Open Journal of Blood Diseases
Vol.3 No.1(2013), Article ID:29319,5 pages DOI:10.4236/ojbd.2013.31012
Does DKC1 Mutation Suffice to Define the Phenotype Severity of Hoyeraal-Hreidarsson Syndrome?*
![]()
1Division of Pediatric Oncology, Department of Pediatrics, Bairro Monte Alegre, Ribeirão Preto, Brazil; 2Faculty of Medicine of Ribeirão Preto, University of São Paulo, Ribeirão Preto, Brazil; 3Department of Internal Medicine, Faculty of Medicine of Ribeirão Preto, University of São Paulo, Ribeirão Preto, Brazil; 4Centre for Paediatrics, Blizard Institute, Barts and The London School of Medicine and Dentistry, London, UK.
Email: †elvisvalera@hotmail.com
Received December 13th, 2012; revised January 15th, 2013; accepted February 1st, 2013
Keywords: DKC1 Mutation; Dyskeratosis Congenital; Hoyeraal-Hreidarsson Syndrome; Bone Marrow Failure
ABSTRACT
Both dyskeratosis congenita (DC) and Hoyeraal-Hreidarsson Syndrome (HHS) are rare inherited bone marrow failure conditions. HHS is considered to be a variant of DC in which neurological deficits and immunodeficiencies are also present. We describe a very interesting familial cluster where an invariant point mutation of DKC1 located in the exon 11 is observed in the carrier mother and in two decedent males. The older child developed the classical phenotype of HHS at a very early age. The second affected child remains poorly symptomatic, with only mild haematological changes. Telomere shortening, with different severity, is also present in both cases. This paper discusses the clinical spectrum of inherited BM failure syndromes from the perspective different clinical presentation within a family with a DKC1 mutation.
1. Introduction
Dyskeratosis congenita (DC) is a rare genetic disorder characterized by progressive bone marrow (BM) failure, associated with nail dystrophy, skin pigmentation and mucosal leukoplakia. Several different genetic forms of inheritance have been described, which include autosomal recessive, autosomal dominant and X-linked traits [1,2]. The Hoyeraal-Hreidarsson syndrome (HHS) is considered to be a much more severe variant of DC that displays clinical features overlapping DC, with additional T, B and NK immunodeficiency, neurological developmental defects (microcephaly, cerebellar hypoplasia), gastrointestinal disorders and intrauterine growth retardation [3]. HHS affected children are at high risk of death at an early age.
Both genetic disorders are considered telomere-associated diseases, with deficiency in maintaining telomere integrity. These patients frequently display variable degrees of telomere dysfunction, either in telomere length or, as more recently determined, in telomere structure [4]. Mutations in the DKC1 gene are responsible for the X-linked form of the disease, while TERC, TERT and TINF2 mutations cause autosomal dominant forms [5,6]. Interestingly, these genes encode components of either the telomerase complex or the shelterin complex. Moreover, DKC1 gene, mapped on Xq28, is responsible for encoding the protein diskerin. This protein has a dual role as a pseudouridine synthase [4] and ultimately affects telomerase complex, largely with reduced telomerase activity.
In the present article we describe a family with a DKC1 mutation in which the two affected boys display extremely diverse phenotypes, ranging from the classical HHS with early death related to infectious and BM complications to a nearly asymptomatic male screened for the mutation simply because his brother had been diagnosed with this X-linked disease. Possible hypotheses to explain these differences in clinical presentation are discussed and reviewed.
2. Materials and Methods
2.1. Familial X-Linked DKC1 Mutation Report
A two-year-old male (Case #1) was referred for investigation because of a family history of HHS. He had no previous complaints of recurrent infections, diarrhea, or vaccine reactions. His weight was in percentile 3, and his height was in percentile 10. At physical exam, the patient had nail dystrophy and leukoplakia of the tongue. His neurological development was normal, and a magnetic resonance imaging (MRI) of the brain showed only a mild enlargement of magnum cistern, without cerebellar hypoplasia. There were minor changes in blood counts (lymphopenia and mild thrombocytopenia) and the family refused a bone marrow aspirate and biopsy. Endoscopy showed an oesophageal stenosis, which was attributed to HHS. Immunological evaluation showed normal levels of immunoglobulins and IgG subclasses. Preand post-immunization levels of IgG to pneumococcal polysaccharides measured by ELISA were low for all tested serotypes. Natural killer cells and lymphocyte subpopulations CD4+, CD8+, CD19+, measured by flow cytometry, were low, as well CD4+/CD8+ ratio. The patient is now 5 years old and remains asymptomatic.
