Advances in Bioscience and Biotechnology
Vol.4 No.3(2013), Article ID:28784,8 pages DOI:10.4236/abb.2013.43053
HCS strategy targeting dysregulation of the VHL/HIF pathway for drug discovery*
![]()
Screening and Target Validation Department, Fundación Medina, Granada, Spain
Email: #bastien.cautain@medinaandalucia.es
Received 2 January 2013; revised 12 February 2013; accepted 20 February 2013
Keywords: High Content Screening; Natural Products; HIF; Drug Discovery
ABSTRACT
One-third of top-selling drugs are derived from natural products. When only a fraction of the bioactive natural products diversity has been explored, huge opportunities still remain for discovering novel leads for the development of new drugs. Clear cell renal cell carcinoma (ccRCC) is a highly vascular tumour arising from epithelial elements. Mutations in the Von Hippel-Lindau (VHL) gene are responsible for VHL disease and arise in the majority of Renal Cell Carcinoma (RCC) as well as in other types of cancer. Renal carcinoma cell lines with naturally occurring VHL mutations (RCC4 VA) and their genetically matched wild-type VHL (RCC4 VHL) counterparts were seeded onto 96-well plates and allowed to attach overnight. Fungal extracts were tested on both cell lines. Clinically useful antitumor agents were used as positive controls and as reference points to establish the efficacy and selectivity of the new compounds. Renal cell carcinoma cell lines expressing VHL or not were treated with Carboxyfluorescein succinimidyl ester (CFSE). The day after cell inoculation, extracts were added and during the following days of incubation, fluorescence intensity was measured as a surrogate marker for cell viability. The most promising extracts selectively inhibited growth of pVHL-deficient cells but not of wild-type VHL cells. We used High Content Bio-imaging, a complete cellular imaging workflow that integrates instruments and software to acquire and analyze images, to evaluate their effect. Cell imaging can reveal effects that would be overlooked by other cell assay approaches. This target-based whole cell screen is a new strategy, which ensures cell permeability and target selectivity especially in natural product screening where natural product purification is a labour of extensive work. This approach permitted a dynamic study where fluorescence was measured without affecting cell viability and enabling a better detection of cytotoxic effects such as autophagy, senescence or late apoptosis.
1. INTRODUCTION
Natural products have largely been and still are considered an extraordinary valuable platform for the discovery of new drugs against diverse pathologies, as well as a tool for the characterization of novel therapeutic targets. Such value is partly due to their higher complexity and chemical diversity as compared to those of synthetic and combinatorial compounds [1].
Von Hippel-Lindau (VHL) syndrome is a rare genetic disorder characterized by an increased risk of developing tumors [2]. The VHL gene is ubiquitously expressed in normal tissues throughout the body; thus, loss of VHL expression is unique to tumor pathology. The high frequency of pVHL inactivation in Renal Cell Carcinoma (RCC) makes it nearly universal to the disease state, and tumor suppression following reintroduction of VHL into RCC cells underscores the crucial role of pVHL in malignancy. A molecular defect that is crucial to the malignant phenotype, unique to diseased tissue, and nearly universal to the disease state serves as an ideal target for therapeutic intervention. Pharmacologic agents that are toxic in the context of pVHL disruption should then have minimal effect on normal tissue [3]. Reintroduction of wild-type VHL into VHL-deficient clear cell Renal Cell Carcinoma (ccRCC) cells inhibited the ability of these cells to form tumors in nude mice [4].
Image acquisition using robotic fluorescent microscopy and automated image analysis, generally referred to as High Content Screening (HCS), has become an essential tool in early drug discovery programs. High content cellular imaging has increasingly met the challenges of high throughput needs and facilitates the integration of disease relevant screens at early stages of the drug discovery process [5].
Early adoption of HCS by the pharmaceutical industry to characterize preclinical drug candidates further fuelled technology development. HCS technology has now expanded throughout all the different stages of the drug development process and is today considered a mainstream technology in the pharmaceutical industry [6]. HCS strategies attempt to deal with the complexity of an intact cell, which is reflected in the wealth of phenotypic information present in HCS datasets. The digital acquisition of quantitative and qualitative information, such as number, intensity, size, morphology, texture and spatial distribution of objects, can be archived for flexible computational analysis with different variables and combinations of parameters, according to the needs of the particular drug discovery or basic research program [7].
