Case Reports in Clinical Medicine
Vol.3 No.3(2014), Article ID:43716,4 pages DOI:10.4236/crcm.2014.33037
Phenotypic Variability in a Family with Aicardi-Goutières Syndrome Due to the Common A177T RNASEH2B Mutation
Victoria Tüngler1*, Franziska Schmidt1, Steve Hieronimus1, Claudio Reyes-Velasco2, Min Ae Lee-Kirsch1
1Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
2General Practice, Ratingen, Germany
Email: *victoria.tuengler@uniklinikum-dresden.de
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 9 January 2014; revised 8 February 2014; accepted 2 March 2014
ABSTRACT
Aicardi-Goutières syndrome (AGS) is a rare inflammatory encephalopathy mimicking in utero acquired viral infection. Cardinal findings comprise leukodystrophy, basal ganglia calcifications and cerebral atrophy along with cerebrospinal fluid lymphocytosis and elevated interferon-α. In the majority of cases AGS is inherited as an autosomal recessive trait and caused by mutations in six genes including RNASEH2A, RNASEH2B, RNASEH2C, TREX1, SAMHD1 and ADAR1, all of which encode enzymes acting on nucleic acid species. Most patients present with first neurological signs in early infancy and experience severe global developmental delay. Here, we report on the unusual divergent phenotype of two siblings who both carry the most frequent AGS causing p.A177T (c.529G > A) RNASEH2B mutation in the homozygous state. While one sibling showed a typical AGS presentation with early onset and severe statomotor and mental impairment, the older sibling was intellectually completely normal. She was only diagnosed because of mild spasticity of the legs and serological signs of autoimmunity. These findings highlight the phenotypic variability of AGS and suggest that AGS may be underdiagnosed among children with mild cerebral palsy.
Keywords:Aicardi-Goutières Syndrome; RNASEH2B; Interferon-α; Autoimmunity

1. Introduction
The classical presentation of AGS consists of an inflammatory encephalopathy characterized by dystonic movements, irritability and/or seizures as well as sterile pyrexic episodes [1] . Parents of affected children typically describe inconsolable crying and insomnia [2] . Some patients present with hepatomegaly and thrombocytopenia [3] . In addition, cutaneous inflammatory lesions at acral locations, also referred to as chilblain lesions, represent a highly distinctive feature of AGS [4] . The lesions are typically cold-induced and may occur later during the course of the disease. In most cases the symptoms begin during the first months of life following a period of normal development. Neuroimaging findings include basal ganglia calcifications, white matter disease and atrophy leading to progressive microcephaly [1] . In addition, lymphocytosis and raised interferon-α levels in CSF are commonly observed during the early encephalitic phase [5] . The disease usually results in profound developmental delay.
All genes currently known to cause AGS, namely TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1 and ADAR1, encode enzymes that process nucleic acid species including DNA, RNA or DNA precursors [6] . Indeed, the elucidation of the genetic causes of AGS has provided novel insight into the role of the intracellular nucleic acid metabolism for the maintenance of immune tolerance. Recognition of foreign nucleic acids of viral origin by innate immune receptors is an integral aspect of the immune defense against pathogens. The limited capacity of these immune receptors to distinguish between endogenous and foreign nucleic acids requires appropriate mechanisms to dispose of nucleic acid metabolites arising during cell metabolism. Mutations in the AGS-causing genes are thought to result in the inability to dispose of such endogenous nucleic acids. As a consequence, these nucleic acid metabolites accumulate and initiate an interferon-α-mediated innate immune response leading to inflammation and autoimmunity [6] .