His older brother (Case #2) had previously been followed by our group, with a history of several recurrent and severe infections since his first month of life. This index case was very symptomatic, with evident neurological delay, transfusion dependent bone marrow failure, cellular and humoral immunodeficiency and the complete clinical phenotype of HHS. Cerebellar hypoplasia was also observed at MRI of brain. Hierogram representing the familial cluster and the main clinical and laboratorial characteristics on these two index cases are show in Figure 1. Additional clinical, immunological and genetic details on this case were previously published [7]. This patient died because of infectious complications at age of nine.
2.2. Laboratorial Studies and Genetic Tests
For mutation analysis of the DKC1 exon 11 the Polymerase Chain Reaction (PCR) was performed on genomic DNA using the following primers: forward 5’-GCACCCTCTTAGTGAATGAACC-3’ and reverse 5’-CCCAGTACCACTTACCTTTGGA-3’. In brief, the final concentration of reagents in the PCR were 0.2 mM each dNTP, 1.5 mM MgCl2, 1 U of HotStartTaq DNA Polymerase (Qiagen), 1 × PCR buffer (Qiagen), 1× QSolution (Qiagen), and 150 nM of each primer. The thermal cycling profile started with incubation at 95˚C for 15 minutes to activate the Taq polymerase, followed by 30 cycles of 94˚C for 45 seconds, 56˚C for 45 seconds and 72˚C for 45 seconds. Approximately 200 ng PCR products were sequenced with 100 nM each of forward and reverse primer using the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems). We performed an initial denaturation at 96˚C for 2 minutes, and 35 cycles of 96˚C for 45 seconds, 50˚C for 30 seconds and 60˚C for 4 minutes. Sequence analysis was performed on an ABI 3130 Prism Sequence detection system (Applied Biosystems). Sequences were identified using the sequencing analysis tools from the CLC Main Workbench
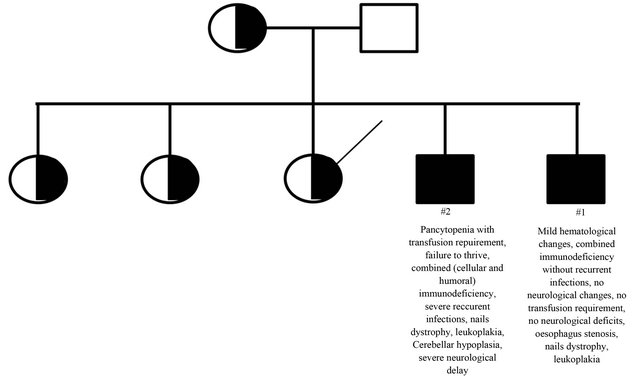
Figure 1. Hierogram representing the familial cluster showing the main clinical differences between Cases #1 and #2.
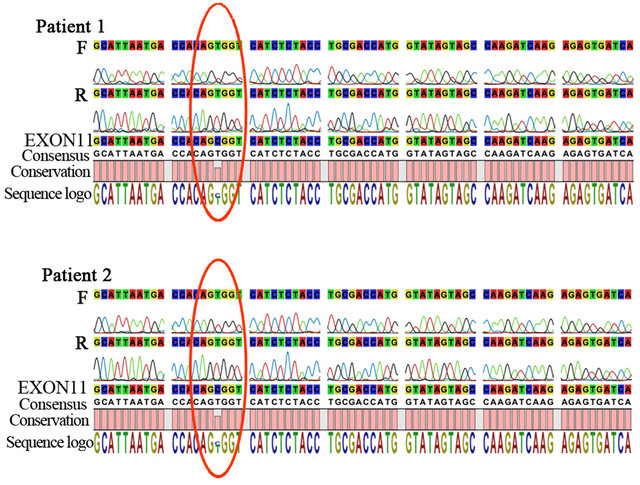
Software 5 (CLC bio). Both patients were hemizygous for a mutation in DKC1 gene, c.1058C > T, responsible for the p.Ala353Val amino acid substitution, (Figure 2(a)). Moreover, the three older sisters and their mother are heterozygous for the same point mutation in DKC1.
