Journal of Cancer Therapy
Vol.3 No.5(2012), Article ID:23449,13 pages DOI:10.4236/jct.2012.35072
Gemcitabine Inhibits Murine Head and Neck Squamous Cell Carcinoma Growth via Proteasome-Dependent Degradation of Chk1 Leading to Cell Cycle Arrest and Apoptosis
![]()
1Department of Diagnostic and Biomedical Sciences, School of Dentistry, University of Texas Health Science Center at Houston, Houston, USA; 2Medical Student, Washington University School of Medicine in St. Louis, St. Louis, USA.
Email: Nadarajah.vigneswaran@uth.tmc.edu
Received August 21st, 2012; revised September 23rd, 2012; accepted October 5th, 2012
Keywords: Chk1; Gemcitabine; Head and Neck Cancer; Apoptosis; siRNA
ABSTRACT
Head and neck squamous cell carcinoma (HNSCC) is a common tumor worldwide that often presents at an advanced stage with poor prognosis. Advanced HNSCC frequently exhibits resistance to chemotherapy limiting its efficacy. Gemcitabine is a pyrimidine-based analog that is currently used for the treatment of metastatic pancreatic cancer. In this study, we examined the anti-tumor effects of gemcitabine in a highly aggressive murine model of HNSCC (LY2). In vitro cell viability and in vivo tumor growth inhibitory assays were carried out to determine the sensitivity of LY2 cells to gemcitabine. Immunohistochemical, Western blotting, RT-PCR and RNAi-mediated silencing assays were used to characterize effects of gemcitabine cell proliferation, DNA synthesis, apoptosis, pro-survival and DNA damage response signaling pathways. LY2 cells treated with gemcitabine undergo apoptosis mediated by the activation of both caspase-3 and -9. Gemcitabine on treatment induces rapid phosphorylation of checkpoint kinase 1 (Chk1) in LY2 cells and subsequent degradation in a time and dose dependent manner. Proteasome inhibitor MG132 blocks Chk1 degradation and decreases LY2 cells susceptibility to gemcitabine. Inhibition of Chk1 function, either using inhibitor PD 407824 or small interfering RNA (siRNA), increases the sensitivity of LY2 cells to gemcitabine. Gemcitabine treatment resulted in significant reduction in tumor growth relative to saline-treated control in a syngeneic orthotopic murine model of HNSCC. Gemcitabine-induced DNA replication stress in LY2 cells activates Chk1 by phosphorylation and promotes Chk1 degradation via the ubiquitin-proteasome pathway. Depletion of Chk1 terminates S-phase check point in LY2 cells resulting in apoptotic cell death. Our data provides an important rationale for integrating gemcitabine to optimize chemotherapeutic efficacy in HNSCC.
1. Introduction
Preservation of the genomic DNA from extrinsic or intrinsic damage is crucial for maintaining normal cellular function and to prevent cellular transformation. When cellular DNA is damaged, it can be repaired to its original sequence; if the DNA damage is extensive and cannot be repaired, however, the cells are triggered to undergo programmed cell death [1,2]. Several partially overlapping DNA repair pathways are involved in the maintenance of genomic integrity [3]. DNA checkpoint pathways are activated in mitotically active cells in response to DNA damage caused by extrinsic (i.e. radiation or chemotherapeutic agents) or intrinsic (i.e. DNA replication stress) factors. Phosphoinositide 3-kinase related kinases (PI3K) namely; ataxia telangiectasia and rad-3-related kinase (ATR) and ataxia-telangiectasia mutated (ATM) coordinate DNA damage responses via activation of Chk1 and Chk2, respectively [4-6]. ATR/ Chk1 are activated by UV and other intrinsic factors that induce DNA replication stress whereas the activation ATM/Chk2 signaling is trigged by DNA double strand breaks [7]. The ATM or ATR cascades become activated within minutes of a DNA damage signal resulting in phosphorylation and activation of the transcription factor p53, either directly or indirectly, via the activation of Chk2 [8]. Activation of p53 induces the transcription of p21 (also known as cyclin-dependent kinase-2 inhibitor or CDKN1a) which arrests the cells at G1 and prevents them from entering into S-phase [8]. Cells with damaged DNA that have escaped G1 checkpoint are halted at G2 phase by the activation of Chk1 which phosphorylates Cdc25 leading to its sequestration in the cytoplasm [8]. Cytoplasmic sequestration Cdc25 prevents the activation of cyclin-dependent kinase 1 (Cdk1) and blocks the cells from entering into the mitosis (M) phase until the damaged DNA is repaired [8]. Hence, ATM/Chk2 pathway primarily regulates the G1 checkpoint whereas ATR/ Chk1 signaling acts on the G2 checkpoint of the cell cycle [8]. Chk1/Chk2 induced cell cycle arrest allows the cells to reenter cell cycle after DNA repair or to undergo apoptosis if the DNA damage cannot be properly repaired [4,6]. Recent findings suggest that cell cycle checkpoints are functional but are deregulated in cancer cells [8]. Cancer cells exploit these deregulated DNA repair pathways for checkpoint overrides resulting in genetic instability and resistance to DNA-damage-inducing chemotherapeutic drugs [8,9]. Hence, inhibition of checkpoint pathways is being explored as a means to sensitize tumor cells to chemotherapy [8,10,11].
Gemcitabine (2’2’-difluorodeoxycytidine) is a pyrimidine-based analog that has antitumor activity both in vitro and in vivo [12,13]. For more than a decade, gemcitabine has been used for the treatment of metastatic pancreatic cancer [14]. Recent clinical studies have shown favorable therapeutic responses and toxicity profiles of gemcitabine treatment in patients with advanced HNSCC [15,16]. Gemcitabine increases tumor cell sensitivity to radiation-induced apoptosis and hence is used as a radiation sensitizer in solid malignancies [14,17,18]. Antitumor activity of gemcitabine is mediated by multiple mechanisms dependent upon tumor types [19-21]. Gemcitabine treatment induces DNA-replication stress leading to the activation of Chk1 and Chk2 [22].
In a recent study, we used LY2 cells to develop an aggressive orthotopic mouse model of HNSCC which demonstrates rapid growth rate with regional (~100%) and distant (lung ~40%) metastasis within 3 - 4 weeks of inoculation [23]. LY2 cells were resistant to apoptosis under hypoxic condition both in vitro and in vivo [23]. These tumor cells survive under hypoxia by invoking an autophagic response [23]. Moreover, LY2 cells were not sensitive to cytotoxic activity of cisplatin and docetaxel, chemotherapeutic drugs that are commonly used in HNSCC-patients (unpublished data). In-contrast, -LY2 cells are highly susceptible for gemcitabine-mediated killing both in vitro and in vivo in a dose equivalent to that used for clinical dosing in patients. The purpose of the current study was to investigate the signal transduction pathways involved in the antitumor effect of gemcitabine against LY2 cells both in vitro and in vivo. Our studies reveal that gemcitabine treatment depletes Chk1 in LY2 cells in a timeand dose-dependent manner. Gemcitabine treatment induces phosphorylation of Chk1 in LY2 cells leading to its ubiquitination and subsequent proteasome degradation. Proteasome inhibitor suppresses gemcitabine-mediated depletion of Chk1 and decreases LY2 cells susceptibility to gemcitabine mediated-killing. Inhibition of Chk1 function, either using a small molecular inhibitor or siRNA mediated silencing enhances the sensitivity of LY2 cells to gemcitabine.
2. Materials and Methods
2.1. Reagents
Gemcitabine was purchased from ChemieTek® (Indianapolis, IN) and dissolved in PBS (12.5 mg/mL = 41.7 mM) and stored in aliquots at −20˚C for in vitro studies. Gemcitabine was dissolved in saline (20 mg/mL) and used for in vivo animal tumor model experiments. Antibodies against cleaved caspase-9 (Asp353, mouse specific), cleaved caspase-3 (Asp175) (5A1E), phosphoChk1 (Ser345) (133D3) and Chk2 were purchased from Cell Signaling Technology (Beverly, MA). Antibodies against β-actin and Chk1 were obtained from Sigma (St. Louis, MO) and Eptomics (Burlingame, CA), respecttively. The proteosomal inhibitor MG132 was purchased from Cayman Chemical (Ann Arbor, MI) and dissolved in DMSO to make 10 mM stock solution. The Chk1 inhibitor PD-407824 was purchased from Sigma Aldrich (St. Louis, MO) and dissolved in DMSO to make 10 mM stock solution.
2.2. Cell Culture
LY2 cells were kindly provided by Dr. Carter Van Waes (NIDCD/NIH). The LY2 cell line was isolated from lymph node metastases that developed in BALB/c mice after inoculation of murine squamous cell carcinoma cell line PAM 212 [24]. LY2 cells were maintained as monolayers in DMEM/F12 medium supplemented with 10% fetal calf serum (Life Technologies, CA), 2 mM L-glutamine, 100 µg/mL penicillin G, 100 µg/mL streptomycin, and 0.25 µg/mL amphotericin B at 37˚C with 5% CO2.
2.3. Cell Viability Assay
The cytotoxic activity of gemcitabine against LY2 cells was determined using Cell Counting Kit-8 (CCK-8) (Dojindo Molecular Technologies Inc, Rockville, MD), which is a highly sensitive colorimetric assay used for the determination of cell viability. Briefly, LY2 cells were plated in 96-well plates (or a plate) under standard culture conditions, next day treated with serial dilutions of gemcitabine (0.12 - 1000 nM) for 48 hours. After the drug treatment, the number of viable cells was determined using CCK-8 according to the manufacturer’s instruction. The 50% inhibitory concentration (IC50) resulting in a 50% reduction in cells compared to the vehicle-treated control cells was calculated by logistic analysis. The assay was done in triplicate, and the mean IC50 value was obtained from the results of two independent assays.
2.4. Immunoblotting Analysis
Immunoblotting assays were performed to determine the apoptotic (PARP, Caspase-3, and -9) and DNA damage response pathways that are activated by gemcitabine treatment. LY2 cells were exposed for 2 hours with 1 µM gemcitabine and total cellular proteins were extracted using M‑PER™ mammalian protein extraction reagent (Pierce, Rockford, IL) at different time points. Total cellular proteins (20 µg) from control and experimental samples were resolved by 10% sodium dodecyl sulfate polyacrylamide gel and transferred to nitrocellulose membranes. These membranes were blocked with Odyssey Blocking Buffer (LI-COR, Lincoln, NB) for 1 hour, and then incubated with primary antibodies overnight at 1:500-1000 dilutions. Specific protein bands were detected using infrared emitting dye-conjugated secondary antibodies (IRDye® 800CW goat anti-rabbit IgG; 1:8,000 dilution or IRDye® 680LT goat anti-mouse secondary antibody; 1:10,000 dilution, LI-COR, Lincoln, NB) and quantified using Odyssey® Infrared Imaging System (LI-COR, Lincoln, NB). Densitometric measurements of immune-reactive bands were carried out using the Odyssey Application software (version 1.2) from LI-COR Biosciences.
2.5. Immunofluorescence Staining
In order to detect gemcitabine-mediated Chk1 phosphorylation, LY2 cells were grown as monolayers on Lab-Tek II chamber slide overnight and exposed to gemcitabine (1 µM) for 15 to 105 minutes. Cells were washed, fixed in 2% PFA and permeabilized with 0.5% saponin and used for immunohistochemical detection of phosphorylated Chk1. Cells were incubated with a primary antibody against phosphorylated mouse Chk1 (1:100) overnight, followed by Alexa Fluor® 555 conjugated anti-rabbit IgG F(ab’)2 secondary antibody (1:1000 dilution) for 1 hour. Cells were washed, counterstained with ProLong® Gold antifade reagent with DAPI (Invitrogen, Eugene, OR). Immunofluorescence staining was viewed and images were captured using a Nikon Eclipse E400 epifluorescence microscope (Nikon Instruments Inc., Tokyo, Japan) equipped with a CoolSNAP EZ CCD camera.