2. Case Reports
2.1. Patient 1
Patient 1 is the 4-year-old forth child of a consanguineous Moroccan couple. He was born at 31 weeks of gestation due to preterm labor following an uneventful pregnancy with a normal birth weight (50th percentile) and a normal head circumference (50th percentile) for his gestational age. Post-partum a mild intracranial hemorrhage was noticed which resolved within 2 months. His newborn screening and newborn hearing tests were unremarkable. He developed normally until the corrected age of 11 months. At this time the parents noted increased irritability, jerky movements and an abrupt regression of developmental milestones. He had previously reached full head control, learned to sit unsupported and was able to raise objects. Upon examination at that age his weight, length and head circumference were at the 10th percentile. His head circumference further declined and was at the 1st percentile at 4 years of age. Neurologically he demonstrated signs of global developmental delay, truncal hypotonia and reappearance of primitive reflexes. Magnetic resonance (MR) imaging performed at the age of 14 and 23 months, respectively, revealed ventricular enlargement due to cortical atrophy with symmetrical hyper-intensive (T2w) signals of the white matter and progressive demyelination of previously myelinated regions including the posterior limb of the capsula interna, cerebellum and subcortical regions (Figure 1). A cranial computer tomography to look for calcifications was not undertaken. A lumbar puncture revealed mild pleocytosis, raised levels of neopterine and interferon-a (6 IU/ml) in the cerebrospinal fluid (CSF). In addition, the child experienced frequent febrile episodes that led to several medical consultations. However, all laboratory findings were unremarkable and no infectious cause could be identified. Further diagnostic work-up revealed no evidence for metabolic or endocrinologic disorders. An immunofluorescence screening test (IFT) demonstrated an anti-nuclear antibody (ANA) titer of 1:160 with a dense fine speckled pattern. Genetic testing revealed homozygosity for the common AGS-associated p.A177T (c.529G > A) mutation of the RNASEH2B gene.
2.2. Patient 2
Patient 2 is the 20-year-old sister of patient 1 and the oldest sibling of the family. Patient 2 was born at term after an uncomplicated pregnancy with weight, length and head circumference within normal range. She achieved all developmental milestones adequately until the age of 21 months. At this time, following intramuscular administration of the MMR (measles, mumps, rubella) vaccine into the thigh, the parents noted a weakness of the ipsilateral leg. The reduction in muscle strength further developed into a partial spastic paralysis of both legs.
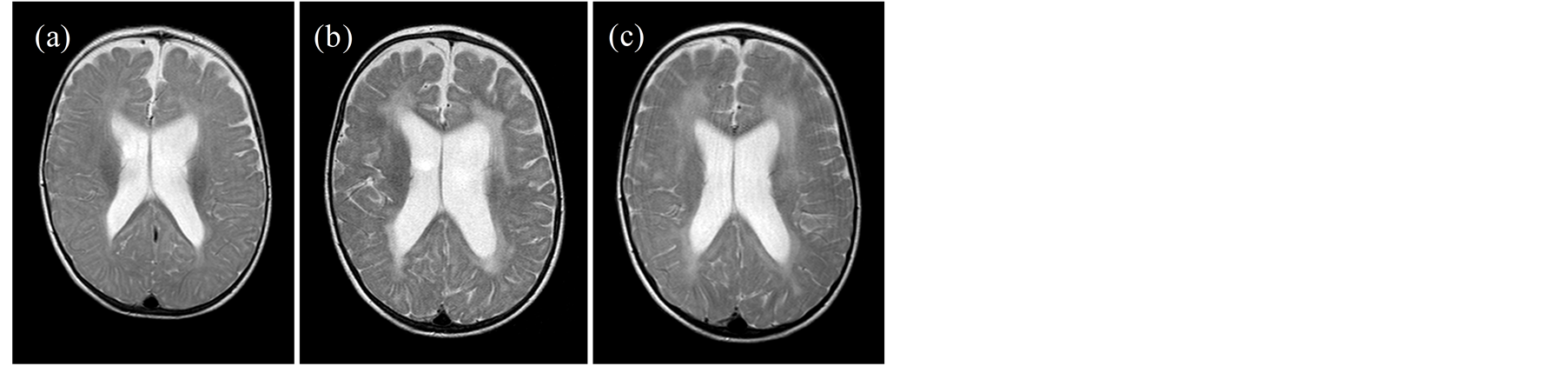
Figure 1. Bilateral periventricular hyperintensities on T2-weighted axial MR imaging at 14 months (b) and 23 months of age (c) indicating pathological white matter changes of previously physiologically myelinated areas at 9 months of age (a), particularly affecting the frontoand occipito-temporal cortex. In addition, enlargement of the ventricles is noted.
