8,9].
Conventional peptide synthesis was employed to build compounds 1a-d. 4-Methyl benzhydrylamine (MBHA)
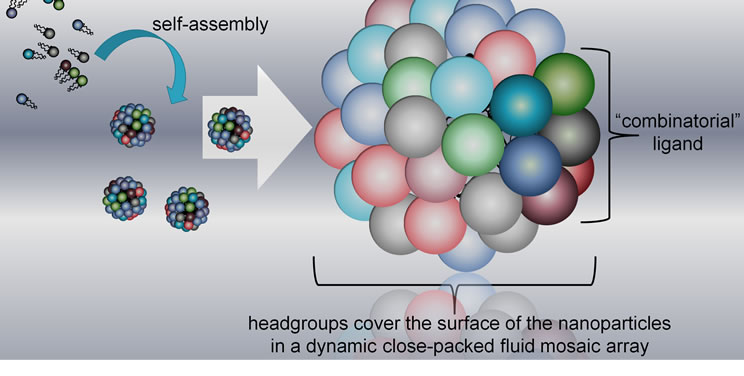
Figure 1. New concept of combinatorial discovery technology, in which the building blocks are brought together by non-covalent interactions.
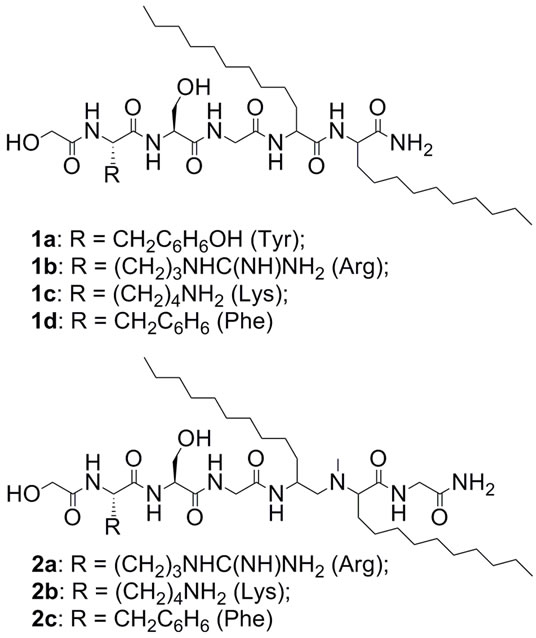
Figure 2. General structure of compounds 1 and 2.
resin was swelled in dimethylformamide (DMF), couplings were performed with Boc-protected amino acids activated by 2-(1H benztriazole-1-yl)-1,2,2-tetra-methyluronium hexafluoro phosphate (HBTU) and N,N-diidopropylethylamine (DIPEA), while neat trifluoroacetic acid (TFA) was employed for each Boc deprotection step. The peptides were cleaved from the resin with HF.
In compounds 2a-c, the amide bond between two lipoamino acids was reduced and the resulting amine function was methylated. N-methylated precursor 8 was synthesized using 2-(R/S)-[(tert-butoxycarbonyl) amino]-dodecanoic acid (3) (Scheme 1). Compound 8 was coupled to the phenacyl (Pac) protected glycine derivative 10 (Scheme 2). The Pac group was chosen for its high stability to acidic hydrolysis [10]. Trityl (Trt) protection of the glycolic acid was achieved in the reaction with trityl chloride [11].
Lipopetides 1a-d possessed only natural peptide bonds, whereas the lipopeptide derivatives (2a-c) were synthesized with N-methylated dilipidic residue to increase the biological stability. In both cases, to prevent the degradation of the building blocks by exopeptidases, glycolic acid was attached to the N-terminus and C-terminus was
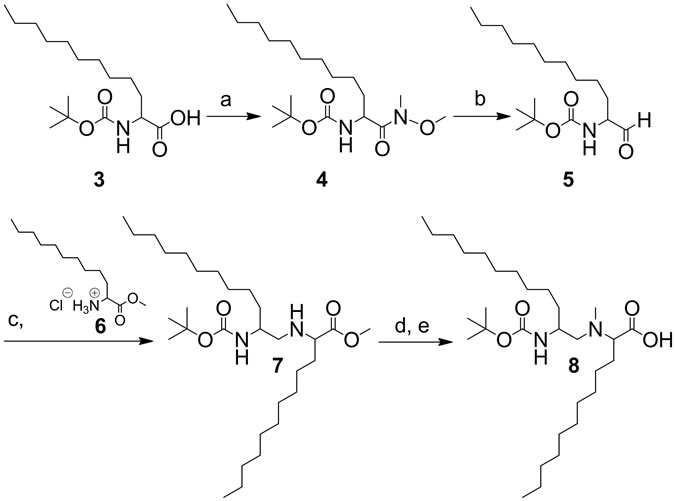
Scheme 1. Synthesis of peptidomimetic precursor (8). Reagents: (a) N,O-dimethyl-hydroxylamine, DIPEA, HBTU, DMF; (b) Lithium aluminium hydride (LAH), THF, KHSO4- ice; (c) NaCNBH3, Na2SO4, DMF; (d) Paraformaldehyde, NaCNBH3, THF, H2O; (e) NaOH, dioxane-H2O.