2.6. RNA Isolation and QuantitativeRT-PCR
LY2 cells were plated in 6-well plates and treated with gemcitabine (1 µM) for 2 hours, then replaced with drugfree media and samples were collected at various time points. Control cells were treated with regular media without the drug. Total RNA from experimental and control cells were isolated using RNeasy Mini Kit (Qiagen, MA), digested with DNase I (Invitrogen, CA), and reverse-transcribed to cDNA using the Super Script III Reverse Transcriptase (Invitrogen, CA). Expression of Chk1 (NCBI gene ID: 12649) transcript was evaluated using the primers, forward: 5’-ATGGCAGGGGTGGTTTATCTT-3’ and reverse 5’-GCGTTCACGATTATTATGCCGAA-3’ (Primer Bank ID: 22902453a3). Murine glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (NCBI gene ID: 100042025) was used as a reference gene and amplified using the primers, forward 5’-TGACCACAGTCCATGCCATC-3’ and reverse 5’-GACGGACACATTGGGGGTAG-3’ (Primer Bank ID: 6679937a3). Following gelelectrophoresis for confirmation of PCR product size, quantitative RT-PCR measurement of Chk1 transcripts was performed using Power SYBR Green PCR Master Mix on the StepOnePlus Real-Time PCR System (Applied Biosystems, CA). Chk1 mRNA levels were calculated by comparative cycling threshold (ΔΔCT) method normalized to the GAPDH expression levels.
2.7. Proteasome Inhibitor MG132 Treatment
We examined the expression level of Chk1 and cell viability in LY2 cells when exposed to gemcitabine with various concentrations of the proteasome inhibitor MG132. LY2 cells were plated into a 6-well culture plate and treated with either gemcitabine (50 nM) alone, or with different concentrations of MG132 (0, 125, 250, and 500 nM) for 12 and 24 hours. At the end of the treatment, total protein extracts were prepared for immunoblotting analysis of Chk1 as described above. For cell viability assays, LY2 cells were cultured in a 96-well plate, pretreated with MG132 (0, 125, 250, and 500 nM) for 2 hours and then co-treated with gemcitabine at different concentrations (0, 12.5, 25, and 50 nM) for 48 hours. Cell viability after drug treatment was determined using the CCK-8 method as described above and expressed as the percentage of vehicle-treated control cells.
2.8. Chk1-siRNA and Chk1 Inhibitor PD 407824 Treatments
The SMARTpoolÒ siRNA targeting murine Chk1 (CHk1 siRNA-SP) (Gene ID# 12649; Cat# M-058640-02-0005) and non-target control siRNA (siRNA-NT) (Cat#: D-001206-13-20) were obtained from Dharmacon RNA Technologies (Lafayette, CO) and reconstituted into 20 µM stock solution according to the manufacturer’s instructions and stored at −20˚C. LY2 cells were transfected with 200 nM Chk1 siRNA-SP or siRNA-NT (200 nM) using NucleofectorÒ II kit with cell line solution L (Amaxa Biosystems, Germany). The siRNAand mocktransfected cells were split and plated into 6- and 96-well plates to asses Chk1 silencing and gemcitabine mediated cytotoxicity, respectively. Total cellular protein and RNA of siRNA-transfected and control cells were isolated at 24 and 48 hours post-transfection and used for immunoblotting and quantitative RT-PCR analysis of Chk1 expression. Control and siRNA transfected cells were grown in 96-well plates for 24 hours and then treated with gemcitabine for 48 hours at the various concentrations (12.5, 25, and 50 nM). Cell viability after gemcitabine treatment was determined using CCK-8.
For Chk1 inhibitor study, LY2 cells were plated in 96-well plate, grown overnight and pre‑treated with PD 407824 (0.5 µM) for 24 hours. Pre-treated cells were co-incubated with PD 407824 (0.5 µM) and gemcitabine at different concentrations (12.5, 25, and 50 nM) for 48 hours and experimental and control cells viability was determined using CCK-8.
2.9. Orthotopic HNSCC Model and Drug Treatment
Female BALB/c mice (6 - 8 weeks old) were purchased from Harlan (Indianapolis, IN). Orthotopic tumor cell implantation and drug treatments were performed according to the animal protocol approved by the Animal Welfare Committee, University Texas Health Science Center at Houston. LY2 cells (0.5 × 106 cells/mouse) were injected via intraoral route into the mandibular buccal sulcus of the mice (n = 16). One week after injecttion of tumor cells, mice were divided randomly into two groups: control group (n = 8) and treatment group (n = 8). Mice in the treatment group were treated with intraperitoneal (IP) injection of gemcitabine (100 mg/Kg body weight) on the 1st and 4th days of the week for 3 weeks. Mice in the control group were treated with IP injections of saline with the same frequency as gemcitabine treatment. Mice were sacrificed two weeks after completion of the drug treatment. The primary tumor size was measured immediately after the mouse was sacrificed. Tumor volume was calculated using the equation: tumor volume = (A × B2/2 mm3) (A represents the longer and B represents shorter diameters of the tumor). Tumors were frozen in liquid nitrogen immediately and stored at −80˚C for further studies. Frozen tumors from control and drug-treated mice were sectioned and fixed in acetone and used for immunofluorescence staining of Chk1.
3. Results
3.1. Gemcitabine Treatment Markedly Reduces LY2 Cell Viability in Vitro
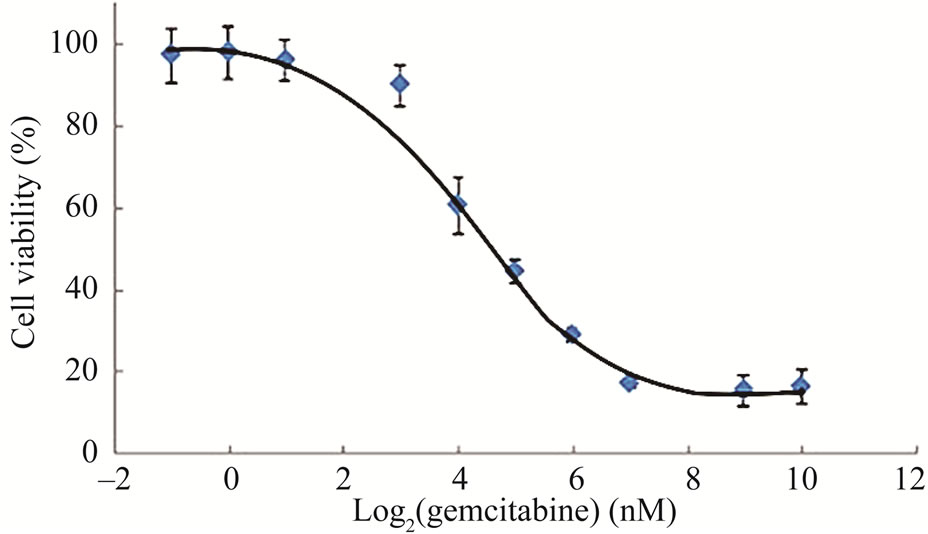
First, we evaluated the growth inhibitory effect of a panel of chemotherapeutic drugs against LY2 cells and noticed that LY2 cells are resistant to most of the chemotherapeutic drugs tested except for gemcitabine (Data not shown). Next, we treated LY2 cells with a range of gemcitabine doses for 48 hours to determine its IC50 value. It was evident that increasing concentrations of gemcitabine in the culture medium inhibited the growth of LY2 cells in a dose-dependent manner (Figure 1(a)). Gemcitabine inhibited the growth of the LY2 cells with an IC50 value of 24.93 nM (Figure 1(a)). Since growth inhibitory effect of gemcitabine against tumor cells can be mediated by apoptosis or by triggering cell cycle arrest [13,21,25], we examined the effect of gemcitabine on induction of apoptosis in LY2 cells.
We treated LY2 cells with gemcitabine for 2 hours and analyzed for various apoptotic markers by immunoblotting. Gemcitabine treatment induced activation of caspases-3, -9 and PARP as shown by the appearance of their cleavage products in the immunoblotting (Figure 1(b)).
 (a)
(a)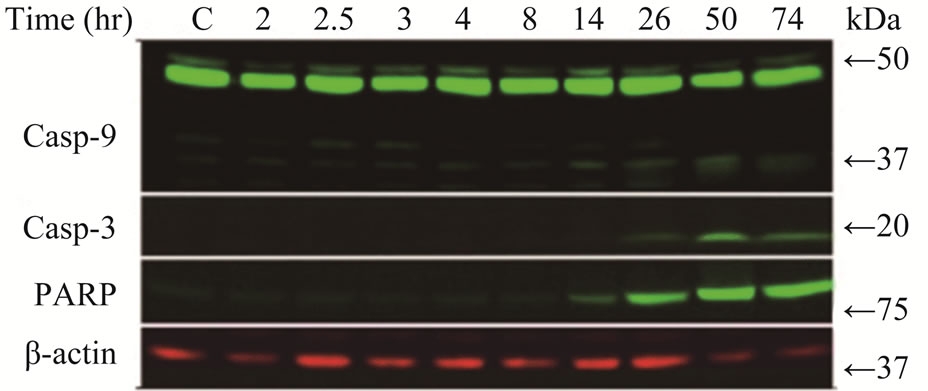 (b)
(b)
Figure 1. Gemcitabine inhibits LY2 cell growth in vitro by inducing apoptosis (a) Dose response curve of gemcitabine mediated-cytotoxicity against LY2 cells.LY2 cells were exposed to various concentrations of gemcitabine (1000 to 0.12 nM in a series of 2-fold dilution) for 48hours (hr) and cells viability was determined using CCK-8 kit. Gemcitabine treatment reduces LY2 cells survival with IC50 value of 24.93 nM; (b) Gemcitabine treatment activates caspases-9, -3 and PARP in LY2 cell as demonstrated by the appearance of their respective cleaved products in representative immunoblots. LY2 cells were treated with 1 µM gemcitabine for 2 hours. Protein samples were collected at indicated time points and subjected to immunoblotting.