High Content Screening complete cellular imaging workflow integrates instruments and software to acquire and analyze images, then processes, stores, and manages the experimental data. All these screening solutions offer the advantages of intuitive acquisition, automated image analysis, turnkey cellular informatics, and the seamless integration of the hardware and software components, which eliminates manual data manipulation or transfer found in other less sophisticated imaging workflows. Compared with single-measurement well-based assays, multi-parameter cell based HCS assays yield data with much higher biological information content. Whereas high throughput screening (HTS) is used as a fast primary screen to identify hits for further testing, HCS can be used to identify leads from hits. The ability to obtain quantitative data from multiple endpoints, from both individual cells and cell populations, greatly enhances the information obtained from whole-cell screens in drug discovery. This could provide new insights into cell function and mode-of-action studies that were previously labour intensive and difficult to interpret [8]. This target-based whole cell screen technology ensures that the hits have an anticancer activity as well as the up-front target selectivity.
2. MATERIALS AND METHODS
2.1. Cell Culture
Human Renal Cell Carcinoma Cell line RCC4 stably transfected with an empty vector, pcDNA3 (ECACC N-03112702; called RCC4-VA) conferring neomycin resistance, or with pcDNA3-VHL (ECACC N-0312703; called RCC4-VHL) conferring neomycin resistance and encoding the VHL tumor suppressor gene product pVHL. The original renal carcinoma cell line RCC4 is VHLdeficient. The RCC4 plus vector alone cell line serve as a negative control cell line to study the effect of pVHL expression from pcDNA3-VHL [9,10].
The human renal carcinoma cell lines RCC4-VA and RCC4-VHL were cultured at a density of 1.0 × 106 cells/ flask in BDFalconTM Tissue Culture Flasks in Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal bovine serum (Origin: Australia GIBCO), 0.01% L-Glutamine 200 mM, 100 × (GIBCO), 0.01% Penicilin Streptomycin (GIBCO), 0.001% Geneticin G418 0.5 mg/ml (GIBCO). Cell cultures were maintained in a humified incubator at 37˚C with 5% CO2 and passaged when confluent using TrypLETM Express 1 × (GIBCO). Cells were counted by means of trypan blue and hemocytometer.
2.2. Screening Process
The automated screening process was carried out following the established conditions during the piloting for this bioassay “Figure 1”.
2.3. MTT Assay
MTT (3-4,5-Dimethyl-2-thiazolyl-2,5-diphenyl-2H-tetrazolium bromide) assay has been widely used as a colorimetric approach based on the activity of mitochondrial dehydrogenase enzymes in cells. MTT assay was performed 24 hours after treatment. For this purpose, MTT solution was prepared at 5 mg/ml in PBS and then diluted at 0.5 mg/mL in MEM without phenol red. Then, 100 μl of MTT were added into each well with Multidrop. Cells were incubated for 3 hours at 37˚C with 5% CO2. After 3 hours, the MTT solution was removed and replaced with 100 μl of DMSO. The plate was further incubated for 5 minutes at room temperature and agitation. The optical density of the wells was determined using a plate reader, named Victor (Beckman Coulter), at a wavelength of 570 nm.
Extracts from actinomycetes and fungi were tested. Cells were seeded at a density of 5.0 × 104 cells/ml into 96-well microplates (BD Falcon) using a Multidrop automatic dispenser. The final volume of the cell suspension was 200 μl in each well. After incubation at 37˚C with 5% CO2 for 24 hours, medium was replaced with a final volume of 195 μl. Then 5 μl of each extract and 5 μl of controls were transferred from the mother plates to the assay plates using a robotic workstation (Biomek® FX, Beckman Coulter).
Controls are on the righthand column. There are four points of doxorubicin and four points of rotenone with an initial concentration of 10 mM and dilutions of 1/2.
2.4. Microbial Natural Products Collection
A collection of 18,320 fungal extracts from Fundación MEDINA were tested. Preparation of test samples from

Figure 1. Screening strategy. Starting from a cell culture flask of VA and another of VHL cell lines, we analyze compounds with HCS technology. Regions of Interest (ROIs) are determined with re-analysis. Masks corresponding to the nucleus and the cytoplasm are created. Selecting the intensity and the size of the ROI we can constrain data, eliminating those spots that are not interesting for the analysis.
fermentation was carried out by the addition of organic solvents in order to extract secondary metabolites. Thus, we added 800 μl of acetone (Merck) to 800 ml of each broth in the actual fermentation plate together with 40 μl of DMSO. The presence of DMSO during the evaporation of organic solvent prevents precipitation of seconddary metabolites to enhance the solubility of the components of the extract. We then stirred it in a Kühner thermostated incubator at 200 rpm for 1 hour and centrifuged under vacuum for 12 hours on a computer Genevac HT-24 for controllably evaporated acetone.