Chromosome preparations for cytogenetic studies were obtained from phytohemaglutinin-stimulated peripheral blood lymphocyte cultures. Metaphase cells were arrested with colcemid after 20 minutes incubation. The cells were pelleted by centrifugation, and resuspended in hypotonic medium (0.075 M KC1) for 20 min at 37˚C and fixed three times with methanol:acetic acid (3:1). The subsequent analysis by GTG-banding showed a normal male karyotype, 46, XY for Case #1.
In situ hybridization (FISH) with Cy3-conjugated PNA (peptide nucleic acid) oligonucleotide (CCCTAA)3 was performed according to Peitl et al. [8]. FISH analysis on peripheral blood metaphases showed telomere shortening in lymphocytes of both cases (Figure 2(b)). As previously reported, significant genomic instability was evident in Case #2, with pseudodiploid complements (numerical changes and structural rearrangements in the form of non-clonal deletions), chromatid breaks and a
 (a)
(a)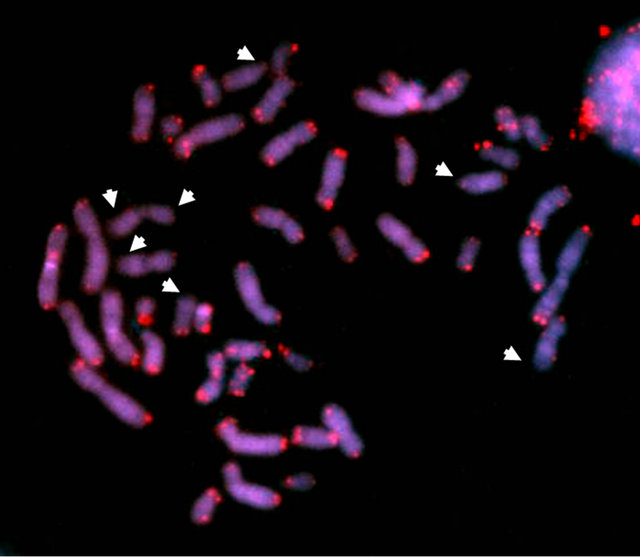 (b)
(b)
Figure 2. (a) Sequencing of DKC1 exon 11 of Patients 1 and 2 using forward (F) and reverse (R) primers; (I would show only the relevant section, not the extended trace); (b) FISH analysis of peripheral lymphocyte metaphases with Cy3- conjugated PNA probes. Note that signals corresponding to telomeres were weak for some chromosomes (arrowheads).
dicentric chromosome. Conversely, no genomic instability was observed in the less symptomatic Case #1.
3. Discussion
Recent interest in the BM failure syndromes DC and HHS has largely focused on genetics. The HoyeraalHreidarsson syndrome is a severe variant of DC and can be caused by mutations in either the X-linked gene DKC1 or the TINF2 gene, responsible for an autosomal dominant disease which usually arises de novo. Although DNA sequencing has widely facilitated the diagnosis of both DC and HHS, recent interest has been focused on the fact that patients with the same mutation can present with an extremely variable phenotype.
In the present report we describe 2 male siblings carrying an identical missense mutation of the DKC1 gene (c.1058C > T) and dissimilar clinical presentations. While the older brother developed the classical phenotype of HHS at early age with BM failure and immunodeficiency leading to premature death, the second brother remains poorly symptomatic, with immunodeficiency but without recurrent infections and only mild haematologycal changes.
Previous reports have demonstrated that the 48 DKC1 mutations described so far seem to cluster in two separate regions of the dyskerin protein [9], although no correlation between the location of the mutation and the severity of the disease has yet been established. A review of the mutation profiles among more than 200 DC families registered at the Hammersmith Hospital in London showed that the c.1058C > T mutation that leads to an A353V substitution, accounts for more than 40% of X-linked cases and there is considerable variation in clinical severity even within this sub-group [1]. This spectrum of disease manifestation therefore indicates that other environmental or genetic factors must also participate in the disease phenotype.