3.2. Gemcitabine Treatment Stimulates Chk1 Activation and Subsequent Depletion in LY2 Cells
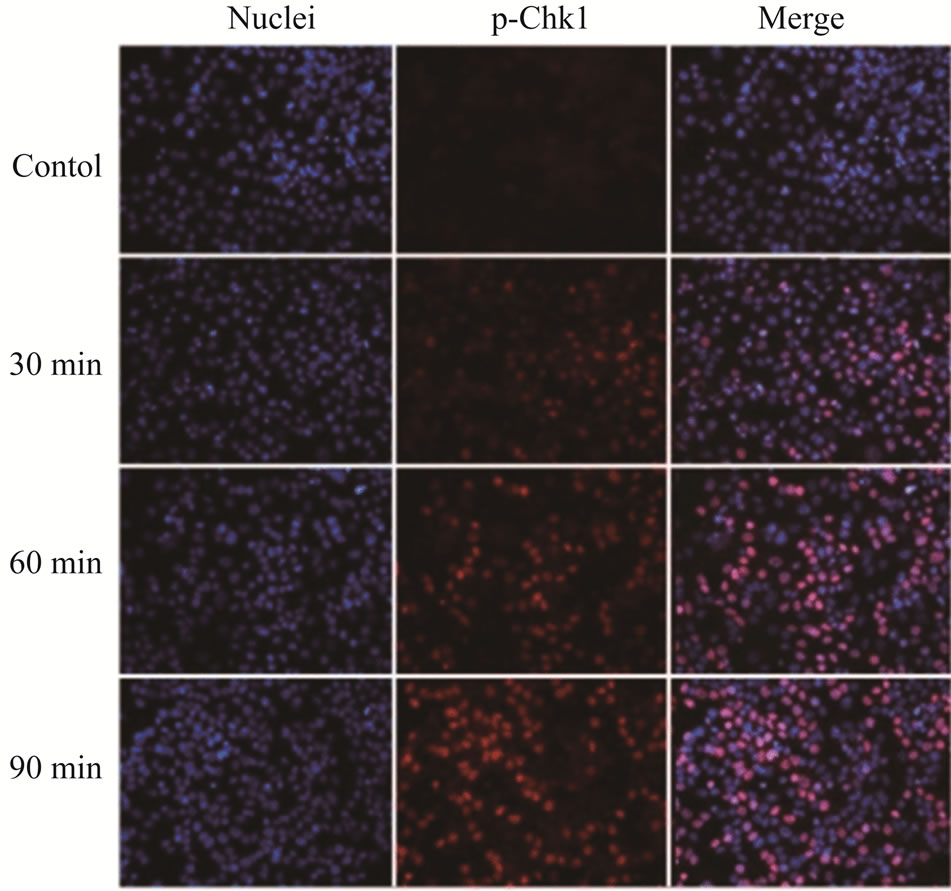
Since the antitumor activities of chemotherapeutic drugs depend primarily on the DNA damage responses, we first tested the activation status and cellular level of a key DNA damage response signaling molecule, Chk1, in LY2 cells. LY2 cells were grown on chamber slides and treated with gemcitabine and the activation (phosphorylation) status of Chk1 at different time points were examined using an antibody that detects phosphorylation of Chk1 at serine345. The phosphorylated Chk1 (p-Chk1) was detected in the nuclei of LY2 cells treated with gemcitabine but not in vehicle treated control cells (Figure 2(a)). Approximately 20% of LY2 cells revealed activetion of Chk1 within 30 minutes of gemcitabine treatment and the percentage of LY2 cells with p-Chk1 increased to almost 50% within 2 hours of gemcitabine treatment (Figures 2(a) and (b)). To further examine how long the activation of Chk1 persists, LY2 cells were exposed to gemcitabine for 2 hours and then replaced with drug-free media and the Chk1 phosphorylation was assessed by western blot at different time points (Figure 2(c)). Activation of Chk1 by gemcitabine was a relatively early event, with the maximal increase in the p-Chk1 protein level occurring within 2 hours of gemcitabine treatment. The p-Chk1 was not detected in untreated control cells (Figures 2(c) and (d)). With increasing time, p‑Chk1 level decreased gradually and reached its lowest level 12 hours after gemcitabine treatment (Figures 2(c) and (d)). To assess the effect of gemcitabine treatment on the expression level of total Chk1, we re-probed the same blot with antibody against Chk1 which revealed that the total
 (a)
(a)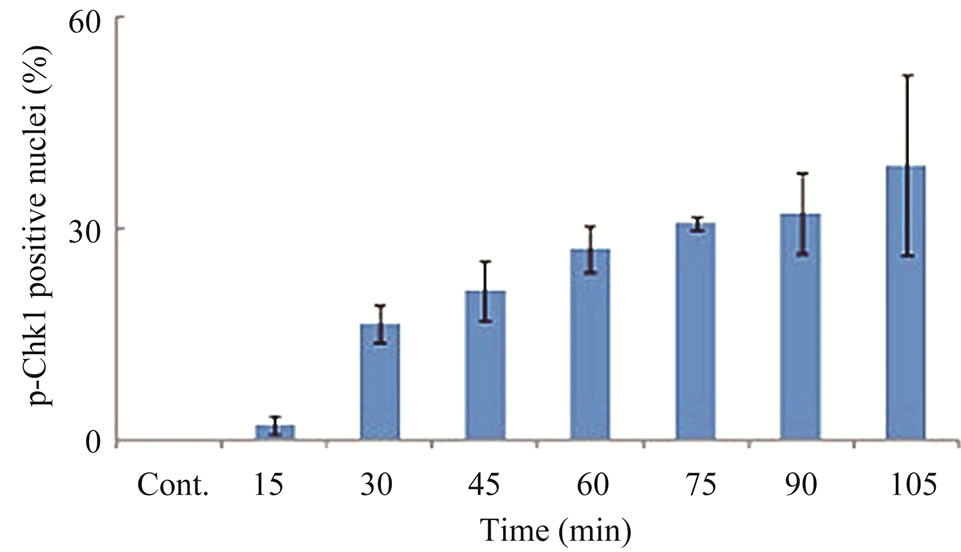 (b)
(b)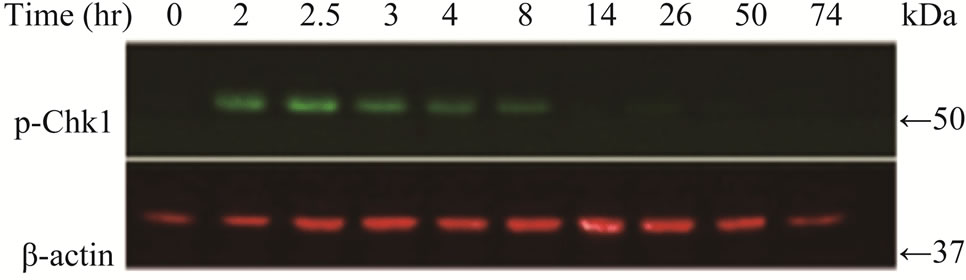 (c)
(c)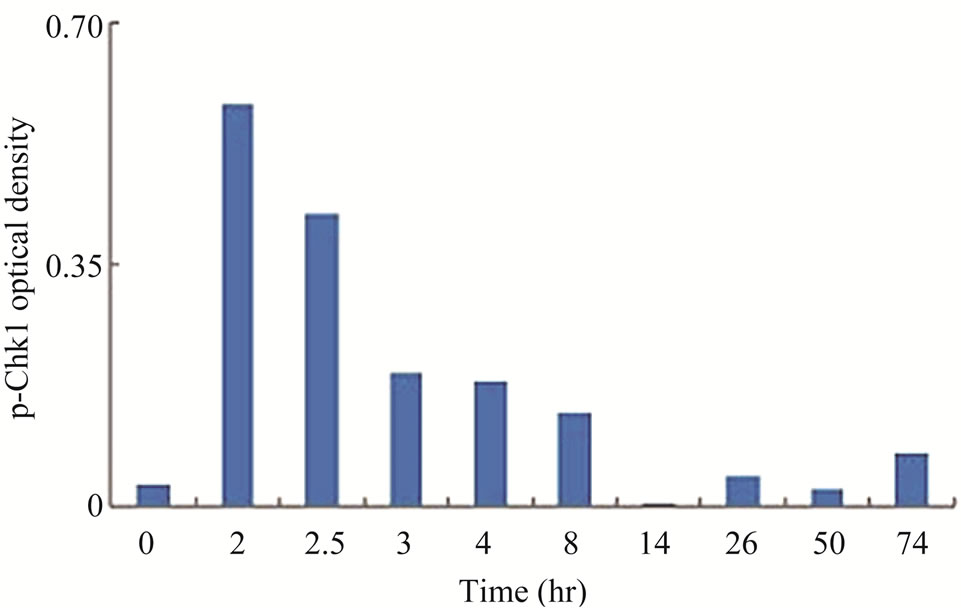 (d)
(d)
Figure 2. Gemcitabine treatment induces rapid activation of Chk1 in LY2 cells. LY2 cells were treated with 1 µM gemcitabine for indicated time periods (up to 105 minutes) and were fixed and used for immunofluorescence staining of phosphorylated Chk1 (Ser345). (a) Representative immunofluorescence images show the expression of p-Chk1 in control and treated LY2 cells at different time points. Nuclear DNA was labeled with DAPI (blue) and rhodamine (red) labeling was used for the detection of p-Chk1. (b) Histograms reveal rapid increase of p-Chk1 positive cells in following gemcitabine treatment. Cells were counted from three representative areas and were expressed as the % of cells positive for p-Chk1. Columns and error bars indicates mean ± SD. (c)-(d) LY2 cells were treated with 1 µM gemcitabine for 2 hours. Total cellular protein of control and treated cells were collected at indicated time points and used for immunoblotting analysis of p-Chk1. (c) Representative immunoblotreveals the presence of p-Chk1-specific band 2 hours after gemcitabine treatment. (d) Bar graph depicts the densitometric measurements of p-Chk1 levels in treated and control cells at different time points after gemcitabine treatment. Each treatment group is normalized to the β-actin loading control.
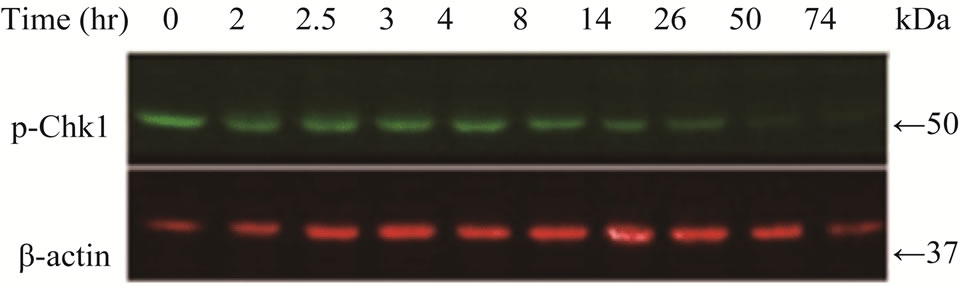
Chk1 level was dramatically reduced in LY2 cells treated with gemcitabine (Figures 3(a) and (b)). The Chk1 protein level declined rapidly by 50% compared to control cells within 2 hours of gemcitabine treatment and it became undetectable 50-hours post-treatment (Figures 3(a) and (b)). Furthermore, gemcitabine treatment of LY2 cells caused reduction in their Chk1 protein level in a dosedependent manner (Figure 3(c)). Gemcitabine treatment reduces the Chk1 level within 2 hours (Figure 4), whereas the caspase-3 and -9 are activated at 12 hours after treatment (Figure 1(b)), suggesting that reduction in Chk1 level is the cause rather than the consequence of apoptosis.
Next, we assessed whether Chk1 mRNA synthesis was affected by gemcitabine treatment using quantitative RTPCR. LY2 cells were pre-treated with gemcitabine for 2 hours and then total RNA was isolated at varying time points and analyzed for Chk1 mRNA levels (Figure 3(d)). Although RNA levels of Chk1 decreased immediately after gemcitabine treatment, Chk1 mRNA levels gradually increased and reached a level comparable to the untreated controls within 24 hours of gemcitabine treatment (Figure 3(d)). Gel-electrophoresis analysis of the Chk1-specfic PCR product further confirmed that Chk1 mRNA levels are not remarkably different between gemcitabine-treated and control cells (Figure S1). In contrast, Chk1 protein levels continue to decrease during this time period (Figures 3(a) and (b)). These findings suggest that gemcitabine does not suppress Chk1 expression through inhibition of its transcription; raising the possibility that gemcitabine reduces Chk1 protein expression via enhancing its degradation or inhibiting its translation.