The final extracts, four equivalents of the fermentation broth, were concentrated to 200 μl with a maximum percentage of 20% DMSO in water to avoid problems of toxicity testing. A percentage above 20% DMSO usually has interference. The sample plates were prepared, lastly, in an automated pipetting station (Biomek FX, Beckman Coulter) by transferring an aliquot of 160 μl of each extract to a well of an ABgene coded plate of 800 μl in order to assess their potential antitumoral activity.
2.5. HCS Assays
Cells were seeded at a density of 5.0 × 104 cells/ml into black-wall clear-bottom 96-well microplates (Greiner) using a Multidrop automatic dispenser. The final volume of the cell suspension was 200 μl in each well. After incubation at 37˚C with 5% CO2 for 24 h, medium was replaced with a final volume of 195 μl. Then 5 μl of each compound was transferred from the mother plates to the assay plates using a robotic workstation (Biomek® FX, Beckman Coulter). Cells were incubated in the presence of the compounds and the controls.
After 48 hours treatment, the culture medium was removed; cells were washed in Annexin V buffer and stained with Annexin V and Propidium Iodide (PI; a non-permeable non-specific nucleic acid dye), adding 5 μl of Annexin V and 1 μl of Propidium Iodide to each 1 ml of PBS. In apoptotic cells, the membrane phospholipid phosphatidylserine (PS) is translocated from the inner to the outer leaflet of the plasma membrane, exposing PS to the external cellular environment. Annexin V has high affinity for PS and binds to these cells. In conjunction with PI we can identify early apoptotic cells (PI negative, Annexin V positive). If cells are Annexin V and PI positive, they are in late apoptosis or already dead. Annexin V is conjugated with Fluorescein Isothiocyanate (FITC).
In the case of CFSE analysis, cells were labeled before compound treatment. CFSE passively diffuses into cells. The acetate groups are cleaved by intracellular esterases to yield highly fluorescent carboxyfluorescein succinimidyl ester. The succinimidyl ester group reacts with intracellular amines, forming fluorescent conjugates that are well retained and can be fixed with aldehyde fixatives. It can be used for in vivo tracing. The label is inherited by daughter cells after either cell division or cell fusion and the intensity decreases. These assays were run as live cells assays allowing possibility to modify the incubation time. This method allows reading of the plate without washing ensuring a complete observation of all the cells. The robustness of a high volume screening assay is generally assessed by calculating the  -factor defined from the mean (μ) and standard deviations (δ) of the positive and negative controls in a screen
-factor defined from the mean (μ) and standard deviations (δ) of the positive and negative controls in a screen 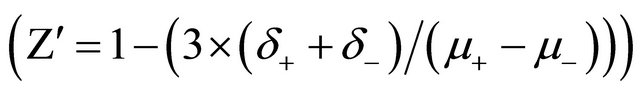 . For each plate a group of 8 wells is dedicated to positives controls (cells treated with a known active extract) and a group of 8 wells to negative control (0.1% DMSO final) randomly disposed in the plates.
. For each plate a group of 8 wells is dedicated to positives controls (cells treated with a known active extract) and a group of 8 wells to negative control (0.1% DMSO final) randomly disposed in the plates.
CellTraceTM CFSE 5 mM (Invitrogen, Life Technology Ltd. UK) was prepared by dissolving the contents of one vial in 18 μl of DMSO. After trypsinitation, the cells are resuspended in prewarmed PBS/0.1% BSA at a final concentration of 1 × 106 cells/ml. 2 μl of 5 mM stock CFSE solution per milliliter of cells were added for a final working concentration of 10 μM.
Cells are centrifuged to obtain a cell pellet and the supernatant is removed. 5 mM CFSE stock solution was diluted in phosphate-buffered saline (PBS) to a final concentration of 0.5 - 25 μM, cells were resuspended in it and incubated for 15 minutes at 37˚C. After that, the solution was centrifuged at 1600 rpm for 10 minutes and resuspended in fresh prewarmed medium. Cells were incubated overnight before compounds treatment. For each plate a group of 8 wells is dedicated to positives controls (cells treated with a known active extract) and a group of 8 wells to negative control (0.1% DMSO final) randomly disposed in the plates.