Except for a temporary delay in gross motor skills due to the spastic paraparesis, the patient developed normally. Her language skills, intellectual and cognitive abilities were within normal range. Owing to extensive physiotherapy and orthopedic surgery she gradually learned to walk again. She successfully completed secondary school and has received a driver’s license. The underlying cause of the mild spastic palsy could not be explained at that time. MR imaging of the brain and spine, somatosensory evoked potentials as well as extensive metabolic, infectious and endocrinologic laboratory studies were unremarkable. An IFT performed at 20 years of age showed an ANA titer of 1:320 with centromere pattern. Upon further serologic examination she tested positive for auto-antibodies against U1-RNP/O (32.2 U/ml, positive) and CENP-B (15 U/ml; weakly positive). Genetic testing confirmed that patient 2 was also homozygous for the same RNASEH2B mutation found in her affected brother. Her two 11- and 16-year-old sisters were healthy and did not carry the A177T RNASEH2B mutation. Both parents were heterozygous carriers.
3. Discussion
The clinical findings of patient 1 who presented with an early-onset non-infectious inflammatory encephalopathy featuring a raised interferon-α level in CSF represent the typical findings seen in patients with AGS. The fever episodes observed during the sub-acute onset of the disease have also commonly been reported in AGS patients and are thought to reflect the early inflammatory phase of the encephalopathy [2] [3] . This phase is followed by the appearance of progressive neurodegenerative signs including loss of skills, psychomotor retardation and microcephaly [1] [3] . Indeed, the diagnosis of AGS in this patient could be confirmed by genetic testing. The identified A177T mutation in the RNASEH2B gene represents the single most common mutation found in AGS patients to date and has been found both in the homozygous or compound heterozygous state across different ethnic populations suggesting that it represents a mutation hot spot rather than a founder mutation [2] [3] [7] .
Previous studies have extended the phenotypic spectrum of the disorder originally described by Aicardi and Goutières [1] . In addition to chilblain lesions, AGS patients commonly develop signs of systemic autoimmunity including hematopoietic abnormalities, antinuclear antibodies or other autoantibodies [2] [4] . Autoimmune phenomena may be present early on, but often appear later during the course of the disease. RNASEH2B encodes one of the three subunits of ribonuclease 2, which degrades the RNA moiety of RNA:DNA hybrids or removes single ribonucleotides embedded within a DNA duplex [7] . Indeed, chronic activation of the antiviral interferon axis induced by immune recognition of unmetabolized endogenous nucleic acids is thought to result in inflammation and autoimmunity [6] .
While the breadth of the clinical presentation of AGS is only emerging, a correlation of certain AGS phenotypes with mutations in distinct AGS genes has previously been recognized. Thus, a severe AGS presentation with neonatal onset or even intrauterine brain calcifications is associated with high morbidity and mortality and is more commonly seen in patients carrying TREX1 mutations [3] . In contrast, a later onset occurring after several months of normal development or beyond the first year of life is associated with a lower mortality rate and more frequently due to RNASEH2B mutations [2] [3] . While cutaneous chilblain lesions have been observed in AGS patients harboring mutations in any of the known AGS genes, arthritis or arthropathy has so far only been described in patients with SAMHD1 mutations [8] [9] .
The here reported siblings who both carry the same homozygous RNASEH2B mutation show an extraordinary intrafamilial phenotypic variability with one sibling being severely affected and the other showing only mild neurologic impairment with normal intelligence. To our knowledge there is only one report of a comparable intrafamilial variable phenotype observed in a family carrying a mutation in the RNASEH2C gene which encodes another subunit of ribonuclease H2 [10] . While the phenotypic variability may be solely genetically determined, our observations also raise the question of whether there are yet unidentified non-genetic trigger factors that could impact disease initiation and outcome. Moreover, these findings suggest that the prevalence of AGS may be much higher than previously estimated. Thus, AGS should be considered in any patient with an unexplained mild form of cerebral palsy.