 (a)
(a)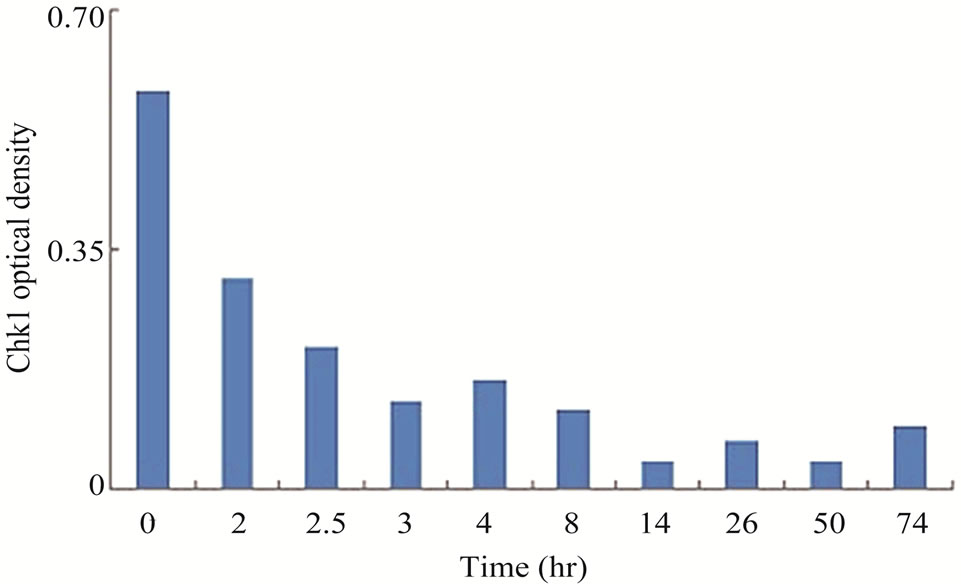 (b)
(b) (c)
(c)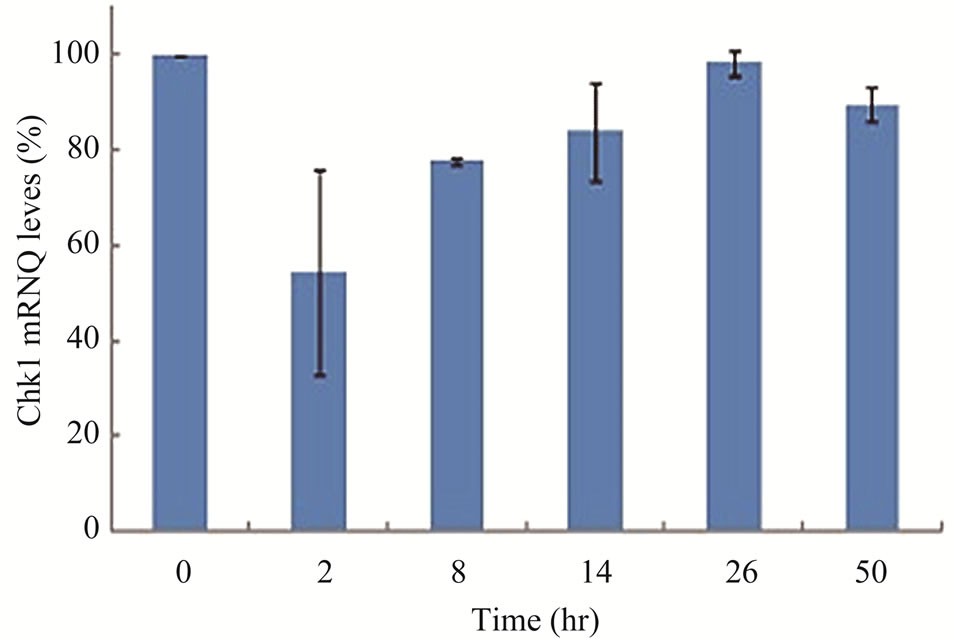 (d)
(d)
Figure 3. Gemcitabine treatment depletes total Chk1 protein in a timeand dose-dependent manner. LY2 cells were treated with 1 µM gemcitabine for 2 hours and cellular extracts were collected at indicated time points and analyzed for Chk1 protein by Western blotting. (a) Western blot reveals the gradual loss of Chk1 protein in gemcitabine treated cells compared to control cells (0 hr). (b) Bar graphs, obtained by densitometric analysis of Western blot data, show the decrease of Chk1 protein levels (after normalizetion to β-actin) in gemcitabine treated cells in a time dependent manner. (c) LY2 cells were exposed to various concentrations of gemcitabine and cellular extracts were collected after 6- and 24-hours and analyzed for Chk1 protein by Western blotting. Representative Western blots reveal the reduction in Chk1 protein levels in gemcitabine treated cells in a dose dependent manner. (d) Bar graph summarizes the results of a quantitative real-time RT-PCR analysis of Chk1 mRNA expression in control (0 hr) and gemcitabine-treated cells at various time points. There was a transient decrease in the Chk1 mRNA level 2 hours after gemcitabine treatment but the relative Chk1 mRNA levels were not remarkably different between treated and control cells at a later time points. Relative Chk1 mRNA levels were depicted as % of control cells, normalized to the GAPDH mRNA levels.
3.3. Proteasome Inhibitor MG132 Blocks Gemcitabine-Mediated Depletion of Chk1 in LY2 Cells
The multicatalytic ubiquitin-proteasome proteolytic pathway plays a critical role in the post-translational
 (a)
(a)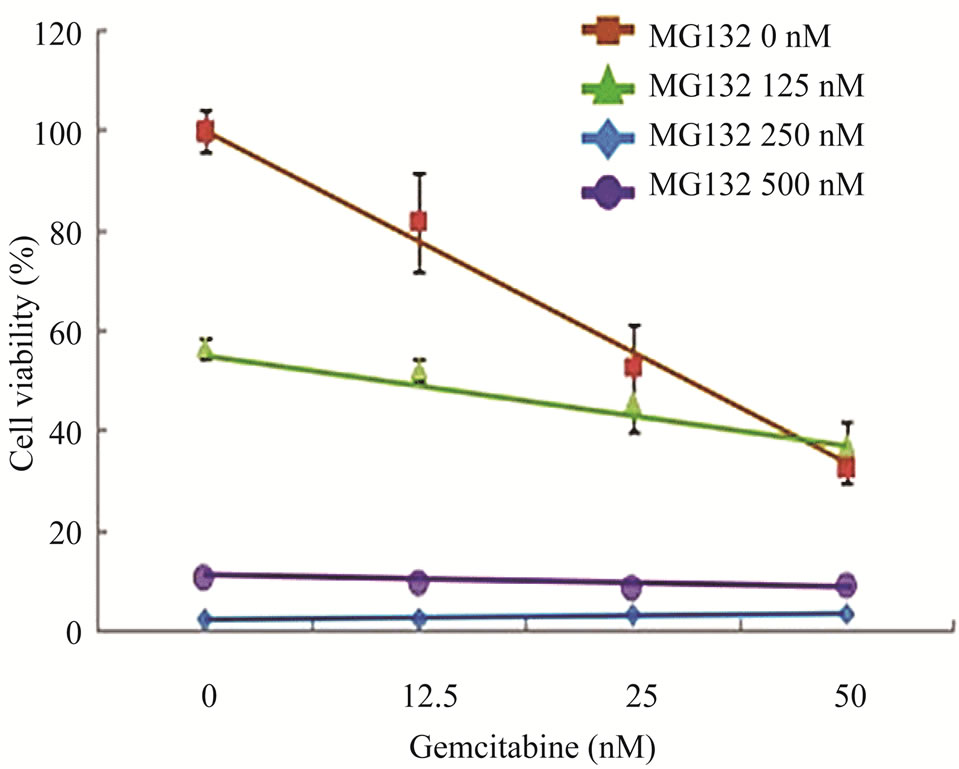 (b)
(b)
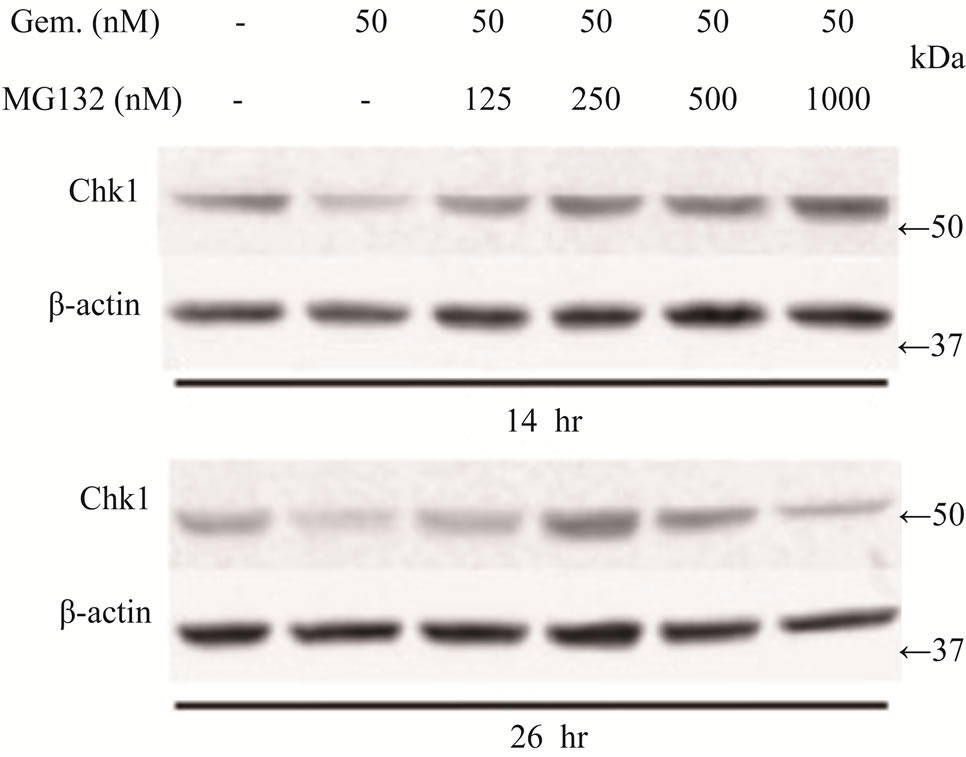
Figure 4. Effects of proteasome inhibition using inhibitor MG132 on gemcitabine-mediated killing of LY2 cells (a) Western blot analyses showing the loss of Chk1 in LY2 cells treated with gemcitabine alone but its presence in LY2 cells pre-treated with MG132. LY2 cells were pre-treated with various concentrations of MG132 for 2 hours and then co-treated with MG132 and gemcitabine (50 nM). Control cells were left untreated or treated with gemcitabine (50 nM) alone. Total cellular proteins were extracted at 14 and 26 hour time points and used for immunoblotting. (b) Proteasome inhibitor MG132 diminishes the sensitivity of LY2 cells to gemcitabine. LY2 cells pre-treated with MG132 for 2 hours and then co-treated with various concentration of gemcitabine for 48-hours.Cells viability was measured after 48 hours and the percentage of viable cells relative to the mock-treated control was plotted. Survival curves of LY2 cells co-treated with gemcitabine and MG132 (125 nM) demonstrate plateau phase which is absent in the survival curve of LY2 cell treated with gemcitabine alone (brown line) indicating that MG132 antagonizes gemcitabine mediated killing of LY2 cells. It should be noted that MG132 is cytotoxic to LY2 cells at higher concentrations (250 nM & 500 nM).