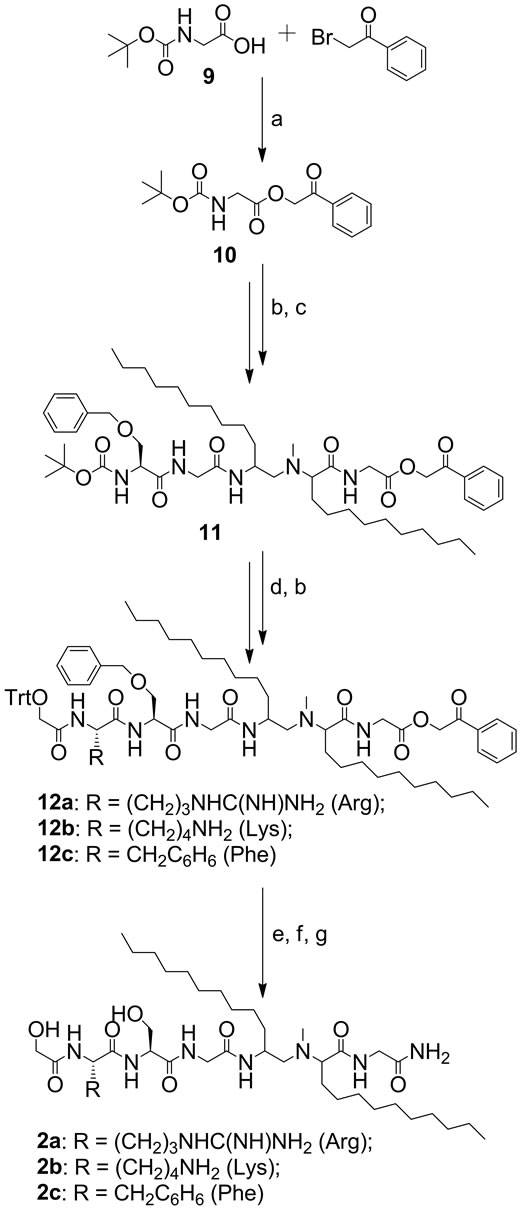
Scheme 2. Synthesis of compounds 2a-c. Reagents: (a) Et3N, DMF; (b) TFA/CH2Cl2; (c) Compound 8, Boc-Gly-OH, BocSer(OBn)-OH, HBTU, DIPEA, DMF; (d) Boc-Arg(Tos)-OH, Boc-Lys(ClZ)-OH, or Boc-Phe-OH and trityl glycolic acid, HBTU, DIPEA, DMF; (e) NH3/MeOH; (f) TFA/CH2Cl2/ triisopropylsilane; (g) HF, scavenger.
amidated. While solid phase synthesis was used to produce compounds 1a-d, solution-phase synthesis was favored for compounds 2a-c.
The synthesized amphiphilic peptides were designed to aggregate spontaneously to form micelles when codispersed in aqueous solution. The ability of the amphiphiles to form supra-molecular assemblies was assessed by isothermal titration calorimetry (ITC), dynamic light scattering (DLS) and then was confirmed by transmission electron microscopy (TEM).
ITC is a sensitive, routine method which allows determining the critical micelles/aggregates concentration (CMC/CAC) and provides access to thermodynamic parameters such as enthalpy (ΔH), entropy (ΔS) and Gibbs free energy (ΔG) of micellization/aggregation [12,13]. Titration of concentrated mixture of compounds 1a-d or 2a-c into water allowed disaggregation and formation of new particles (aggregates/micelles). The ITC results for both mixtures 1a-d and 2a-c were similar (for 1a-d the CAC was 0.0043 ± 0.0002 mM, and for 2a-c the CAC was 0.0041 ± 0.0002 mM). The low CAC value suggested a good aptitude of these amphiphiles for self-assembly. The same similarities were observed for the thermodynamics, the ΔH was 9.00 ± 1.00 kcal·mol–1 (37.7 ± 4.2 kJ·mol–1) for the mixture 1a-d and 11.7 ± 0.3 kcal·mol–1 (49.2 ± 1.0 kJ·mol–1) for 2a-c. The particles (mixed micelles) formation was thermodynamically favored as the negative values of Gibbs free energy were found for both mixtures, ΔG = –10.1 ± 0.0 kcal·mol–1 (–42.2 ± 0.1 kJ·mol–1) for 1a-d, and ΔG = –10.1 ± 0.0 kcal·mol–1 (–42.3 ± 0.02 kJ·mol–1) for 2a-c. The determination of ΔG and ΔH led to the calculation of the entropy of micellization. ΔS was 61.6 ± 3.2 cal·K–1·mol–1 (258 ± 14 J·K–1·mol–1) for the mixture 1a-d and 70.3 ± 1.0 cal·K–1·mol–1 (294 ± 5 J·K–1·mol–1) for 2a-c. Since in the both cases the enthalpy of spontaneous micellization/ aggregation was endothermic, this process was entropydriven.