Acknowledgements
We thank the family for participation in the study. We thank Peggy Binkenstein for excellent technical assistance. We thank Franz-Georg Kaup, Florence-Nightingale-Krankenhaus, Düsseldorf, for MR images. This work was supported by the Deutsche Forschungsgemeinschaft (VT 421/2-1 to V.T., LE 1074/4-1 to M.L.-K.). V.T. is recipient of a MeDDrive grant of the Medical Faculty, TU Dresden. We acknowledge support by the Deutsche Forschungsgemeinschaft and the Open Access Publication Funds of the TU Dresden.
References
- Aicardi, J. and Goutieres, F. (1984) A Progressive Familial Encephalopathy in Infancy with Calcifications of the Basal Ganglia and Chronic Cerebrospinal Fluid Lymphocytosis. Annals of Neurology, 15, 49-54. http://dx.doi.org/10.1002/ana.410150109
- Ramantani, G., Kohlhase, J., Hertzberg, C., Innes, A.M., Engel, K., Hunger, S., et al. (2010) Expanding the Phenotypic Spectrum of Lupus Erythematosus in Aicardi-Goutieres Syndrome. Arthritis & Rheumatology, 62, 1469-1477. http://dx.doi.org/10.1002/art.27367
- Rice, G., Patrick, T., Parmar, R., Taylor, C.F., Aeby, A., Aicardi, J., et al. (2007) Clinical and Molecular Phenotype of Aicardi-Goutieres Syndrome. American Journal of Human Genetics, 81, 713-725. http://dx.doi.org/10.1086/521373
- Tolmie, J.L., Shillito, P., Hughes-Benzie, R. and Stephenson, J.B. (1995) The Aicardi-Goutieres Syndrome (Familial, Early Onset Encephalopathy with Calcifications of the Basal Ganglia and Chronic Cerebrospinal Fluid Lymphocytosis). Journal of Medical Genetics, 32, 881-884. http://dx.doi.org/10.1136/jmg.32.11.881
- Lebon, P., Badoual, J., Ponsot, G., Goutieres, F., Hemeury-Cukier, F. and Aicardi, J. (1988) Intrathecal Synthesis of Interferon-Alpha in Infants with Progressive Familial Encephalopathy. Journal of the Neurological Sciences, 84, 201- 208. http://dx.doi.org/10.1016/0022-510X(88)90125-6
- Lee-Kirsch, M.A., Wolf, C. and Gunther, C. (2014) Aicardi-Goutieres Syndrome: A Model Disease for Systemic Autoimmunity. Clinical Experimental Immunology, 175, 17-24. http://dx.doi.org/10.1111/cei.12160
- Crow, Y.J., Leitch, A., Hayward, B.E., Garner, A., Parmar, R., Griffith, E., et al. (2006) Mutations in Genes Encoding Ribonuclease H2 Subunits Cause Aicardi-Goutieres Syndrome and Mimic Congenital Viral Brain Infection. Nature Genetics, 38, 910-916. http://dx.doi.org/10.1038/ng1842
- Dale, R.C., Gornall, H., Singh-Grewal, D., Alcausin, M., Rice, G.I. and Crow, Y.J. (2010) Familial Aicardi-Goutieres Syndrome Due to SAMHD1 Mutations Is Associated with Chronic Arthropathy and Contractures. American Journal of Medical Genetics A, 152A, 938-942. http://dx.doi.org/10.1002/ajmg.a.33359
- Ramantani, G., Hausler, M., Niggemann, P., Wessling, B., Guttmann, H., Mull, M., et al. (2011) Aicardi-Goutieres Syndrome and Systemic Lupus Erythematosus (SLE) in a 12-Year-Old Boy with SAMHD1 Mutations. Journal of Child Neurology, 26, 1425-1428. http://dx.doi.org/10.1177/0883073811408310
NOTES

*Corresponding author.