Data analysis: The BD (BD Biosciences, San Jose, CA) Pathway 855 software attovision 1.7 conjugate with Genedata Screener software were used for this purpose. After the first analysis, a re-analysis was made with Attovision 1.7 from BD (Figure 1). With re-analysis we determined the Regions of Interest (ROIs). Masks corresponding to the nucleus and the cytoplasm are created and then each mask is associated to its corresponding dye. Nucleus mask is associated with Hoechst and PI, and cytoplasm mask is associated with FITC.
The BD Pathway Bioimager data outputs are in standard text files. Data were imported into the data analysis software BD Image Data Explorer. A ROI summary text file is created with the intensity of the Regions of Interest in each well, as well as another text file with the Plate Map treatment. BD Image Data Explorer is an Excel file that combines both text files and shows if the results are appropriate for the analysis.
3. RESULTS
3.1. MTT Assay
Cells were incubated with the extracts and controls. Once the screening process over the whole collection was completed, positive activities from the primary assay were selected. Selection was made according to their general toxicity and their ability to inhibit growth of pVHL-deficient cells but not of cells with wild-type pVHL.
Compounds that prove to be positive in the screening are analyzed in detail to corroborate the hit/s (active extracts). We assessed whether the antitumor active extracts are already known, in order to dismiss them as new hits (not shown). Prior to the study of any extract, LC/MS profiling is performed using low resolution MS (positive and negative mode) against an in-house database of microbial natural products. For those extracts containing potential new metabolites, a high throughput approach is tackled, aimed at standardizing basic fractionation systems by using automated low pressure chromatography. At the same time, standard protocols are developed, depending on the physicochemical features of original samples, to obtain enriched fractions of active components. Orthogonal fractionation, considering more than one physicochemical property at a time, allows us to obtain arrays of biomolecules sorted out by size and polarity, and generating plates that represent each active compound which, once rescreened (using the same high throughput screening tests), delimits, to a large extent, the different components of the original mixture, allowing a simpler purification process and less consumption of resources (not shown).
A large number of extracts, 20,320 extracts from actinomycetes and fungi specimens were tested “Figure 2”. The assay results were analyzed with Data Management and Genedata® Screener obtaining percent activity in each well of the plate.
3.2. High Content Screening (HCS Assay)
3.2.1. Carboxyfluorescein Succinimidyl Ester (CFSE) Label
We have compared RCC4-VA and RCC4-VHL cell lines after treatment with fraction positively selected by MTT results in the presence of CFSE. CFSE passively diffuses into cells where the acetate groups are cleaved by intracellular esterases to yield highly fluorescent carboxyfluorescein succinimidyl ester.
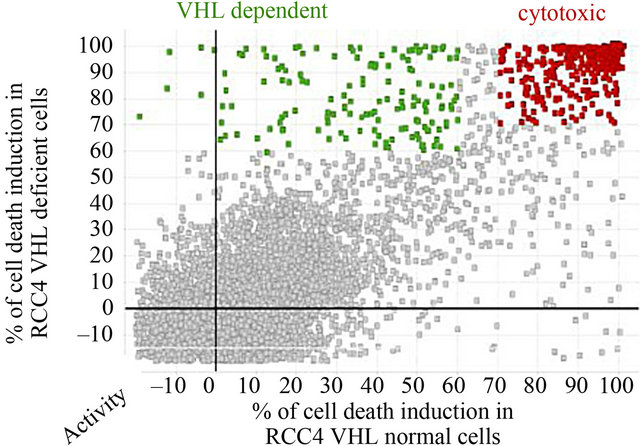
Figure 2. Cell death induction in both cell lines. We tested 20,320 extracts. Some of them are cytotoxic, killing both cell lines. The X axis represents activity percent of the extract in the RCC4-VHL cell line. The Y axis represents activity percent of the extract in the RCC4-VA cell line. Activity grows to the right on the X axis and upwards on the Y axis. The interesting extracts are the green ones, which keep VHL cells alive and kill VA cells which are VHL deficient. An extract on the lower lefthand area of the graph kills no cell lines, while those extracts located on the upper righthand area of the graph are cytotoxic, killing both cell lines. When the activity is negative, it means that the extract stimulates the growth of the cell line.
The succinimidyl ester group reacts with intracellular amines, forming fluorescent conjugates that are well retained and can be fixed with aldehyde fixatives. The label is inherited by daughter cells after cell division and the intensity decreases linearly loosing half of the label intensity in each division.