The particles formed during the ITC experiments were used for the DLS analysis. We detected two sets of size distributions, the first was in the range of 30 - 110 nm for the mixture 1a-d and of 25 - 100 nm for 2a-c, and the second was in the range from 200 to over 1 μm for both mixtures.
TEM experiments confirmed the results obtained from the DLS measures and confirmed the formation of supramolecular structures (Figure 3). TEM images of the mixture 1a-d showed nanoparticles (20 - 60 nm) with tendency to aggregate to larger particles. Images of the mixture 2a-c showed nanoparticle formation in the size ranges from 20 - 40 nm to particles of hundreds nm.
The aim of the approach described here was to facilitate the studies of ligand-receptor interactions and to provide a new tool for drug discovery. We developed the

Figure 3. Transmission electron microscopy photographs of the mixture 1a-d (left) and 2a-c (right) in water (bar 200 nm), stained with ammonium molybdate 10%.
described library of lipopeptides to address the structural diversity and variability of short peptide sequences. The lipopeptide building blocks were designed to form micelles on which surface the headgroups/amino acids are displayed in such manner that they are mimicking a random peptide sequence. Thus, a variety of amino acids were incorporated into the building blocks. It was desirable to keep the length of the peptide to a minimum to avoid eliciting unwanted immune response. We focused on the development of chemical bonds linking the head groups to the lipidic tails. Lipopetides 1a-d were synthesized with natural peptide bonds exhibiting a considerable degree of flexibility, while lipopeptide 2a-c were synthesized with N-methylated dilipidic residue to study its influence on peptide conformation and biological activity. The mixtures of synthesized compounds were able to form nanoparticles. However, an extensive aggregation was also observed. The tendency to form large aggregates can be explained by the relatively high hydrophobicity of synthesized amphiphiles [13,14]. To obtain more uniform particles, the nanoparticle might be selfassembled by dialysis of a solution of the lipopeptides in organic solvent against water [15,16].
3. Conclusion
New fragment-based lipopeptide building blocks were designed, synthesized and characterized. The lipid-modified peptidic amphiphiles were capable of forming a supramolecular assemblies bound by weak non-covalent associations. These compounds will be applied for the study of the receptor-ligand interactions. Biological evaluation of these lipopeptidic constructs is underway, and results will be reported elsewhere. Applying our dynamic concept might provide the new generation of therapeutics.
4. Experimental
Peptide synthesis solvent, N,N-dimethylformamide (DMF), and reagents, trifluoroacetic acid (TFA) and diisopropylethylamine (DIPEA), were purchased from Auspep (Melbourne, VIC, Australia). O-Benzotriazole-N,N,N’, N’-tetramethyl-uronium-hexafluoro-phosphate (HBTU) and di-tert-butyl dicarbonate were obtained from GL Biochem Ltd (Shanghai, China). N-Boc-protected amino acids and pMBHA resin were supplied by Novabiochem (Läufelfingen, Switzerland). Palladium (10% wt on carbon) was purchased from Lancaster Synthesis (Lancashire, England). Ultra pure gases (N2, H2, Ar) were supplied by BOC Gases (Brisbane, QLD, Australia). Silica for flash chromatography (Silica gel 60, 230 - 400 mesh) was obtained from Lomb Scientific (Taren Point, NSW, Australia). Deuterated solvents (d1-CDCl3 and DMSO-d6) were manufactured by Cambridge Isotope Laboratories Inc. (Andover, MA, USA). All other reagents were purchased in analytical grade or higher purity from SigmaAldrich (Castle Hill, NSW, Australia) or Merck Pty Ltd (Kilsyth, VIC, Australia). Solvents were freshly distilled and dried prior to use and all moisture-sensitive reactions were carried out under inert atmosphere (N2/Ar) using oven-dried glassware.
