Open Journal of Anesthesiology
Vol.3 No.1(2013), Article ID:27634,4 pages DOI:10.4236/ojanes.2013.31012
Subarachnoid and Peripheral Nerve Block in a Patient with Charcot-Marie-Tooth Disease
![]()
Department of Anesthesiology and Pain Medicine, University of Washington, Seattle, USA.
Email: stinr@uw.edu
Received October 3rd, 2012; revised November 8th, 2012; accepted December 11th, 2012
Keywords: Charcot-Marie-Tooth Disease; CMT; Regional Anesthesia; Neuraxial Anesthesia
ABSTRACT
Charcot-Marie-Tooth disease (CMT) is a hereditary peripheral neuropathy characterized by progressive distal muscle weakness and wasting. If conservative treatment fails, or is not appropriately initiated, deformity, immobility and chronic pain may result. In severe cases, surgical intervention may be required. With the exception of case reports and case series, limited safety and efficacy data exists regarding the use of neuraxial and regional anesthesia for patients with CMT. This paper describes an anesthetic case report of a patient with CMT, and also provides a review of general and regional anesthetic considerations for this cohort. The purpose of this report is to highlight the potential benefits of neuraxial and regional anesthesia in patients with neuromuscular disorders, especially in settings where intraand post-operative resources may be limited.
1. Introduction
Charcot-Marie-Tooth (CMT) disease is a type of hereditary sensory-motor neuropathy caused by defects in myelin structure, maintenance and formation (CMT1, CMT3, and CMT4), or due to damage to axons themselves (CMT2) [1]. The consequence of this myelin and axonal degradation is reduced conduction of sensory and motor nerve impulses. This can lead to progressive distal muscle weakness and wasting as well as sensory loss, which is often coupled with neuropathic pain.
Affecting as many as 1 in 2500 people in the US and 1 in 10,000 people worldwide, it is the most common type of inherited neuropathy [2]. There is no sexual or racial predilection in CMT. When recognized and treated appropriately patients are able to lead relatively normal lives. However, in resource-poor settings diagnosis and treatment is often delayed. This can lead to the need for surgical correction of soft-tissue contractures and bony deformities.