In case of normal cell division there is a moderate decrease in the intensity due to slow release of CFSE; when the metabolism is stationary, there is no cell growth and the intensity of CFSE does not change with respect to the moment of incubation, whereas a high decrease of intensity reflects an induction of cell death (Figure 3). The fluorescence was followed during several days. We focused our study on the CFSE intensity differences between both cell lines. Assessment of assay quality resulted in a  -factor value of 0.75 and 0.69 for RCC4- VA and RCC4-VHL respectively. Since a satisfactory
-factor value of 0.75 and 0.69 for RCC4- VA and RCC4-VHL respectively. Since a satisfactory  -factor is above 0.5, the calculated
-factor is above 0.5, the calculated 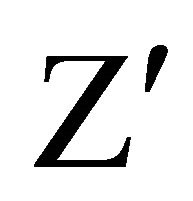 -factors levels indicated that our assay showed sufficient robustness for its development.
-factors levels indicated that our assay showed sufficient robustness for its development.
This approach allowed us to identify several extracts that act on VHL/VA cell growth in a different way “Figure 3”. RCC4-VHL cell line degrades HIF, unlike RCC4-VA cell line in which accumulates HIF, therefore extract acts in RCC4-VA, focusing it action in VHL/HIF pathway. We focused our attention on a particular extract that induces cell death preferentially in VA cells. Then, we studied the apoptosis induction in both cell lines after treatment by Annexin V staining.

Figure 3. CFSE label. Comparison between RCC4 VA and RCC4 VHL cell death. a) Cells are labeled with CFSE. Left, RCC4 VA; right, RCC4 VHL; b) Different possibilities of interest. Left, RCC4 VA; right, RCC4 VHL. After treatment, RCC4-VA cells dye (few damaged cells and low intensity) or cellular metabolism is stopped (no cell growth and the same intensity as the labeled day). RCC4-VHL cells, after 24 hours, have grown and consequently the intensity has decreased.
After treatment, RCC4-VA cells were few damaged cells and therefore have low intensity, or their cellular metabolism was stopped, there is no cell growth and the same intensity as the labeled day. RCC4-VHL cells, after 24 hours, have grown and consequently the intensity has decreased.
3.2.2. Annexin V, PI Assay
After 48 hours’ treatment (single point) with the active fraction, cells were stained with Annexin V and Propidium Iodide. The plate was analyzed with BD Biosciences PathwayTM 855 Bioimager and observing that early and late apoptosis were higher in RCC4 VA than in RCC4 VHL. Images were acquired in the FITC and PI channels of each well using a statement 20X/0.75NA air objective lens. Plates were exposed in 0.33 ms (Gain 0) to acquire FITC images and 0.5 ms (Gain 0) for PI.
Images show that the active fraction acts differently in RCC4-VHL and RCC4-VA. Analysis of dye intensity in each cell allows obtaining two graphs, each one corresponding to one dye “Figure 4”. FITC intensity, which is Annexin V intensity, is higher in RCC4-VA than in RCC4-VHL; this means that there is more apoptotic induction in cells without pVHL. In PI chart, RCC4-VHL and RCC4-VA are represented in the same way; again PI intensity is higher in RCC4-VA than in RCC4-VHL; so most RCC4-VA cell lines are in late apoptotic state. These results show that the selected fraction induces apoptosis in a more efficient way in RCC4-VA than in

Figure 4. HCS AnnexinV PI staining. Left, RCC4-VHL; right, RCC4-VA Merge: FITC Annexin V, green; PI, red. Active fraction causes more damage to RCC4-VA cells line than to VHL cell line, with an increase in apoptosis (left) and death (right). On the graphs, the X axis represents the wells and the Y axis represents the intensity. On the X axis, left, RCC4-VHL; right, RCC4-VA, FITC intensity (first graph), and PI (second graph) show a difference between RCC4-VA and RCC4-VHL cell lines.
RCC4-VHL (Figure 4). These results show a difference in the growth of VA and VHL, suggesting that this extract can be used to study further the metabolic pathway. Results show that the active fraction acts for almost 48 hours.
4. DISCUSSION
The molecular basis of human cancers has been illuminated greatly by the identification of familial cancer syndrome genes [7]; one of these is VHL gene. To study this gene, we focused our study on RCC4 cell line. The high frequency of VHL inactivation in RCC makes it nearly universal to the disease state and tumor suppression following reintroduction of VHL into RCC cells underscores the crucial role of VHL in malignancy. A molecular defect that is crucial to the malignant phenotype, unique to diseased tissue, and nearly universal to the diseased state serves as an ideal target for therapeutic intervention. Pharmacologic agents that are toxic in the context of VHL disruption should then have minimal effect on normal tissue [9].