1H NMR spectra were recorded at 297 K using a Bruker spectrometer at 400 MHz and 500 MHz. The instrument operating at 400 MHz and 500 MHz for 1H used CDCl3, CD3OD or Acetone-d6 as solvent and tetramethylsilane as an internal standard, unless stated otherwise. Coupling constants were provided in Hz. 13C spectra were measured at 100.62 MHz and 125.77 MHz and referenced to CDCl3 (77.0 Hz), CD3OD (49.0 Hz) or Acetone-d6 (30.5 Hz). Homoand heteronuclear 2D NMR spectroscopy was performed using Bruker standard software.Thin layer chromatography (TLC) was performed on Merck pre-coated aluminum sheets (Silica Gel 60F254); spots were detected by spraying with H2SO4. Amine derivatives were detected by ninhydrin spray, followed by heating the sheets. Column chromatography was performed on silica gel columns (size A, 28 ´ 2; B, 30 ´ 2.5; and C, 43 ´ 4 cm; silica gel 0.040 - 0.063 mm). Analytical RP-HPLC was performed on a Shimadzu instrument (LC-10AT liquid chromatograph, SCL-10A system controller, SPD-6A UV detector, a SIL-6B auto injector with a SCL-6B system controller, and a C18- HPLC-column using an acetonitrile/water/0.1% TFA gradient as well as an isopropanol/water/0.1% TFA gradient. HPLC purification was done on a Waters HPLC system (Model 600 controller, 490E UV detector, F pump, and 0.46 × 15 cm Vydac RP-C18 column with 0.005 mm particle size) using a acetonitrile/water/0.1% TFA gradient. He-gas was applied for degassing of HPLCsolvents.Mass spectra were obtained on a quatropolelectrospray-MS (Perkin Elmer API 3000 instrument) in the positive ion mode. Concentration of solutions was performed at reduced pressure and at temperatures < 40˚C.
High-resolution mass spectrometry (HRMS) data were obtained on a QStar Pulsar instrument (Applied Biosystems) operating in positive-ion electrospray mode.The thermodynamic interactions were calculated using a MicroCal VP-ITC Microcalorimeter (Northampton, MA, USA) with Origin 5.0 software and VPViewer 2000.The particle size measurements were done by using a Zetasizer Nano ZP instrument (Malvern Instruments, UK) with DTS software.
Transmission electron microscopy photographs were done on a JEOL-1010 microscope operating at an acelerating voltage of 100 kV. The sample images were taken and analyzed using the AnalySIS® software (Soft Imaging Systems, Megaview III, Munster, Germany).
4.1. Synthesis
2-(R/S)-Aminododecanoic acid and its Boc-protected intermediate 3 were synthesized according to the published method [17,18]. Tritylation of glycolic acid was performed by reaction of glycolic acid with tritylchloride and 4-dimethylaminopyridine (DMAP) in pyridine according to the literature [11].
4.1.1. Synthesis of Peptides 1a-d
General Protocol for the synthesis of peptides 1a-d (SPPS method):
MBHA resin (4-Methyl benzhydrylamine, substitution ratio: 1.03 mmol/g, 3.0 mmol scale) was swelled in dimethylformamide in a sintered glass peptide synthesis vessel for 90 min. Each amino acid coupling cycle consisted of Boc-deprotection with neat TFA (2 × 1 min), a 1 min DMF flow wash, followed by 20 min coupling with the pre-activated amino acid. An activation mixture consisting of Boc-amino acid (3 eq. per mol amino-group), HBTU (2-(1H benzotriazole-1-yl)-1,3,3-tetramethyl-uronium hexafluoro phosphate, 0.5 M in DMF, 3 eq.) and DIPEA (0.442 mL, 4 eq.) was shaken for 12 min. Coupling efficiency was monitored by a quantitative ninhydrin test (³99.8%). Upon completion of the synthesis and removal of the terminal Boc groups, the resin was washed with DMF, methanol and DCM. The resin was dried to constant weight over KOH in vacuo. The peptides were cleaved from the resin using HF, and p-cresol as a scavenger. The cleaved peptides were precipitated, filtered and washed thoroughly with ice-cold diethyl ether. The crude peptide was obtained as an amorphous powder after redissolving the cleaved peptides in acetonitrile-water (1:1) with 0.1% TFA and lyophilisation. 200 mg of each crude peptide was separated using Sephadex LH-20 (70 × 3 cm) and acetonitrile-water (1:1) as a solvent. Peptide-positive fractions were determined on silica gel by spraying with ninhydrin-reagent. Analytical RP-HPLC (C4, 25 cm Vydac C4, C18 column with 5 nm pore size and 4.6 mm internal diameter) was performed using two different solvent systems in order to check the peptides’ purity. Solvent system 1 comprised; Solvent A (H2O, 0.1% TFA), and Solvent B (90% CH3CN, 10% H2O, 0.1% TFA); and Solvent system 2 comprised; Solvent A (H2O, 0.1% TFA), Solvent B (90% Methanol, 10% H2O, 0.1% TFA). The gradient was 0% - 100% B within 20 min; the flowrate was 1 mL/min; and the wave length was 214 nm. The purity of all peptides analyzed by both systems on HPLC was over 95%. Peptides were also characterized using ESI-MS. The resulting peptides were used as diastereomeric mixtures.
Compound 1a:
HPLC, system A, tR, (min): 18.74 and 19.07; system B, tR, (min): 17.46 and 17.70; ESI-MS, m/z: 777.7 [M + H]+, 799.9 [M + Na]+; HRMS calcd for C40H68O9N6Na 799.4940, found: 799.4932.
Compound 1b:
HPLC, system A, tR, (min): 17.91 and 18.20; system B, tR, (min): 16.94 and 17.15; ESI-MS, m/z: 771.1 [M + H]+; HRMS calcd for C37H72O8N9 770.5498, found: 770.5499.