Figure S1.. Gemcitabine treatment does not affect Chk1 mRNA expression by RT-PCR.
regulation of important signaling molecules and kinases involved in cell cycle control and apoptosis [26]. The proteasome inhibitor MG132 is a membrane-permeable peptide aldehyde which blocks the chymotrypsin-like activity of the proteasome [27]. Proteasome inhibitors block the breakdown of cellular proteins targeted for degradation by proteasomes [27]. We examined the effect of MG132 on gemcitabine mediated depletion of Chk1 protein and killing of LY2 cells. LY2 cells were treated with gemcitabine (50 nM) alone with increasing concentrations of MG132 for 14 and 26 hours and then analyzed for Chk1 protein levels. Immunoblotting studies revealed that gemcitabine-induced depletion of Chk1 was blocked in a dose-dependent manner by MG132 (Figure 4(a)). Since MG132 inhibits gemcitabine induced degradation of Chk1, we sought to determine the effect of MG132 on gemcitabine-induced killing of LY2 cells. We pretreated LY2 cells with a range of concentrations of MG132 (0 - 500 nM) and then co-treated with a range of gemcitabine concentrations (0 - 50 nM) and determined cell viability at 48 hours post-treatment. The data obtained from these experiments are complicated by the fact that MG132 itself is cytotoxic to the LY2 cells (Figure 4(b)), however, the comparison of survival curves for different treatment combinations reveals remarkable difference in the cell killing between LY2 cells treated with gemcitabine alone and gemcitabine and MG132 (Figure 4(b)). The slope of the cell viability curve attains a plateau in gemcitabine-mediated killing of LY2 cells with an increasing dose of MG132 (Figure 4(b)). These findings imply that the proteasome inhibitor MG131 antagonizes gemcitabine-mediated killing of LY2 cells by blocking proteosomal degradation of Chk1. This prompted us to investigate whether silencing Chk1 activity by siRNA or Chk1 inhibitor would increase the sensitivity of LY2 cells to gemcitabine-mediated killing.
3.4. Repression of Chk1 Activity by siRNA or Chk1 Inhibitor Increases the Sensitivity of LY2 Cells to Gemcitabine
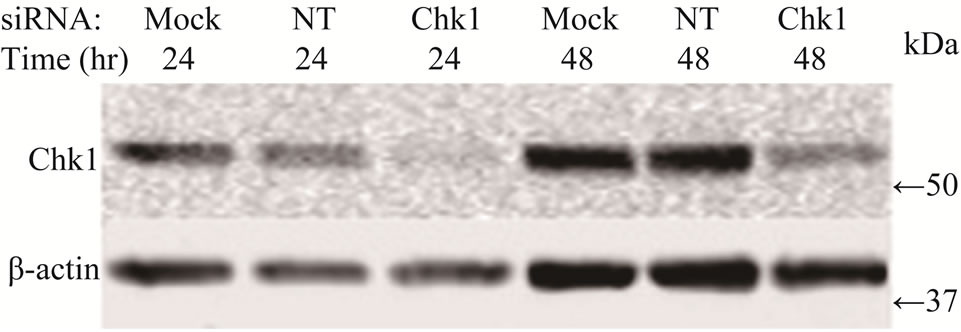
To determine whether suppression of Chk1 activity can be exploited therapeutically to increase the chemosensitivity of LY2 cells, we first examined the effect of siRNA-mediated Chk1 knockdown on the susceptibility of LY2 cells to gemcitabine-mediated killing. LY2 cells were transfected with Chk1 siRNA-SP consisting of a pool of 4 sets of siRNAs targeting different regions of Chk1 mRNA. Chk1 protein level was reduced by more than 70% compared with cells transfected with siRNANT (Figures 5(a) and (b)). Quantitative RT-PCR analysis also confirmed the reduction of Chk1 mRNA levels in Chk1 siRNA-SP treated LY2 cells compared with siRNA-NT treated cells (Figure S2). RNAi-mediated silencing of Chk1 did not impact the viability of LY2 cells. Next we analyzed the effect of gemcitabine on LY2 cell viability following siRNA-mediated silencing of Chk1. LY2 cells were transfected with Chk1 siRNA-SP for 24 hours and then treated with different concentrations of gemcitabine. As shown in Figure 5(c), Chk1 siRNA-SP treated cells revealed significantly increased sensitivity to gemcitabineat 12.5 nM (p < 0.1) and at both 25 and 50 nM gemcitabine concentrations (p < 0.001) compared to mockand siRNA-NT-transfected
 (a)
(a)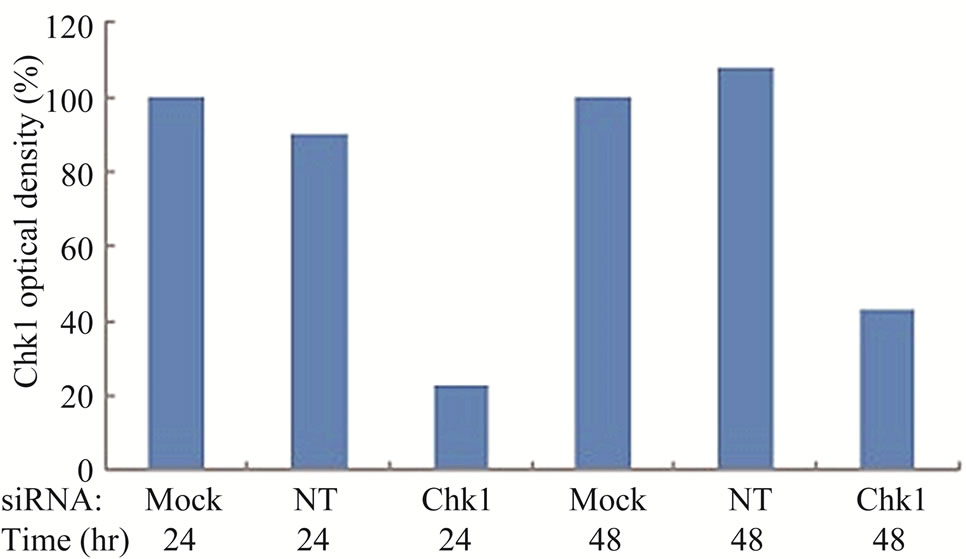 (b)
(b)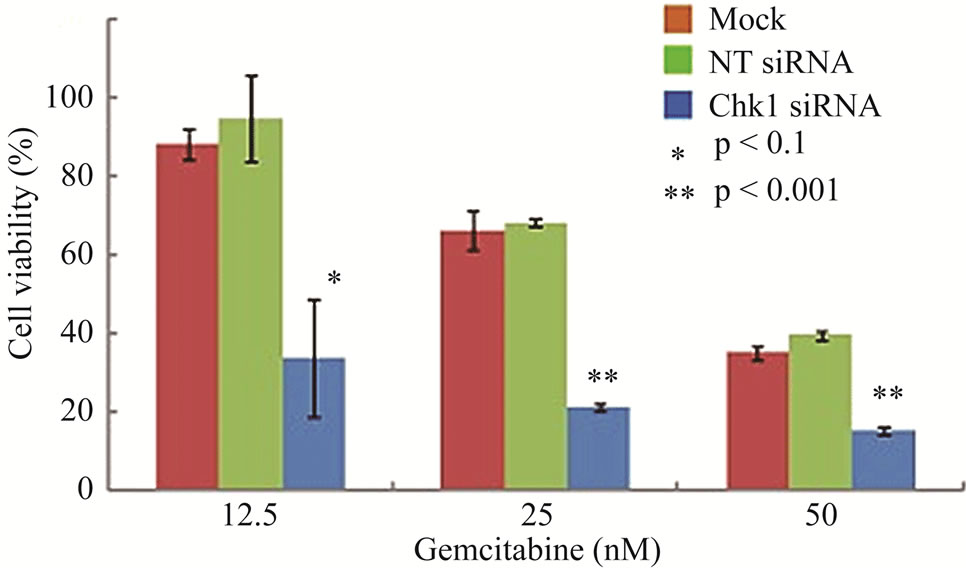 (c)
(c)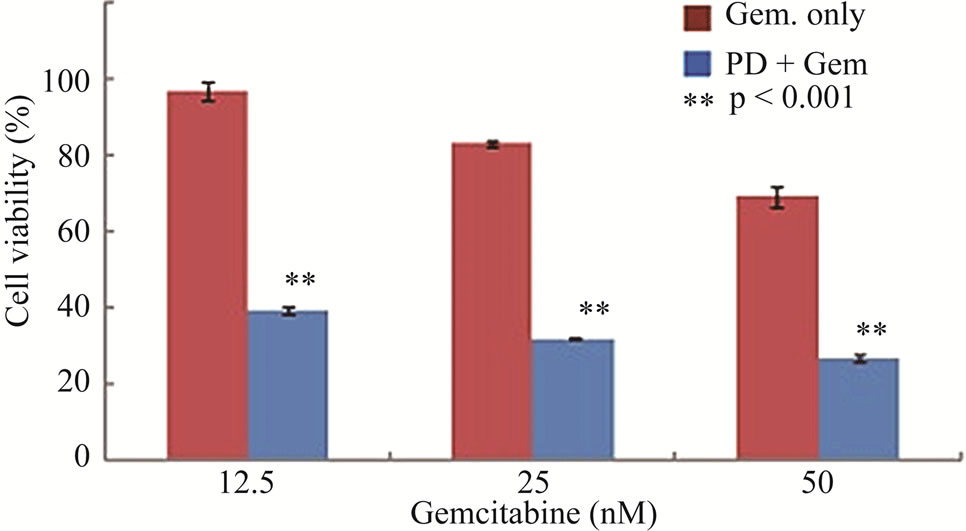 (d)
(d)
Figure 5. Repression of Chk1 activity by siRNA or inhibitor PD 407824 increases the sensitivity of LY2 cells to gemcitabine. (a) Western blot shows the knock-down of Chk1 24- and 48-hours post siRNA transfection. LY2 cells were treated with transfection agent without siRNA (Mock), non-targeting siRNA (NT; 200 nM) and Chk1 siRNA-SP (Chk1, 200 nM) and cellular extracts were collected after 24 and 48 hours and used for immunoblotting; (b) Bar graph shows the densitometric measurements of Chk1 protein levels in Chk1-siRNA treated and control cells after 24 and 48 hours of siRNA transfection; (c) Control and Chk1 siRNA-transfected LY2 cells were grown for 24 hours and then treated with gemcitabine for 48 hours, and the % of viable cells was determined. Bar graphs demonstrate that siRNA-mediated silencing of Chk1 significantly (p < 0.001) increases the cell killing by gemcitabine compared to control groups. D. Similarly, inhibition of Chk1 activity by Chk1 inhibitor PD 407824 sensitizes LY2 cells to gemcitabine killing. LY2 cells were first exposed to 0.5 µM PD 407824 for 24 hours and then co-treated with gemcitabine (12.5 - 50 nM) ± 0.5 µM PD 407824) for another 24-hours before determining the cell viability.