VHL status should weigh heavily on the rational design of therapies to combat late-stage clear cell disease. The strategy was thus to validate the concept of specifically targeting the loss of the VHL tumor suppressor gene though pharmacologic means [11-14].
Genetic studies of VHL disease led to the identification and cloning of the VHL gene in 1993. Tumor formation in VHL disease follows the Knudson two-hit model, whereby a defective copy of the VHL gene is inherited, whereas somatic inactivation occurs in the remaining allele, leading to malignancy. Clear cell renal cell carcinoma (CC-RCC) is a prominent feature in hereditary VHL disease, suggesting an etiologic role of the VHL gene in sporadic CC-RCC. Gnarra et al. [15] found that VHL is mutated in 57% of CC-RCC. An additional 10% to 20% of cases are due to inactivation of the VHL gene through hypermethylation. Thus, loss of pVHL function occurs in a significant fraction of sporadic CC-RCC, approaching 70% to 80% of all cases [16], suggesting that genetic abnormalities involved in inherited RCC syndromes (and subsequent alterations in downstream intracellular signaling cascades) may also play a central role in sporadic RCC [17]. The strong association of pVHL inactivation in sporadic and hereditary CC-RCC indicates a causative role for the loss of VHL in the molecular pathogenesis of CC-RCC.
The aim of this article is to use metabolites of microbial origin to screen for new molecules that can specifically target cells with a defect in pVHL activity, an important oncogene involved in kidney cancer.
As a matter of fact, the strategy of looking for active molecules in pVHL deficient cells has been successfully applied to the identification of a compound, STF-62247, which was selectively cytotoxic towards pVHL-deficient, cells in vitro and in vivo [18]. In the present study, while we used the same cell type, the strategy has been considerably modified because, for the first time, optimized labeling with CFSE staining allows for a successful outvome. This new strategy allowed us to follow the cell response on compound treatments without additional genetic manipulation of the cells. Furthermore, the CFSE staining provides information on cell division and cell cycle that GFP labeled cells normally does not provide on its own [19].
There are new advancements and developments in the treatment of renal cell carcinoma based on mTOR pathway that regulates the expression and stability of HIFα, and on transcription of VEGF and PDGF when HIFα is not degraded. Some known compounds are: pazopanib, a multitargeted tyrosin kinase inhibitor (TKI) which targets and potently inhibits VEGF-R1-3, PDGFR-α and -β, and c-kit approved for use in metastatic RCC in October 2009; sunitinib, a receptor tyrosine kinase inhibitor, that has become the most commonly employed agent in the first-line treatment of RCC [20-23]; Bevacizumab, an anti-VEGF antibody, evaluated in patients with advanced clear cell renal carcinoma [24]. Sorafenib, a small molecule inhibitor which targets the tyrosine kinase domain of VEGFR and PDGFR, and the intracellular signaling enzyme, raf kinase [25]; it is frequently used as a second line agent in patients in combination with bevacizumab [26]. Temsirolimus and everolimus target mTOR correlate with the block in translation of mRNA encoding HIF1α [27,28]. Everolimus may be considered in patients who have failed VEGF-targeted agents [29].
The recent advancement of HCS represents the next step in the ongoing evolution of large-scale, systemslevel biological research. HCS-based strategies are a natural part of systems biology, preclinical drug testing and, perhaps, personalized medicine [7]. Through this HCS, we have generated the first results to select a group of hits from fungal extract that selectively kill cells with VHL defects, providing further validation of the concept of targeting the loss of tumor suppressor genes. A hit that shows a difference in the growth of VA and VHL was found. Although this difference is not too large, it can be useful to study how it acts and the metabolic pathway showing the target metabolite. Although the difference in the growth between both cell lines in vitro was not very significative.
The next approach will be to perform a fractionation of the crude extract guided by the biological activity and, elucidate the structure of the corresponding isolated active compound. Later steps will include a validation using western blot, Q-PCR, knock down analysis using si and sh RNA screening strategy, live-cell imaging and quantitative single-cell analysis will be developed to determine the specific target of the active compound. This approach will allow investigating the metabolic target, and ruling out any interference with other metabolic pathways. If it targets in a key metabolite of the molecular pathway, it could be an interesting compound which, might be developed to be used in combination with other therapies like chemotherapy or radiotherapy, and focused on eradicating cells without pVHL or possibly extend or improve the quality of the patient’s life.