Compound 1c:
HPLC, system A, tR, (min): 17.91 and 18.17; system B, tR, (min): 16.85 and 17.05; ESI-MS, m/z: 743.2 [M + H]+; HRMS calcd for C37H72O8N7 742.5437, found: 742.5426.
Compound 1d:
HPLC, system A, tR, (min): 19.55; system B, tR, (min): 18.33; ESI-MS, m/z: 762.0 [M + H]+, 784.2 [M + Na]+; HRMS calcd for C40H68O8N6Na 783.4991, found: 783.4972.
4.1.2. Synthesis of Peptides 2a-c
Tert-butyl 1-oxododecan-2-ylcarbamate (5):
To a stirred solution of 3 (998 mg, 3.17 mmol), DIPEA (16.5 mL, 95 mmol), HBTU (12.6 g, 33 mmol) and N, O-Dimethylhydroxylamine hydrochloride (3.21 g, 33 mmol) in dry dimethylacetamide (150 mL) were added and stirred for 3 h. The mixture was concentrated, co-evaporated with toluene (3x, each 70 mL), diluted with 150 mL ethyl acetate and washed with ice-cold aqueous HCl-solution (5%, 2 × 50 mL), saturated with NaHCO3-solution (2×, each 50 mL), washed with brine (3×, each 30 mL), and dried (MgSO4). The crude Weinreb-amide 4 (tert-butyl- 1-(methoxy(methyl) amino)-1-oxododecan-2-ylcarbamate) was dissolved in dry THF (180 mL), LiAlH4 (1.2 g, 0.0318 mmol) was added at 0˚C within 10 min and then stirred for 2.5 h at room temperature. The mixture was then poured into a solution of 1 M KHSO4/ice (250 mL) and when the ice had melted, it was re-extracted with ethylacetate (4×, each 200 mL), dried (MgSO4) and concentrated. Aldehyde 5 was obtained as white crystals (8.2 g, 86% yield, C17H33NO3, MW 299.46 g·mol–1). ESI-MS, m/z: 300.31 [M+H]+, 599.9 [2M+H]+, 621.71 [2M + Na]+. NMR: 1H NMR (Acetone-d6): d 7.35 - 7.32 (m, 2 H, arom. H), 6.66 - 6.68 (m, 2 H, arom. H), 3.37 - 3.23 (m, 4 H, CH2), 2.77 (s, 2 H, NH2). 1H-13C HMQC Correlation (d6-Acetone): Crosspeaks: 7.35-7.32 and 122.2 (aromat. H(A) and aromat. C (A), 2C), 6.66 - 6.68 and 103.5 (aromat. H (B), 2H, and aromat. C (B), 2C)), and between 3.37 - 3.23 (m, 4 H, CH2) and 31.8 (CH2, 2 C).
Methyl 2-aminododecanoate hydrochloride (6):
To a solution of 3 (10.2 g, 32.4 mmol, 2.84 mmol) in dry methanol (30 mL) thionylchloride (2.4 mL, 33 mmol) was added at 0˚C and then refluxed for 24 h. Co-evaporation with CH3OH (4×, each 100 mL) and recrystallization from methanol, afforded compound 6 as white crystals (6.8 g, 79% yield, C13H27NO2, MW 229.37 g·mol–1). ESI-MS, m/z: 230.3 [M + H]+, 459.5 [2M + H]+.
2-(2-(tert-butoxycarbonylamino)dodecyl)(methyl)amino dodecanoic acid (8):
Compound 6 (5.3 g, 20 mmol), compound 5 (5.96 g, 20 mmol), MgSO4 (2.9 g, 24 mmol), acetic acid (0.5 mL) and NaCNBH3 (6.3 g, 100 mmol) were stirred in dry DMF (100 mL) for 16 h at room temperature. After 16 h compound 5 (1.8 g, 6.8 mmol), NaCNBH3 (1 g, 17 mmol) and MgSO4 (1 g, 8 mmol) were added again and stirred continuously for 36 h. The suspension was filtered and concentrated at high-vacuum. The clear residue was dissolved in ether (500 mL), washed with water (200 mL), 1 M KHSO4 (100 mL), brine (100 mL) and dried (MgSO4). The dried organic layer afforded a yellow-orange oily residue (9.8 g, 95%) which was compound 7 (Methyl 2-(2-(tert-butoxy-carbonylamino) dodecylamino) dodecanoate). To a solution of compound 7 (6.16 g, 12.0 mmol) in THF (200 mL), NaCNBH3 (6.3 g, 100 mmol), paraformaldehyde (8.8 g) and acetic acid (2.5 mL) were added slowly at 0˚C under N2 and stirred for 16 h. Et3N (1 mL) was then added and the mixture was concentrated at high-vacuum. The organic residue was then dissolved in ether (300 mL), washed with water (200 mL), ice-cold 1 M aqueous KHSO4-solution (2 × 100 mL), saturated NaHCO3-solution (2 × 100 mL), brine (3 × 30 mL), and dried (MgSO4). The organic layer was concentrated and the residue was purified by flash-chromatography (B, 7:1 hexane-EtOAc ® EtOAc, solvents containing 