Remarkably, approximately one half of all DC patients remain genetically uncharacterized, a significant proportion of whom present the most severe form, HHS [1]. In such cases, efforts have been made to survey mutation candidates such as NOP10, NHP2 and GAR1 genes which are also components of the telomerase holoenzyme complex [10-13]. Although additional mutations in other telomerase subunits (NOP10, NHP2 and hTR) have been found in DC patients [14,15], they have not been seen in HHS. Also, homozygous mutations in hTERT [10,11], dominant mutations in TINF2 [16] and a splice variant in Apollo [17] have been associated with HHS demonstrating that other disease-causing mutations might also converge to the same phenotype.
Telomerase deficiency and accelerated telomere shortening are hallmarks of DC and HHS [18], and patients with the more severe form tend to have even shorter telomeres. Therefore, some authors suggest that telomere length could be treated as a surrogate marker for DC but only in the context of BM failure [1,19]. FISH using a peptide-nucleic acid (PNA) probe showed telomere shortening in peripheral lymphocytes from both our patients when compared to normal counterparts (#2 published in [7]), however, the extent of shortening was milder in asymptomatic boy. Although this is consistent with the knowledge that defective dyskerin results in impaired telomerase activity, a study performed by Lamm et al. [20] in a HHS family in which the genes implicated in other cases of DC and HHS had been excluded and normal telomerase activity was observed, showed that the telomeric 3’ overhangs were diminished in blood cells and fibroblasts derived from the patients, suggesting that the cause for telomere shortening lies not in the telomerase catalytic core but rather in its recruitment or activation. Therefore, when identical mutations lead to different manifestations it is likely that other homozygous or heterozygous genetic alterations not yet known might also contribute to HHS.
Our familial cluster exemplifies how diverse the clinical phenotype can be between relatives carrying the same DKC1 point mutation. This clinical information may be important for counseling, as affected families may be very concerned about the potential clinical outcome of their offspring affected by this serious disease. Genetic studies on a larger cohort of families with HHS may provide additional information in relation to the factors that determine disease severity.
REFERENCES
- T. J. Vulliamy, A. Marrone, S. W. Knight, et al., “Mutations in Dyskeratosis Congenita: Their Impact on Telomere Length and the Diversity of Clinical Presentation,” Blood, Vol. 107, No. 7, 2006, pp. 2680-2685. doi:10.1182/blood-2005-07-2622
- R. T. Calado and N. S.Young, “Telomere Maintenance and Human Bone Marrow Failure,” Blood, Vol. 111, No. 9, 2008, pp. 4446-4455. doi:10.1182/blood-2007-08-019729
- F. Berthet, R. Caduff, U. B. Schaad, et al., “A Syndrome of Primary Combined Immunodeficiency with Microcephaly, Cerebellar Hypoplasia, Growth Failure and Progressive Pancytopenia,” European Journal of Pediatrics, Vol. 153, No. 5, 1994, pp. 333-338. doi:10.1007/BF01956413
- I. Dokal, “Dyskeratosis Congenita,” American Society of Hematology Education Program, Vol. 2011, No. 1, 2011, pp. 480-486.
- I. Dokal and T. Vulliamy, “Inherited Bone Marrow Failure Syndromes,” Haematologica, Vol. 95, No. 8, 2010, pp. 1236-1240. doi:10.3324/haematol.2010.025619
- B. P. Alter, P. S. Rosenberg, N. Giri, et al., “Telomere Length Is Associated with Disease Severity and Declines with Age in Dyskeratosis Congenita,” Haematologica, Vol. 97, No. 3, 2012, pp. 353-359. doi:10.3324/haematol.2011.055269
- E. T. Valera, M. S. Brassesco, P. Roxo Jr., et al., “Genomic Instability in Hoyeraal-Hreidarsson Syndrome,” Pediatric Blood & Cancer, Vol. 54, No. 5, 2010, pp. 779- 780. doi:10.1002/pbc.22446
- P. Peitl, S. S. Mello, M. L. Camparoto, G. A. Passos, et al., “Chromosomal Rearrangements Involving Telomeric DNA Sequences in Balb/3T3 Cells Transfected with the Ha-ras Oncogene,” Mutagenesis, Vol. 17, No. 1, 2002, pp. 67-72. doi:10.1093/mutage/17.1.67
- Y. H. Du, M. Bessler and P. J. Mason, “Telomerase Mutations and Prematures Ageing in Humans,” In: K. L. Rudolph, Telomeres and Telomerase in Ageing, Disease and Cancer: Molecular Mechanisms of Adult Stem Cell Ageing, Springer-Verlag, Berlin, 2008, pp. 77-107. doi:10.1007/978-3-540-73709-4_5
- A. Marrone and P. J. Mason, “Dyskeratosis Congenita,” Cellular and Molecular Life Sciences, Vol. 60, 2003, pp. 507-517.