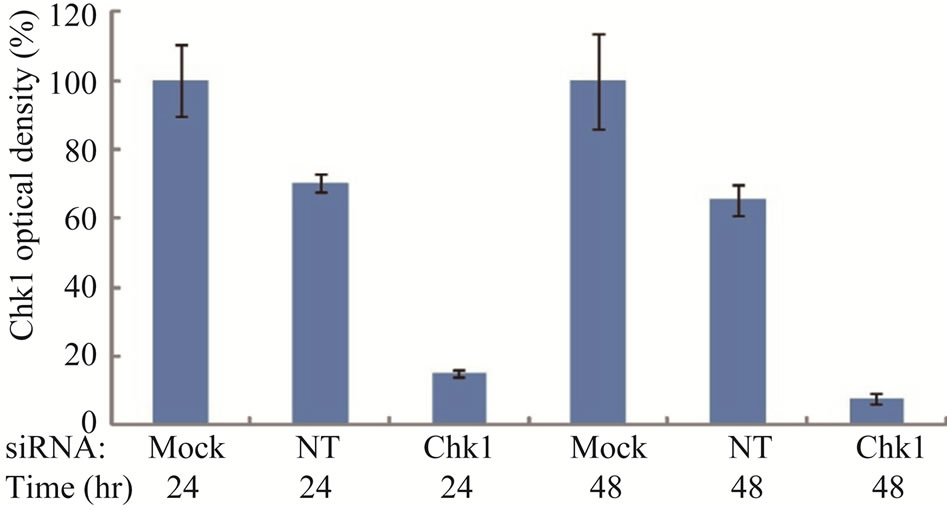
Figure S2. . Chk1-siRNA down-regulates Chk1 at mRNA level by quantitative RT-PCR.
cells. To further validate that blocking Chk1 kinase activity in LY2 cells enhances their sensitivity to gemcitabine, we pretreated LY2 cells with Chk1 inhibitor PD 407824 and then co-treated the cells with a range of concentrations of gemcitabine and PD 407824. Inhibition of Chk1 activity by PD 407824 significantly (p < 0.001) increased the gemcitabine cytotoxicity against LY2 cells (Figure 5(d)).
3.5. Gemcitabine Inhibits LY2 Tumor Growth in Vivo
To determine whether the antitumor activity of gemcitabine against LY2 cells in vitro could be translated into an in vivo, a tumor growth delay assay was performed using an orthotopic murine HNSCC model. LY2 cells
 (a)
(a)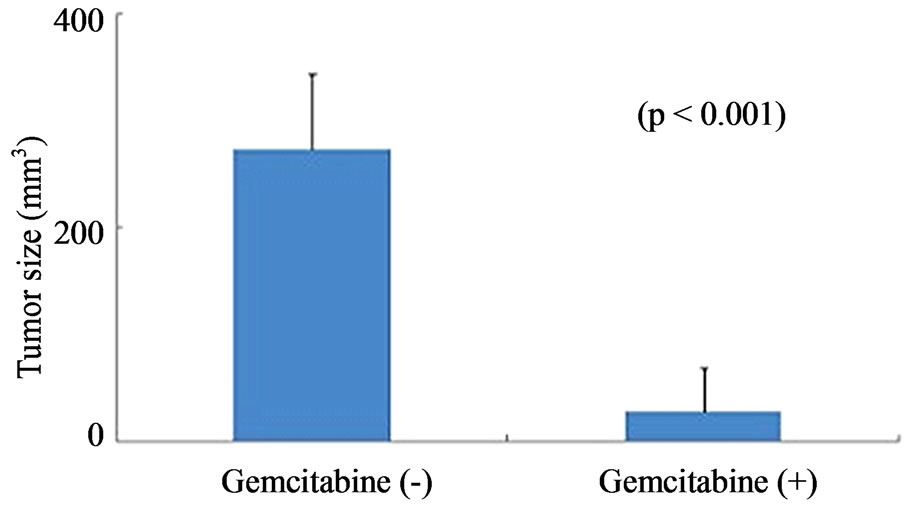 (b)
(b)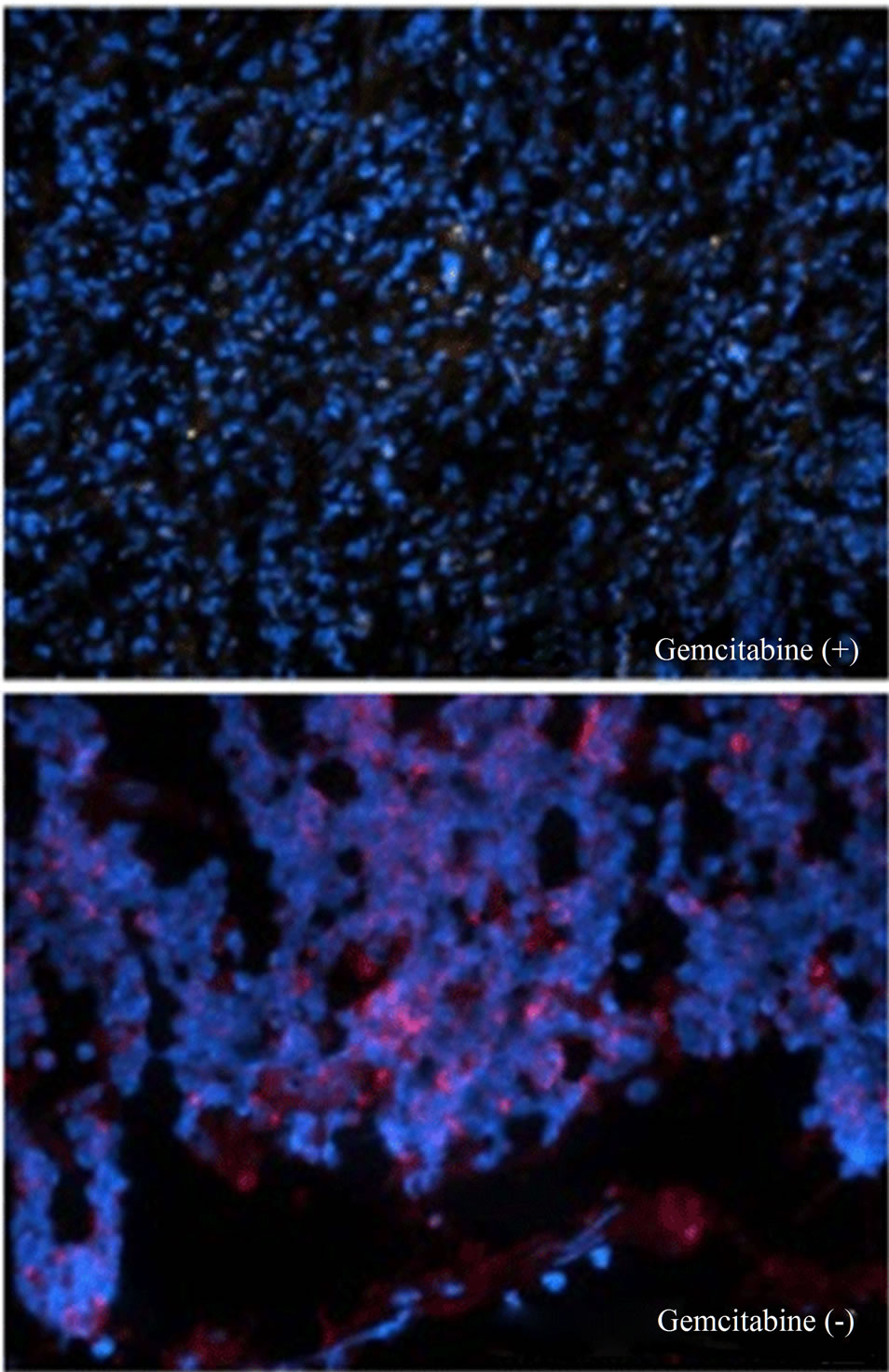 (c)
(c)
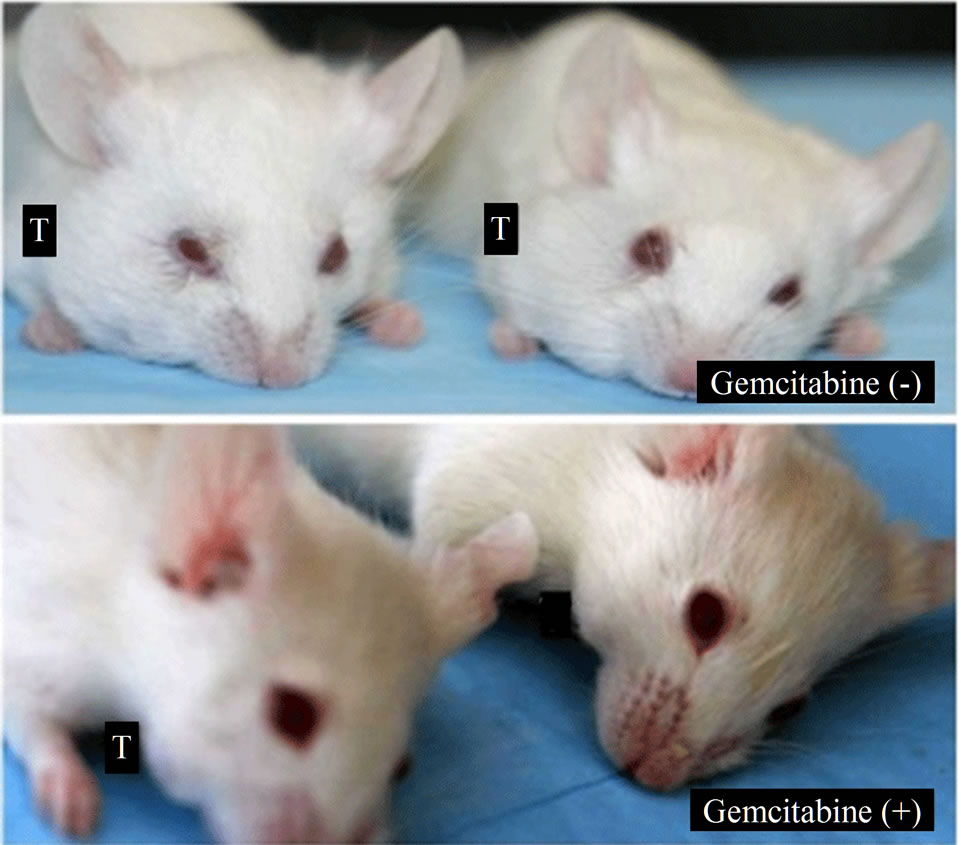
Figure 6. Gemcitabine inhibits tumor growth and depletes Chk1 expression in vivo. LY2 cells (0.5 × 106 cells/mice) were injected via intraoral route into the mandibular buccal sulcus of the mice (n = 16). One week after injection of tumor cells, mice were divided randomly into control group (n = 8) and treatment group (n = 8). Mice in the treatment group were treated with IP injection of gemcitabine (100 mg/Kg body weight) on the 1st and 4th days of a week for 3 weeks. Mice in the control group were treated with IP injections of saline with the same frequency as gemcitabine treatment. (a) Representative pictures are the control [Gemcitabine (−)] and gemcitabine-treated [Gemcitabine (+)] mice bearing HNSCC and tumors at six weeks after tumor cell inoculation; (b) Bar graph demonstrates that the average tumor sizes of control and gemcitabine-treated mice are significantly (p < 0.001) different. The primary tumor size was measured immediately after the mouse was sacrificed. Tumor volume was calculated using the equation: tumor volume = (A × B2/2 mm3) (A represents the longer and B represents shorter diameters of the tumor); (c) Immunofluorescence staining for Chk1 in tumor sections reveals that it is highly expressed in tumors of control animals [Gemcitabine (−)] but not in gemcitabine treated tumor section [Gemcitabine (+)]. Tumor sections were counterstained with DAPI (blue) and rhodamine (red) labeling was used for the detection Chk1 protein.
were inoculated into the mandibular buccal sulcus of the Balb/c mice via intraoral route, and gemcitabine therapy (100 mg/kg, biweekly IP injections for 3 weeks) was initiated 1week after tumor cell injection. Control animals received biweekly IP injections of normal saline. All animals were sacrificed at 6 weeks, when tumorbearing control mice became moribund. Gemcitabine treatment resulted in dramatic inhibition of tumor growth and the average tumor volumes at the end of 6 weeks were significantly smaller in gemcitabine-treated mice compared to controls (p < 0.001) (Figures 6(a) and (b)). We also investigated the effect of gemcitabine treatment on the Chk1 protein levels in tumor tissues in vivo. Consistent with the results obtained with the in vitro studies, Chk1 was mostly negative in gemcitabine-treated tumor tissue compared to its strong expression in control tumors (Figure 6(c)).