0.5% triethylamine) to produce methylated compound 8 (7.91 g, 80%) as a yellow syrup. Then, the methyl ester was stirred with a 1 M NaOH-solution in a mixture of dioxane/water/acetonitrile (4:1:2, v/v) for 48 h. The clear solution was concentrated, suspended in CH2Cl2 (200 mL) and then a pH 5 was obtained by adding concentrated aqueous citric acid-solution. The product was immediately re-extracted with CH2Cl2 (5 × 100 mL), washed with brine (3 × 30 mL), and dried (MgSO4) resulting in an oily residue, 8 (7.05 g, 98%, C30H60N2O4, MW 512.82 g·mol–1). ESI-MS, m/z: 513.7 [M + H]+, 1026.2 [2M + H]+. 1H NMR (CDCl3): d 4.60 - 4.39 (m, 1 H, NHCO), 3.65 - 3.51 (m, 1 H, CH-CH2), 3.10 - 3.25 (m, 1 H, CHCO), 2.70 - 2.45 (m, 2 H, CH2NH), 2.31 (br-s, NCH3), 1.78 - 1.17 (m, 36 H, 16 CH2), 0.85 (br-t, 6 H, 2 CH3CH2). 13C NMR (CDCl3): d 172.88, 155.93 (2 C, C=O), 78.95 (1 C, tButyl), 67.11 (1 C, CHCO), 51.55, 51.07 (3, C, CH3O, CH2NH, CHCO), 38.27 (1 C, NCH3), 36.07 CH2NCH3), 33.59, 33.49 (1 C, CH2, (A,B), 31.89, 29.60, 29.51, 29.45, 26.37, 26. 26, 25.73, 25.65 (18 C, CH2), 22.67 (1 C, (CH3)3C), 14.10 (2 C, CH3CH2).
2-Oxo-2-phenylethyl 2-(tert-butoxycarbonylamino) acetate (10):
A phenylacetylbromide (5 g, 25.2 mmol) and Boc-Gly (9) (4.4 g, 25 mmol) were dissolved in dry DMF (30 mL), and then Et3N (4 mL, 28.7 mmol) was slowly added at 0˚C and the mixture stirred for 3 h at room temperature. After co-evaporation with toluene (3 × 50 mL), the residue was dissolved in DCM (300 mL), washed with water (3 × 300 mL), ice-cold aqueous HCl-solution (5%), saturated NaHCO3-solution (3 × 100 mL), brine (3 × 100 mL), and dried (MgSO4). The organic layer was concentrated and recrystallized from ethanol (6.3 g, 86% yield, C15H19O5N, MW 293.32 g·mol–1). ESI-MS, m/z: 294.1 [M + H]+, 316.21 [M + Na]+, 587.7 [2M + H]+, 609.4 [2M + Na]+. 1H NMR (CDCl3): d 7.89 - 7.46 (m, 5 H, aromat. H), 5.39 (s, 2H, CH2-O), 5.05 (m, 1 H, NH), 4.11 (d, 2 H, CH2NH), 1.44 (s, 9 H, tBu). 13C NMR (CDCl3): d 191.49, (s, 1 C, Ar-CO-CH2), 170.02 (s, 1 C, NH-CO-O), 155.69 (s, 1 C, O-CO-CH2), 134.06, 133.94, 128.92, 127.73 (4 C, aromat.), 80.12 (s, 1 C, C(CH3)3), 66.50 (s, 1 C, CH2-O), 42.27 (s, 1 C, CH2-NH), 28.27 (s, 3 C, (CH3)3).
General Protocol for the synthesis of peptides 2a-c (solution method):
General protocol for Boc-removal:
Boc-protected peptides were dissolved in ice-cold TFA to give 1M solution and stirred for 1 h at room temperature. TFA was removed by co-evaporation with toluene (3 × 50 mL), the residue was washed with toluene (3 × 50 mL) and sonicated frequently to afford a white precipitate as a TFA-salt (98% - 100% yield). Deprotected peptides were analyzed by electrospray MS, dried at high-vacuum over NaOH and used as crude product for the following amino acid-coupling reaction.
General protocol for amino acid coupling:
Crude peptide-TFA-salts were dissolved in dry DMF (3 mL/mmol TFA-salt) and mixed with DIPEA (1 mL/mmol TFA-salt), Boc-amino acid, Boc-dilipid 6, and trityl-glycolic acid, respectively (1.5 equivalent), HBTU (1.5 equivalent) and molecular sieve (4Å, 30 mg/mmol TFA-salt). After overnight stirring the suspension was filtered through celite and the clear solution was coevaporated with toluene (3 × 50 mL) to dryness. The resultant residue was dissolved in CHCl3 (100 mL) and washed with water (150 mL), ice-cold aqueous HClsolution (5%, 3 × 100 mL), saturated NaHCO3-solution (3 × 100 mL), brine (3 × 30 mL), and dried (MgSO4). The resulting peptide was obtained as a yellow oil (yield ~95% per coupling) and only peptidic intermediate 9 was purified by flash-chromatography (B, 1:1 hexane-EtOAc ® 1:2 hexane-EtOAc, solvents containing 1% triethylamine).