2. Case Report
A 19-year-old female from Kathmandu, Nepal, with CMT of unknown type presented to our visiting surgical team with peroneal muscle atrophy and inversion of her left foot. She was scheduled to undergo gastrocnemius recession with anterior tibialis tendon transfer. She was 5 feet 8 inches tall and weighed 29 kilograms. Her symptoms had been progressive and she had untreated severe pain with ambulation. Her exam revealed appropriate strength in the muscles associated with foot inversion, weakness in the muscles of eversion, and an absent left achilles deep tendon reflex. Her legs exhibited the stork leg phenotype and high arched foot characteristic of CMT, and she had abnormal facies with a narrow mandible and a small mouth opening (Figures 1-3). Two years prior she underwent tibia is anterior lengthening on the right with good results. The details of her anesthetic for this procedure were unavailable. The rest of her medical history was unremarkable with the exception of occasional gastritis and headaches.
Her anesthetic for this procedure consisted of a subarachnoid block with 50 mg lidocaine + dextrose followed by a pre-surgical ultrasound-guided popliteal sciatic nerve block with 6 ml 1% ropivicaine + 3 mg dexamethasone. Midazolam 1 mg IV was given for anxiolysis prior to subarachnoid block. Per ultrasound, the anatomic relationships, nerve structure, and size appeared as they do in patients without CMT. She tolerated the procedure well and had excellent post-operative pain control, requiring only 30 mcg intravenous fentanyl in the post-anesthesia care unit. On post-operative day 1 she received one dose of intramuscular tramadol. For the remainder of her hospital stay she required only twice daily oral diclofenac for analgesia, using no breakthrough pain medications. She was discharged home on
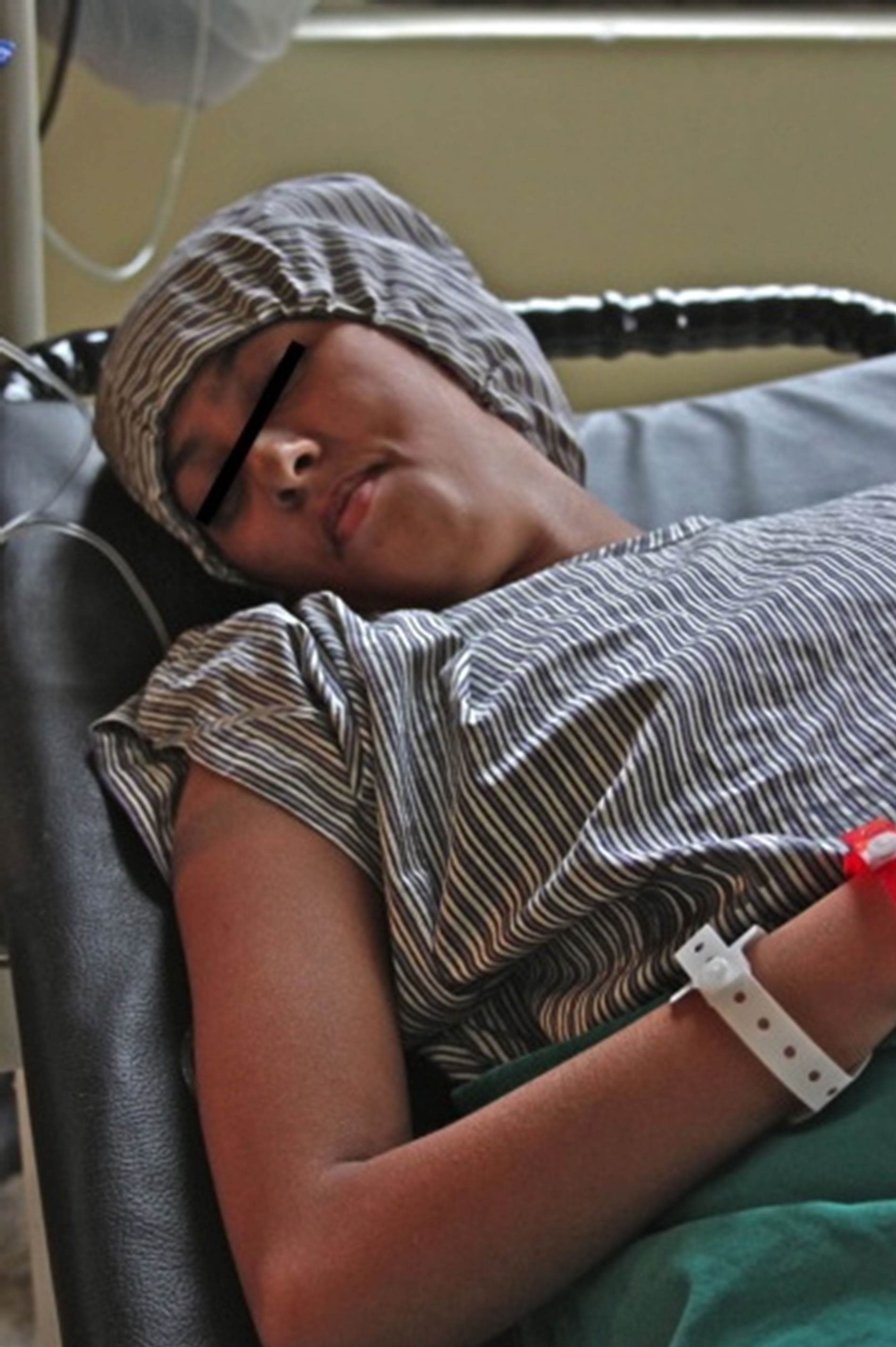
Figure 1. Narrow mandible and small mouth opening of our patient.