4. Discussion
HNSCC accounts for 5% of newly diagnosed cancers and 350,000 cancer-related deaths worldwide [28]. The majority of patients with HNSCC present with locally advanced, stage III or IV disease that requires a combination of chemotherapy, radiation, or surgery [29]. Despite these aggressive combination therapies, 5 year survival rates remain poor in these patients due to locoregional recurrence (60%) and distant metastasis (25%) [28,29]. Hence, there is an unmet demand for chemotherapeutic drugs that confer improved efficacy and better outcome in patients with advanced HNSCC. The nucleoside analogue gemcitabine demonstrates promising antitumor activity against a variety of solid malignancies that include pancreatic, gall bladder, lung and breast cancer [30]. Recent clinical studies have documented the therapeutic efficacy of gemcitabine against advanced head and neck cancer [15,16]. Hence, there is a burgeoning interest in elucidating the molecular basis for the differential sensitivity of head and neck cancer to chemotherapeutic agents including gemcitabine. Murine squamous cell carcinoma cell line LY2 forms a rapidly growing and highly metastatic orthotopic model of HNSCC in syngeneic immunocompetent Balb/c mice [23]. Our preliminary studies revealed that LY2 cells, which are extremely chemoresistant to many of widely used chemotherapeutic drugs, are susceptible to gemcitabine-mediated cytotoxicity at therapeutic concentrations. Our study revealed that LY2 cells treated with gemcitabine undergo apoptosis mediated primarily by an intrinsic (mitochondrial) pathway as evidenced by the activation of both caspase-3 and -9 (Figure 1(b)).
Antitumor activity of gemcitabine is mediated by multiple mechanisms dependent upon tumor types [19]. Gemcitabine is a prodrug which upon entering the tumor cells undergoes sequential phosphorylation resulting in the formation of its active metabolites, gemcitabine diphosphate (dFdCDP) and triphosphate (dFdCTP). Active metabolites of gemcitabine target a number of intracellular enzymes and signaling molecules, which vary considerable between different tumor cell types that have varying sensitivity to gemcitabine [20]. This sensitivity will also vary depending on which molecular mechanism contributes to its tumoricidal activities [20]. The dFdCTP inhibits DNA polymerase and also becomes incorporated into the DNA chain during its synthesis which prevents detection and repair by DNA repair enzymes (chain elongation termination) [20]. The dFdCTP also inhibits the enzymes cytidine triphosphate synthetase and deoxycytidylate deaminase that are critical for DNA synthesis and repair [20]. The dFdCDP inhibits ribonucleotide reductase resulting in the depletion of deoxyribonucleotide pools necessary for DNA synthesis [20]. Active metabolites of gemcitabine can poison topoisomerase I leading to DNA break formation [31]. Gemcitabine causes DNA replication stress and subsequent activation of various DNA repair pathways by inducing DNA damage and/or nucleotide depletion in tumor cells.
In response to DNA replication stress, tumor cells activate ATR and ATM, which in turn phosphorylates their respective downstream targets, namely Chk1 and Chk2, resulting in cell cycle checkpoint activation. Chk1 is the primary mediator of cellular response to DNA-damaging chemotherapeutic drugs such as gemcitabine [4-6]. Gemcitabine-mediated DNA replication fork stalling causes the activation of Chk1 by phosphorylation at Ser317 and Ser345 [22]. Chk1 also activates molecules involved in stabilization of replication forks and spindle cell checkpoints [32,33]. Chk1, by preventing tumor cells with replication stress entering into G2/M transition, protects them from undergoing cell death by apoptosis. Checkpoints in G2 protect tumor cells with damaged DNA and replication stress entering premature and lethal mitosis resulting in apoptosis or mitotic catastrophe [8]. However, activation of Chk1 in tumor cells can also facilitate checkpoint override and illegitimate cell-cycle progresssion resulting in tumor progression with increased genomic instability [34].
Following DNA replication stress, Chk1 is essential for blocking caspase-3 mediated apoptosis, independent of p53 status [35]. Thus, Chk1 has an anti-apoptotic role in the cellular response to DNA replication stress-inducing drugs such as gemcitabine [36]. It should be noted that LY2 cells have mutant p53 [37]. Previous studies have shown that cells lacking Chk1 undergo apoptosis in a caspase-3 dependent and p53 independent manner when exposed to genotoxic drugs [35]. Moreover, inhibition of Chk1 has been shown to promote apoptosis of ultraviolet light B (UVB) damaged keratinocytes [38]. Gemcitabine has been shown to activate Chk1 in tumor cells which prevented caspase-3 mediated apoptosis leading to tumor cell resistance to gemcitabine [22]. Treatment of LY2 cells with gemcitabine induced rapid phosphorylation of Chk1, reaching a maximum at 2 hours post-treatment. Interestingly, however, gemcitabine mediated activation of Chk1 in LY2 cells failed to prevent apoptosis of LY2 cells mediated by caspase-9 and -3. Therefore, we examined whether gemcitabine treatment has any impact on total Chk1 protein level in LY2 cells. Our studies showed an unexpected finding that gemcitabine treatment of LY2 cells results in the loss of Chk1 protein in a time and dose dependent manner. Total Chk1 protein level decreased gradually in gemcitabine-treated LY2 cells after 2 hours with maximal loss noted after 50-hours of treatment. There are two potential mechanisms through which gemcitabine may attenuate the expression of Chk1 in LY2 cells. One possible mechanism is inhibition of Chk1 expression at the transcriptional level. One of the active metabolites of gemcitabine, namely dFdCTP, is known to incorporate into the RNA of tumor cells and inhibit transcription [39]. Hence, we determined the effect of gemcitabine treatment on Chk1 mRNA level in LY2 cells by quantitative RT-PCR. Although there was an initial decrease in Chk1 mRNA levels in gemcitabine-treated LY2 cells, the Chk1 mRNA level recovered in treated cells and attained the same level as untreated control cells 12 hours post treatment. This finding suggests gemcitabine effect Chk1 transcription is transient and cannot be responsible for the total depletion of the Chk1 protein. Another potential mechanism is that gemcitabine treatment has been shown to cause activation and subsequent degradation of epidermal growth factor receptor (EGFR) in human HNSCC cells via ubiquitination along the proteasome/lysosome mediated pathway [40]. In a similar fashion, pretreatment of LY2 cells with proteasome inhibitor MG132 reduced Chk1 protein depletion when exposed to gemcitabine and decreased LY2 cells sensitivity to gemcitabine. Taken together, these findings imply that gemcitabine-mediated depletion of Chk1 in LY2 cells is also mediated by phosphorylation-dependent ubiquitination along the proteosomal pathway.
Our data implies that reduction of Chk1 protein is the primary mechanism that confers greater sensitivity of LY2 cells for gemcitabine. Recent studies have shown that genomic instability coupled with aberrant activation of checkpoint pathways help cancer cells to resist intensive chemotherapy regimens [8,10,11]. Hence, there has been recent interest in targeting DNA damage response signaling pathways to enhance tumor response to cytotoxic chemotherapy and radiation [10,11].
A number of Chk1 inhibitors are being currently developed for chemotherapy sensitization [41,42]. In this study, we show that siRNA-mediated silencing of Chk1 in LY2 cells increases their sensitivity to gemcitabine. Furthermore, pretreatment of LY2 cells with the Chk1 inhibitor PD 407824 markedly increases their killing with extremely low doses of gemcitabine. Our data gives further credence for the development of Chk1-targeted therapies to enhance chemotherapeutic efficacy in head and neck cancers.
Consistent with our in vitro results, we found gemcitabine to have significant anti-tumor activity in an orthotopic and syngeneic HNSCC animal model established using the LY2 cells [23]. Biweekly IP injection of gemcitabine in a dose (100 mg/Kg body weight) equivalent to that used in humans into tumor bearing female Balb/c mice inhibited tumor growth significantly (p < 0.001) compared to control mice treated with saline. Immunofluorescence studies revealed that the Chk1 protein was highly expressed in control tumor tissue but was absent in gemcitabine treated tumor tissue.
Previous studies have shown that Chk1 protects cells arrested in S-phase from undergoing apoptosis during replication stress [35]. Furthermore, siRNA mediated silencing of Chk1 causes cells arrested in S-phase by various replication inhibitors to undergo apoptosis [35]. In addition, we observed that siRNA-mediated silencing of Chk1 markedly increases tumor cell sensitivity to gemcitabine. Based on the published reports and our data, we conclude that gemcitabine inhibits murine squamous cell carcinoma cells growth both in vitro and in vivo by complex mechanisms that appear to involve DNA replication stress, S-phase check point termination and subsequent apoptosis. There is increasing interest in targeting molecules involved in DNA repair such as Chk1 to reverse chemo and radiation therapy resistance of solid malignancies. Findings from this study provide an important rationale for integrating gemcitabine with currently used chemotherapeutic drugs for the treatment of HNSCC to enhance their therapeutic index.
5. Acknowledgements
We thank Dr. Carter Van Waes, M.D., Ph.D. (Chief Head and Neck Surgery Branch) for providing the PAM-LY2 cell line. This study is supported by NIH/NIDCR grant R21DE019956 (N. Vigneswaran).
REFERENCES
- A. Aguilera and B. Gomez-Gonzalez, “Genome Instability: A Mechanistic View of Its Causes and Consequences,” Nature Reviews Genetics, Vol. 9, No. 5, 2008, pp. 204-217. doi:10.1038/nrg2268
- J. A. Pietenpol and Z. A. Stewart, “Cell Cycle Checkpoint Signaling: Cell Cycle Arrest versus Apoptosis,” Toxicology, Vol. 181, 2002, pp. 475-481. doi:10.1016/S0300-483X(02)00460-2
- S. Jalal, J. N. Earley and J. J. Turchi, “DNA Repair: From Genome Maintenance to Biomarker and Therapeutic Target,” Clinical Cancer Research, Vol. 17, No. 22, pp. 6973-6984. doi:10.1158/1078-0432.CCR-11-0761
- R. T. Abraham, “Cell Cycle Checkpoint Signaling through the ATM and ATR Kinases,” Genes & Development, Vol. 15, No. 17, 2001, pp. 2177-2196. doi:10.1101/gad.914401
- D. Branzei and M. Foiani, “Regulation of DNA Repair throughout the Cell Cycle,” Nature Reviews Molecular Cell Biology, Vol. 9, No. 4, 2008, pp. 297-308. doi:10.1038/nrm2351
- B. B. Zhou and S. J. Elledge, “The DNA Damage Response: Putting Checkpoints in Perspective,” Nature, Vol. 408, No. 6811, 2000, pp. 433-439. doi:10.1038/35044005
- H. Zhao and H. Piwnica-Worms, “ATR-Mediated Checkpoint Pathways Regulate Phosphorylation and Activation of Human Chk1,” Molecular and Cellular Biology, Vol. 21, No. 13, 2001, pp. 4129-4139. doi:10.1128/MCB.21.13.4129-4139.2001
- S. Lapenna and A. Giordano, “Cell Cycle Kinases as Therapeutic Targets for Cancer,” Nature Reviews Drug Discovery, Vol. 8, No. 7, 2009, pp. 547-566. doi:10.1038/nrd2907
- B. B. Zhou and J. Bartek, “Targeting the Checkpoint Kinases: Chemosensitization versus Chemoprotection,” Nature Reviews Cancer, Vol. 4, No. 3, 2004, pp. 216-225. doi:10.1038/nrc1296
- N. Bucher and C. D. Britten, “G2 Checkpoint Abrogation and Checkpoint Kinase-1 Targeting in the Treatment of Cancer,” British Journal of Cancer, Vol. 98, No. 3, 2008, pp. 523-528. doi:10.1038/sj.bjc.6604208
- C. Merry, K. Fu, J. Wang, I. J. Yeh and Y. Zhang, “Targeting the Checkpoint Kinase Chk1 in Cancer Therapy,” Cell Cycle, Vol. 9, No. 2, 2010, pp. 279-283. doi:10.4161/cc.9.2.10445
- A. M. Bergman, V. W. Ruiz van Haperen, G. Veerman, C. M. Kuiper and G. J. Peters, “Synergistic Interaction between Cisplatin and Gemcitabine in Ovarian and Colon Cancer Cell Lines,” Advances in Experimental Medicine and Biology, Vol. 370, 1994, pp. 139-143.