Deprotecting procedures:
Removal of Pac-group and Formyl-group: each peptide (0.75 mmol) was stirred in a solution of saturated NH3 in CH3OH (50 mL) for 16 h. The solvent was evaporated under vacuum and the product was dried over NaOH for 16 h. For the removal of the trityl-group, each peptide (0.75 mmol) was stirred in a solution of CH2Cl2 /TFA/TIS (1:94:5, v/v) for 1.5 h. The reagents were removed under reduced pressure and co-evaporated with toluene (2 × 50 mL). The amino acid side-chain protecting groups of the peptides were cleaved using HF, and p-cresol was used as a scavenger. The cleaved peptide was precipitated, filtered and washed thoroughly with ice-cold diethyl ether. The crude peptide was obtained as an amorphous powder after redissolving the precipitate in acetonitrile-water (1:1) with 0.1% TFA and lyophilization.
Compound 2a:
HPLC, system A, tR, (min): 15.55 and 16.03; system B: 17.60 and 17.96; ESI-MS, m/z: 827.8 [M + H]+; HRMS calcd for C40H79O8N10 827.6077, found: 827.6093.
Compound 2b:
HPLC, system A, tR, (min): 15.60 and 16.06; system B: 21.03 and 21.43; ESI-MS, m/z: 799.9 [M + H]+; HRMS calcd for C40H79O8N8 799.6015, found: 799.5997.
Compound 2c:
HPLC, system A, tR, (min): 16.78 and 17.09; system B: 19.24 and 19.68; ESI-MS, m/z: 819.8 [M + H]+; HRMS calcd for C43H75O8N7Na 840.5569, found: 840.5567.
4.2. Analysis of Supra-Molecular Assembly
4.2.1. ITC Experiments
The sample cell and the reference cell (1.5 mL each) contained degassed, deionized water (pH 6.0) and the microsyringe (0.3 mL) contained mixture of compounds 1a-d (the mixture of 1a, 1b, 1c, and 1d) or 2a-c (the mixture of 2a, 2b, and 2c) The mixtures 1a-d or 2a-c were prepared with the same concentration, 0.05 mM each (the compounds from the same mixture were combined at the solid stage and then solubilized in miliQ water finally giving the concentration of 0.05 mM). Experiments were performed in triplicate at 37˚C where mixtures 1a-d or 2a-c were titrated into the sample cell loaded with water using a stirrer/syringe rotating at 450 rpm. Sequences were built as follows: title compounds concentration, 0.05 mM; number of injections, 27; injection duration, 29.9 s; injection volume, 10 µL; spacing 760 sec.
4.2.2. DLS Experiments
Sizes were analysed using a non-invasive back scatter system with a scattering angle of 173˚. DLS analyses were performed after ITC experiments and using the samples from microcalorimeter sample cell. The samples contained mixture 1a-d or 2a-c after self-assembly process (which gave final concentration of 0.0076 mM for each of them). The mixtures 1a-d and 2a-c were analysed at 25˚C using disposable low volume cuvettes. Experiments were performed at least three times.
4.2.3. TEM Experiments
The samples were used after self-assembly process from microcalorimeter sample cell. A drop of mixture 1a-d or 2a-c solution was allowed to air-dry onto a 200 mesh copper grid, and the sample was stained with 10% of ammonium molybdate for 5 minutes.
5. Acknowledgements
This work was supported by ARC Linkage Grant LP0347149. We wish to thank Dr. Tri Le for his advice and help on NMR acquisitions and Graham MacFarlane for his accurate mass measurements.
REFERENCES
- H. Kubinyi, “Chemogenomics” In: H. Kubinyi, Ed., Comprehensive Medicinal Chemistry II, Volume 3: Drug Discoveries Technolgies, Elsevier, New York, 2006, pp 921-937.
- P. Seneci, “Combinatorial Chemistry,” In: H. Kubinyi, Ed., Comprehensive Medicinal Chemistry II, Volume 3: Drug Discoveries Technolgies, Elsevier, New York, 2006, pp 697-760.