The results of this work are the first ones to present indentification of potential lead compounds from natural products libraries with a lethal interaction in the absence of the von Hippel-Lindau tumor suppressor gene).
Moreover, the bioactive compounds identified could possible provide very useful tools for investigating in depth the function of their targets. The achievements of this work contribute to advance our understanding of cancer metabolism regulation in context of this cell model, thereby increasing our basic biological knowledge on VHL dependent cancer and further developing the feasibility of extrapolating data to other types of cancer. The choice of RCC VHL deficient cells as a model system is appropriate because it harbors regulatory mechanisms characteristic of cancer cell metabolism and offers ease of use in handling as well as a well-characterized genome and physiology.
This project demonstrated the feasibility of targeting the loss of the VHL tumor suppressor gene for the development of new targeted therapies from microbial natural products. This is of importance from a clinical perspective, not only to better understand oncological processes dependent on pVHL but also to evaluate these factors for their possible value as targets for therapeutic intervention. Moreover, the results herein obtained could have a great impact on society as they could represent an effective therapeutic option not only for cancer patients with VHL mutations but for all patients with cancer that present a change in cell metabolism.
5. ACKNOWLEDGEMENTS
The authors acknowledge and thank Dr Jun Wang for helpful discussion. This work was funded by the Fundación MEDINA, a publicprivate partnership of Merck Sharp & Dohme de España S.A./Universidad de Granada/Junta de Andalucía. Medina Foundation has received the following grants from the Spanish Ministry of Science and Innovation through its INNPLANTA 2010 program from the National Plan for Scientific Research, Technological Development and Innovation 2008- 2011 and the European Regional Development Fund (ERDF): Project PCT-010000-2010-003 “Strategic Equipment for High Content Screening for use in Oncology and Neurology”.
![]()
![]()
REFERENCES
- Carlson, E.E. (2010) Natural products as chemical probes. ACS Chemical Biology, 5, 639-653. doi:10.1021/cb100105c
- Hu Lonser, R.R., Glenn, G.M., Walther, M., Chew, E.Y., Libutti, S.K., Linehan, W.M., et al. (2003) Von HippelLinda disease. Lancet, 361, 2059-2067. doi:10.1016/S0140-6736(03)13643-4
- Iliopoulos, O., Levy, A.P., Jiang, C., Kaelin Jr., W.G. and Goldberg, M.A. (1996) Negative regulation of hypoxia inducible genes by the von Hippel-Lindau protein. Proceedings of the National Academy of Science of the United States of America, 93, 10595-10599. doi:10.1073/pnas.93.20.10595
- Iliopoulos, O., Kibel, A., Gray, S. and Kaelin Jr., W.G., (1995) Tumour suppression by the human von HippelLindau gene product. Nature Medicine, 1, 822-826. doi:10.1038/nm0895-822
- Zanella, F., Rosado, A., Blanco, F., Henderson, B.R., Carnero, A. and Link, W. (2007) An HTS approach to screen for antagonists of the nuclear export machinery using high content cell-based assays. ASSAY and Drug Development Technologies, 5, 333-341. doi:10.1089/adt.2007.058
- Haney, S.A., LaPan, P., Pan, J. and Zhang, J. (2006) High-content screening moves to the front of the line. Drug Discovery Today, 11, 889-894. doi:10.1016/j.drudis.2006.08.015
- Zanella, F., Lorens, J.B. and Link, W. (2010) High content screening: Seeing is believing. Trends in Biotechnology, 28, 237-245. doi:10.1016/j.tibtech.2010.02.005
- Liptrot, C. (2001) High content screening—From cells to data to knowledge. Drug Discovery Today, 6, 832-834.
- Sutphin, P.D., Chan, D.A., Li, J.M., Turcotte, S., Krieg, A.J. and Giaccia, A.J. (2007) Targeting the loss of the von Hippel-Lindau tumor suppressor gene in renal cell carcinoma cells. Cancer Research, 67, 5896-5905. doi:10.1158/0008-5472.CAN-07-0604
- Turcotte, S., Sutphin, P.D. and Giaccia, A.J. (2008) Targeted therapy for the loss of von Hippel-Lindau in renal cell carcinoma: A novel molecule that induces autophagic cell death. Autophagy, 4, 944-946.