- P. Huang and W. Plunkett, “Induction of Apoptosis by Gemcitabine,” Seminars in Oncology, Vol. 22, No. 4, Suppl. 11, 1995, pp. 19-25.
- M. A. Morgan, L. A. Parsels, J. Maybaum and T. S. Lawrence, “Improving Gemcitabine-Mediated Radiosensitization Using Molecularly Targeted Therapy: A Review,” Clinical Cancer Research, Vol. 14, No. 21, 2008, pp. 6744-6750. doi:10.1158/1078-0432.CCR-08-1032
- J. D. Raguse, H. J. Gath, J. Bier, H. Riess and H. Oettle, “Gemcitabine in the Treatment of Advanced Head and Neck Cancer,” Clinical Oncology, Vol. 17, No. 6, 2005, pp. 425-429. doi:10.1016/j.clon.2005.05.006
- P. M. Specenier, J. Weyler, C. Van Laer, D. Van den Weyngaert, J. Van den Brande, M. T. Huizing, et al., “A Non-Randomized Comparison of Gemcitabine-Based Chemoradiation with or without Induction Chemotherapy for Locally Advanced Squamous Cell Carcinoma of the Head and Neck,” BMC Cancer, Vol. 9, 2009, p. 273. doi:10.1186/1471-2407-9-273
- L. Milas, T. Fujii, N. Hunter, M. Elshaikh, K. Mason, W. Plunkett, et al., “Enhancement of Tumor Radioresponse in Vivo by Gemcitabine,” Cancer Research, Vol. 59, No. 1, 1999, pp. 107-114.
- B. Pauwels, J. B. Vermorken, A. Wouters, J. Ides, S. Van Laere, H. A. Lambrechts, et al., “The Role of Apoptotic Cell Death in the Radiosensitising Effect of Gemcitabine,” British Journal of Cancer, Vol. 101, No. 4, 2009, pp. 628-636. doi:10.1038/sj.bjc.6605145
- H. Ueno, K. Kiyosawa and N. Kaniwa, “Pharmacogenomics of Gemcitabine: Can Genetic Studies Lead to Tailor-Made Therapy?” British Journal of Cancer, Vol. 97, No. 2, 2007, pp. 145-151. doi:10.1038/sj.bjc.6603860
- E. Mini, S. Nobili, B. Caciagli, I. Landini and T. Mazzei, “Cellular Pharmacology of Gemcitabine,” Annals of Oncology, Vol. 17, Suppl. 5, 2006, pp. 7-12. doi:10.1093/annonc/mdj941
- C. Nabhan, D. Gajria, N. L. Krett, V. Gandhi, K. Ghias and S. T. Rosen, “Caspase Activation Is Required for Gemcitabine Activity in Multiple Myeloma Cell Lines,” Molecular Cancer Therapeutics, Vol. 1, No. 13, 2002, pp. 1221-1227.
- L. M. Karnitz, K. S. Flatten, J. M. Wagner, D. Loegering, J. S. Hackbarth, S. J. Arlander, et al., “Gemcitabine Induced Activation of Checkpoint Signaling Pathways that Affect Tumor Cell Survival,” Molecular Pharmacology, Vol. 68, No. 6, 2005, pp. 1636-1644.
- N. Vigneswaran, J. Wu, A. Song, A. Annapragada and W. Zacharias, “Hypoxia-Induced Autophagic Response Is Associated with Aggressive Phenotype and Elevated Incidence of Metastasis in Orthotopic Immunocompetent Murine Models of Head and Neck Squamous Cell Carcinomas (HNSCC),” Experimental and Molecular Pathology, Vol. 90, No. 2, 2011, pp. 215-225. doi:10.1016/j.yexmp.2010.11.011
- Z. Chen, C. W. Smith, D. Kiel and C. Van Waes, “Metastatic Variants Derived Following in Vivo Tumor Progression of an in Vitro Transformed Squamous Cell Carcinoma Line Acquire a Differential Growth Advantage Requiring Tumor-Host Interaction,” Clinical and Experimental Metastasis, Vol. 15, No. 5, 1997, pp. 527-537. doi:10.1023/A:1018474910432
- P. Huang, S. Chubb, L. W. Hertel, G. B. Grindey and W. Plunkett, “Action of 2’,2’-Difluorodeoxycytidine on DNA Synthesis,” Cancer Research, Vol. 51, No. 22, 1991, pp. 6110-6117.
- M. Hochstrasser, “Ubiquitin, Proteasomes, and the Regulation of Intracellular Protein Degradation,” Current Opinion in Cell Biology, Vol. 7, No. 2, 1995, pp. 215-223. doi:10.1016/0955-0674(95)80031-X
- K. L. Rock, C. Gramm, L. Rothstein, K. Clark, R. Stein, L. Dick, et al., “Inhibitors of the Proteasome Block the Degradation of Most Cell Proteins and the Generation of Peptides Presented on MHC Class I Molecules,” Cell, Vol. 78, No. 5, 1994, pp. 761-771. doi:10.1016/S0092-8674(94)90462-6
- J. H. Lorch, M. R. Posner, L. J. Wirth and R. I. Haddad, “Induction Chemotherapy in Locally Advanced Head and Neck Cancer: A New Standard of Care?” Hematology/ Oncology Clinics of North America, Vol. 22, No. 6, 2008, pp. 1155-1163. doi:10.1016/j.hoc.2008.08.004
- R. I. Haddad and D. M. Shin, “Recent Advances in Head and Neck Cancer,” New England Journal of Medicine, Vol. 359, No. 11, 2008, pp. 1143-1154. doi:10.1056/NEJMra0707975
- S. L. Koolen, P. O. Witteveen, R. S. Jansen, M. H. Langenberg, R. H. Kronemeijer, A. Nol, et al., “Phase I Study of Oral Gemcitabine Prodrug (LY2334737) Alone and in Combination with Erlotinib in Patients with Advanced Solid Tumors,” Clinical Cancer Research, Vol. 17, No. 18, 2011, pp. 6071-6082. doi:10.1158/1078-0432.CCR-11-0353
- P. Pourquier, C. Gioffre, G. Kohlhagen, Y. Urasaki, F. Goldwasser, L. W. Hertel, et al., “Gemcitabine (2’,2’- Difluoro-2’-deoxycytidine), an Antimetabolite That Poisons Topoisomerase I,” Clinical Cancer Research, Vol. 8, No. 8, 2002, pp. 2499-2504.
- E. Petermann, A. Maya-Mendoza, G. Zachos, D. A. Gillespie, D. A. Jackson and K. W. Caldecott, “Chk1 Requirement for High Global Rates of Replication Fork Progression during Normal Vertebrate S Phase,” Molecular and Cellular Biology, Vol. 26, No. 8, 2006, pp. 3319-3326. doi:10.1128/MCB.26.8.3319-3326.2006
- G. Zachos, E. J. Black, M. Walker, M. T. Scott, P. Vagnarelli, W. C. Earnshaw, et al., “Chk1 Is Required for Spindle Checkpoint Function,” Developmental Cell, Vol. 12, No. 2, 2007, pp. 247-260. doi:10.1016/j.devcel.2007.01.003
- A. Hoglund, L. M. Nilsson, S. V. Muralidharan, L. A. Hasvold, P. Merta, M. Rudelius, et al., “Therapeutic Implications for the Induced Levels of Chk1 in Myc-Expressing Cancer Cells,” Clinical Cancer Research, Vol. 17, No. 22, 2011, pp. 7067-7079. doi:10.1158/1078-0432.CCR-11-1198
- K. Myers, M. E. Gagou, P. Zuazua-Villar, R. Rodriguez and M. Meuth, “ATR and Chk1 Suppress a Caspase- 3-Dependent Apoptotic Response Following DNA Replication Stress,” PLoS Genetics, Vol. 5, No. 1, 2009, p. e1000324. doi:10.1371/journal.pgen.1000324
- R. Rodriguez and M. Meuth, “Chk1 and p21 Cooperate to Prevent Apoptosis during DNA Replication Fork Stress,” Molecular Biology of the Cell, Vol. 17, No. 1, 2006, pp. 402-412. doi:10.1091/mbc.E05-07-0594
- A. Loercher, T. L. Lee, J. L. Ricker, A. Howard, J. Geoghegen, Z. Chen, et al., “Nuclear Factor-{kappa}B Is an Important Modulator of the Altered Gene Expression Profile and Malignant Phenotype in Squamous Cell Carcinoma,” Cancer Research, Vol. 64, No. 18, 2004, pp. 6511-6523. doi:10.1158/0008-5472.CAN-04-0852
- T. P. Heffernan, M. Kawasumi, A. Blasina, K. Anderes, A. H. Conney and P. Nghiem, “ATR-Chk1 Pathway Inhibition Promotes Apoptosis after UV Treatment in Primary Human Keratinocytes: Potential Basis for the UV Protective Effects of Caffeine,” Journal of Investigative Dermatology, Vol. 129, No. 7, 2009, pp.1805-1815. doi:10.1038/jid.2008.435
- V. W. Ruiz van Haperen, G. Veerman, J. B. Vermorken and G. J. Peters, “2’,2’-Difluoro-Deoxycytidine (Gemcitabine) Incorporation into RNA and DNA of Tumour Cell Lines,” Biochemical Pharmacology, Vol. 46, No. 4, 1993, pp. 762-766. doi:10.1016/0006-2952(93)90566-F
- F. Y. Feng, S. Varambally, S. A. Tomlins, P. Y. Chun, C. A. Lopez, X. Li, et al., “Role of Epidermal Growth Factor Receptor Degradation in Gemcitabine-Mediated Cytotoxicity,” Oncogene, Vol. 26, No. 23, 2007, pp. 3431-3439. doi:10.1038/sj.onc.1210129
- C. C. Chen, R. D. Kennedy, S. Sidi, A. T. Look and A. D’Andrea, “CHK1 Inhibition as a Strategy for Targeting Fanconi Anemia (FA) DNA Repair Pathway Deficient Tumors,” Molecular Cancer, Vol. 8, 2009, p. 24. doi:10.1186/1476-4598-8-24
- Z. F. Tao and N. H. Lin, “Chk1 Inhibitors for Novel Cancer Treatment,” Anti-Cancer Agents in Medicinal Chemistry-Anti-Cancer Agents, Vol. 6, No. 4, 2006, pp. 377-388. doi:10.2174/187152006777698132