- A. Marrone, A. Walne, H. Tamary, et al., “Telomerase Reverse-Transcriptase Homozygous Mutations in Autosomal Recessive Dyskeratosis Congenita and Hoyeraal-Hreidarsson Syndrome,” Blood, Vol. 110, No. 13, 2007, pp. 4198-4205. doi:10.1182/blood-2006-12-062851
- H. Yamaguchi, R. T. Calado, H. Ly, et al., “Mutations in TERT, the Gene for Telomerase Reverse Transcriptase, in Aplastic Anemia,” New England Journal of Medicine, Vol. 352, 2005, pp. 1413-1424. doi:10.1056/NEJMoa042980
- S. A. Savage and A. A. Bertuch, “The Genetics and Clinical Manifestations of Telomere Biology Disorders,” Genetics in Medicine, Vol. 12, No. 12, 2010, pp. 753-764.
- A. J. Walne, T. Vulliamy, A. Marrone, et al., “Genetic Heterogeneity in Autosomal Recessive Dyskeratosis Congenita with One Subtype Due to Mutations in the Telomerase-Associated Protein NOP10,” Human Molecular Genetics, Vol. 16, No. 13, 2007, pp. 1619-1629. doi:10.1093/hmg/ddm111
- T. Vulliamy, R. Beswick, M. Kirwan, A. Marrone, et al., “Mutations in the Telomerase Component NHP2 Cause the Premature Ageing Syndrome Dyskeratosis Congenital,” Proceedings of the National Academy Sciences of the USA, Vol. 105, No. 23, 2008, pp. 8073-8078. doi:10.1073/pnas.0800042105
- S. A. Savage, N. Giri, G. M. Baerlocher, et al., “TINF2, a Component of the Shelterin Telomere Protection Complex, Is Mutated in Dyskeratosis Congenital,” American Journal of Human Genetics, Vol. 82, No. 2, 2008, pp. 501-509. doi:10.1016/j.ajhg.2007.10.004
- F. Touzot, I. Callebaut, J. Soulier, et al., “Function of Apollo (SNM1B) at Telomere Highlighted by a Splice Variant Identified in a Patient with Hoyeraal-Hreidarsson Syndrome,” Proceedings of the National Academy Sciences of the USA, Vol. 107, No. 22, 2010, pp. 10097- 10102. doi:10.1073/pnas.0914918107
- T. J. Vulliamy, S. W. Knight, P. J. Mason and I. Dokal, “Very Short Telomeres in the Peripheral Blood of Patients with X-Linked and Autosomal Dyskeratosis Congenital,” Blood Cells, Molecules, and Diseases, Vol. 27, No. 2, 2001, pp. 353-357. doi:10.1006/bcmd.2001.0389
- B. P. Alter, G. M. Baerlocher, S. A. Savage, et al., “Very Short Telomere Length by Flow Fluorescence in Situ Hybridization Identifies Patients with Dyskeratosis Congenital,” Blood, Vol. 110, No. 5, 2007, pp. 1439-1447. doi:10.1182/blood-2007-02-075598
- N. Lamm, E. Ordan, R. Shponkin, et al., “Diminished Telomeric 3’Overhangs Are Associated with Telomere Dysfunction in Hoyeraal-Hreidarsson Syndrome,” PLoS One, Vol. 4, No. 5, 2009, p. e5666. doi:10.1371/journal.pone.0005666
NOTES
*Financial support: FAPESP Process number 2010/15717-0.
#Valera ET and Brassesco MS contributed equally.
†Corresponding author.