- Dolma, S., Lessnick, S.L., Hahn, W.C. and Stockwell, B.R. (2003) Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell, 3, 285-296. doi:10.1016/S1535-6108(03)00050-3
- Giaccia, A., Siim, B.G. and Johnson, R.S. (2003) HIF-1 as a target for drug development. Nature Reviews Drug Discovery, 2, 803-811. doi:10.1038/nrd1199
- Kaelin Jr., W.G. (2005) The concept of synthetic lethality in the context of anticancer therapy. Nature Reviews Cancer, 5, 689-698. doi:10.1038/nrc1691
- Sutphin, P.D., Chan, D.A. and Giaccia, A.J. (2004) Dead cells don’t form tumors: HIF-dependent cytotoxins. Cell Cycle, 3, 160-163. doi:10.4161/cc.3.2.617
- Gnarra, J.R., Lerman, M.I., Zbar, B. and Linehan, W.M. (1995) Genetics of renal-cell carcinoma and evidence for a critical role for von Hippel-Lindau in renal tumorigenesis. Seminars in Oncology, 22, 3-8.
- Gossage, L. and Eisen, T. (2010) Alterations in VHL as potential biomarkers in renal-cell carcinoma. Nature Reviews Clinical Oncology, 7, 277-288. doi:10.1038/nrclinonc.2010.42
- Milella, M. and Felici, A. (2011) Biology of metastatic renal cell carcinoma. Journal of Cancer, 2, 369-373. doi:10.7150/jca.2.369
- Turcotte, S., Chan, D.A., Sutphin, P.D., Hay, M.P., Denny, W.A. and Giaccia, A.J. (2008) A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell, 14, 90-102. doi:10.1016/j.ccr.2008.06.004
- Robinson, J.P. (1998) Current protocols in cytometry. John Wiley and Sons, Inc., New York, 9.11.1-9.11.9.
- Cowey, C.L., Sonpavde, G. and Hutson, T.E. (2010) New advancements and developments in treatment of renal cell carcinoma: Focus on pazopanib. Journal of OncoTargets and Therapy, 3, 147-155. doi:10.2147/OTT.S12480
- Xu, C.F., Reck, B.H., Xue, Z., Huang, L., Baker, K.L., Chen, M., et al. (2010) Pazopanib-induced hyperbilirubinemia is associated with Gilbert’s syndrome UGT1A1 polymorphism. British Journal of Cancer, 102, 1371- 1377. doi:10.1038/sj.bjc.6605653
- Linehan, W.M., Bratslavsky, G., Pinto, P.A., Schmidt, L.S., Neckers, L., Bottaro, D.P., et al. (2010) Molecular diagnosis and therapy of kidney cancer. Annual Review of Medicine, 61, 329-343. doi:10.1146/annurev.med.042808.171650
- Motzer, R.J., Hutson, T.E., Tomczak, P., Michaelson, M.D., Bukowski, R.M., Oudard, S., et al. (2009) Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. Journal of Clinical Oncology, 27, 3584-3590. doi:10.1200/JCO.2008.20.1293
- Yang, J.C., Haworth, L., Sherry, R.M., Hwu, P., Schwartzentruber, D.J., Topalian, S.L., et al. (2003) A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. New England Journal of Medicine, 349, 427-434. doi:10.1056/NEJMoa021491
- Wilhelm, S.M., Adnane, L., Newell, P., Villanueva, A., Llovet, J.M. and Lynch, M. (2008) Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Molecular Cancer Therapeutics, 7, 3129-3140. doi:10.1158/1535-7163.MCT-08-0013
- Escudier, B., Eisen, T., Stadler, W.M., Szczylik, C., Oudard, S., Staehler, M., et al. (2009) Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. Journal of Clinical Oncology, 27, 3312-3318. doi:10.1200/JCO.2008.19.5511
- Hudson, C.C., Liu, M., Chiang, G.G., Otterness, D.M., Loomis, D.C., Kaper, F., et al. (2002) Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Molecular and Cellular Biology, 22, 7004-7014. doi:10.1128/MCB.22.20.7004-7014.2002
- Thomas, G.V., Tran, C., Mellinghoff, I.K., Welsbie, D.S., Chan, E., Fueger, B., et al. (2006) Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nature Medicine, 12, 122-127. doi:10.1038/nm1337
- Motzer, R.J., Escudier, B., Oudard, S., Hutson, T.E., Porta, C., Bracarda, S., et al. (2008) Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet, 372, 449-456. doi:10.1016/S0140-6736(08)61039-9
NOTES
*All authors are employed by MEDINA foundation (Granada, Spain).
#Corresponding author.