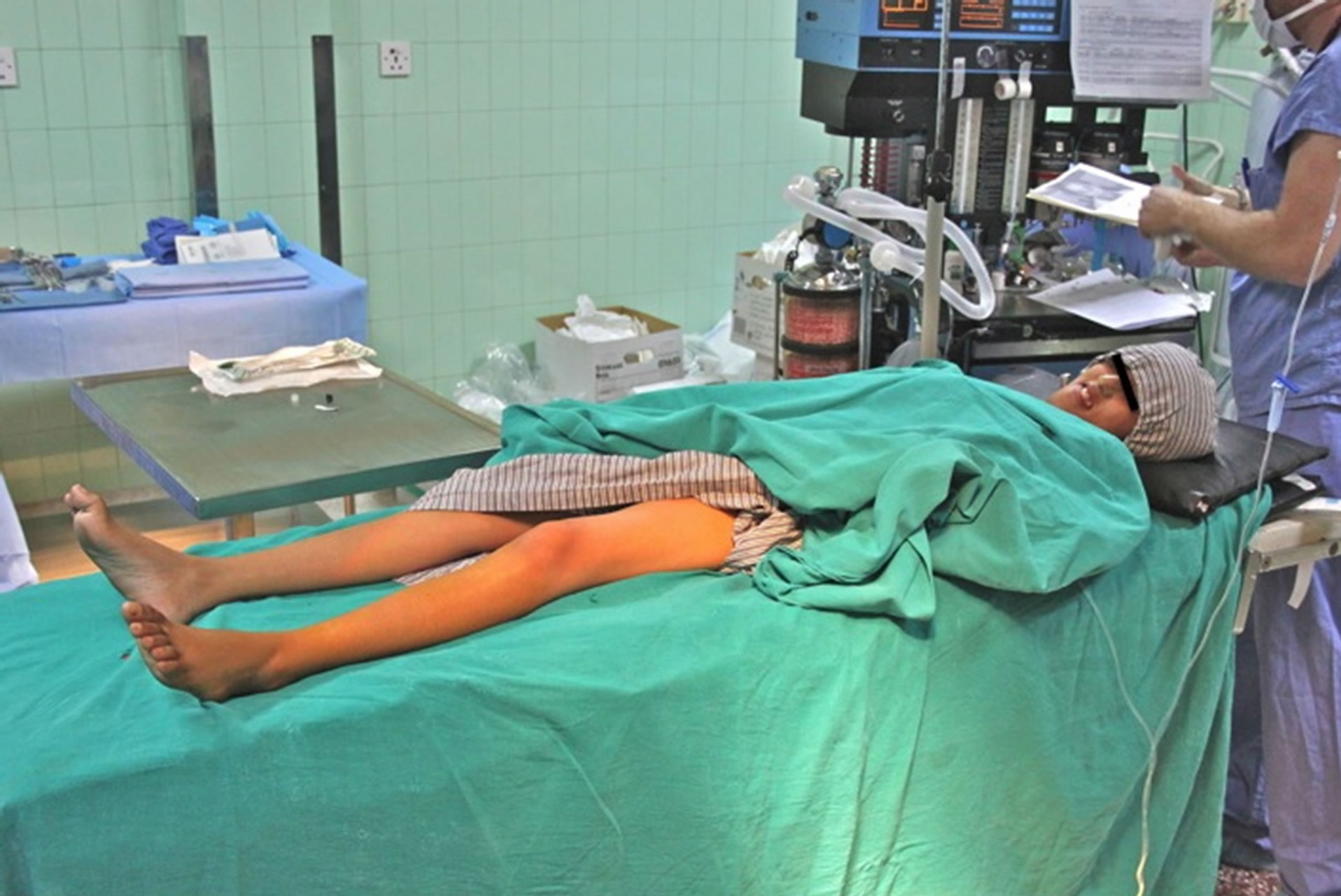
Figure 2. “Stork-Leg” phenotype, due to distal muscle wasting.
post-operative day 4 and was satisfied with her surgical course.
3. Clinical Features
In 1886 Jean Martin Charcot and Pierre Marie of Paris, France, and Howard Henry Tooth of Cambridge, Eng-
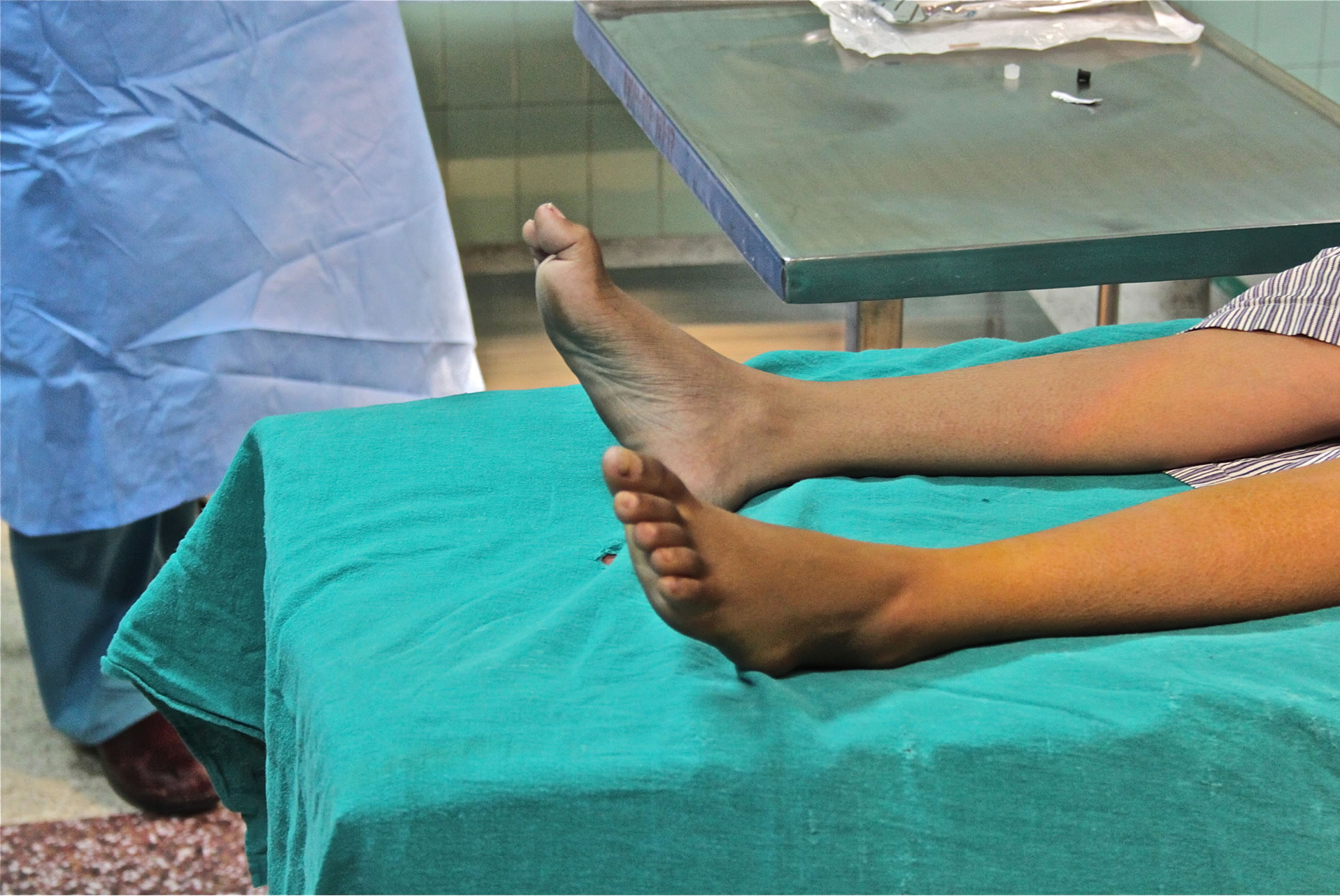
Figure 3. High arched foot, or “Pes Cavus” deformity.
land, first described a specific constellation of neurodegenerative findings that we now know as CMT [3]. Although age of onset can be variable, CMT typically manifests within the first two decades of life, and most often in the teenage years. Clinical phenotype is similar among the different forms of CMT, as are severity and rate of symptom progression [4]. Tripping or clumsiness is frequently the presenting complaint and parents often describe their child as being “un-athletic”. As strength deteriorates, symptoms progress to foot drop owing to peroneal muscle atrophy. These patients are found to have a steppage or “equine gait”, as well as a “champagne bottle” or stork leg phenotype. Intrinsic foot muscle atrophy commonly results in a high arched foot, known as pes cavus. Other bony abnormalities including scoliosis can be seen in long-standing CMT. Weakness of the arms and hands follows onset of lower extremity symptoms, and is demonstrated by poor handwriting and difficulty manipulating small objects such as buttons and zippers [5]. Spinothalamic functions such as pain, temperature, and pressure remain intact, as unmyelinated type C nerve fibers are responsible for these