- S. Nayak, D. Panda and J. Sahoo, “Nanosuspension: A Novel Drug Delivery System,” Journal of Pharmacy Reserach, Vol. 3, No. 2, 2010, pp. 241-246. http://jpronline.info/article/view/1604/1004
- V. Shankarananth and K. Rajasekhar, “Combinatorial Chemistry: A Review,” Drug Invention Today, Vol. 2, No. 1, 2010, pp. 49-52. http://ditonline.info/article/view/1487/963
- V. J. Hruby and J. Vagner, “High Throughput Synthesis of Peptides and Peptidomimetics,” Chimica Oggi, Vol. 24, No. 4, 2006, pp. 18-21. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2447911/
- H. A. Behanna, J. J. M. Donners, A. C. Gordon and S. I. Stupp, “Coassembly of Amphiphiles with Opposite Peptide Polarities into Nanofibers” Journal of American Chemical Society, Vol. 127, No. 4, 2005, pp. 1193-1200. doi:10.1021/ja044863u
- E. Kokkoli, A. Mardilovich, A. Wedekind, E. L. Rexeisen, A. Garg and J. A. Craig, “Self-Assembly and Applications of Biomimetic and Bioactive Peptide-Amphiphiles,” Soft Matter, Vol. 2, No. 3, 2006, pp. 1015-1024. doi:10.1039/b608929a
- W. A. Gibbons, R. A. Hughes, M. Charalambous, M. Christodoulou, A. Szeto, A. E. Aulabaugh, P. Mascagni and I. Toth, “Lipidic Peptides. VIII. Cellular Uptake Studies of Lipidic Amino Acid, Its Oligomers and Highly Lipophilic Drug Conjugates,” Liebigs Annalen der Chemie, Vol. 79, No. 1-3, 1990, pp. 1175-1183. doi:10.1002/jlac.1990199001215
- R. New and I. Toth, “Epitopes Formed by Non-Covalent Association of Conjugates,” PCT Int. Appl. 2001, WO 2001001140.
- C. C. Yang and R. B. Merrifield, “The Beta-Phenacyl Ester as a Temporary Protecting Group to Minimize Cyclic Imide Formation during Subsequent Treatment of Aspartyl Peptides with HF,” Journal of Organic Chemistry, Vol. 41, No. 6, 1976, pp. 1032-1041. doi:10.1021/jo00868a025
- I. Schwope, C. F. Bleczinski and C. Richert, “Synthesis of 3’,5’-Dipeptidyl Oligonucleotides,” Journal of Organic Chemistry, Vol. 64, No. 13, 1999, pp. 4749-4761. doi:10.1021/jo990036d
- A. S. Abdelrahim, Z. M. Ziora, J. A. Bergeon, A. R. Moss and I. Toth, “Design and Synthesis of a Series of Novel, Cationic Liposaccharide Derivatives as Potential Penetration Enhancers for Oral Drug Delivery,” Tetrahedron, Vol. 65, No. 45, 2009, pp. 9436-9442. doi:10.1016/j.tet.2009.08.072
- Z. M. Ziora, N. Wimmer, R. New, M. Skwarczynski and I. Toth, “Synthesis of Glycolipopeptidic Building Blocks for Carbohydrate Receptor Discovery,” Carbohydate Research, Vol. 346, No. 12, 2011, pp. 1439-1444. doi:10.1016/j.carres.2011.03.019
- M. Skwarczynski, B. H. Parhiz, F. Soltani, S. Srinivasan, K. A. Kamaruzaman, I-C. Lin and I. Toth, “Lipid Peptide Core Nanoparticles as Multivalent Vaccine Candidates against Streptococcus Pyogenes,” Australian Journal of Chemistry, Vol. 65, 2012, pp. 35-39. doi:10.1071/CH11292.
- M. Zaman, M. Skwarczynski, J. M. Malcolm, C. N. Urbani, Z. F. Jia, M. R. Batzloff, M. F. Good, M. J. Monteiro and I. Toth “Self-Adjuvanting Polyacrylic NanoParticulate Delivery System for Group A Streptococcus (GAS) Vaccine,” Nanomedicine: Nanotechnology, Biology and Medicine, Vol. 7, No. 2, 2011, pp. 168-173. doi:10.1016/j.nano.2010.10.002
- M. Skwarczynski, M. Zaman, C. N. Urbani, I. C. Lin, Z. F. Jia, M. R. Batzloff, M. F. Good, M. J. Monteiro and I. Toth, “Polyacrylate Dendrimer Nanoparticles: A SelfAdjuvanting Vaccine Delivery System,” Angewandte Chemie-International Edition, Vol. 49, No. 33, 2010, pp. 5742- 5745. doi:10.1002/anie.201002221
- I. Toth, M. Danton, N. Flinn and W. A. Gibbons, “Combined Adjuvant and Carrier System for Enhancing Synthetic Peptides Immunogenicity Utilising Lipidic Amino Acids,” Tetrahedron Letter, Vol. 34, 1993, pp. 3925-3928. doi:10.1016/S0040-4039(00)79265-3
- M. Skwarczynski and I. Toth, “Lipid-Core-Peptide System for Self-Adjuvanting Synthetic Vaccine Delivery,” In: S. S. Mark, Ed., Bioconjugation Protocols: Strategies and Methods, 2nd Edition, Humana Press Inc., Totowa, 2011, pp. 297-308. doi:10.1007/978-1-61779-151-2_18
NOTES
*Corresponding author.