sensations. Vibration and proprioception can be significantly affected and result in an ataxic gait [3]. Although rare, sensorineural hearing loss, vocal cord involvement, or phrenic nerve dysfunction may occur [6-9]. Peripheral nerves can be enlarged and are often times palpable.
Diagnosis of CMT is primarily made through history and physical exam, and can be confirmed with electromyography (EMG), nerve conduction studies (NCS), nerve biopsies and/or genetic testing. EMG or NCS is the first line confirmatory test and can provide differentiation for subtype classification (demyelinating versus axonal). Nerve biopsy, though rarely indicated, will show a characteristic “onion bulb” histology in CMT1 due to demyelination and subsequent multiple layers of remyelination, and in CMT2 will demonstrate axonal loss with Wallerian degeneration [3]. Genetic testing can offer definitive diagnosis of CMT as well as further characterization into specific categories and categorical subtypes; however, not all genetic markers are known. Inheritance patterns for CMT are autosomal dominant, recessive, and xlinked depending on the subtype.
Life expectancy is normal for patients with CMT, but the condition can be extremely disabling. A cure does not exist, and treatment is focused on maintaining mobility and function. Physical and occupational therapy therefore both play an integral role focusing on muscle strengthening, achilles tendon stretching, and ankle-foot orthoses for improved gait stability. Medical therapies that limit or arrest disease progression are non-existent; however, studies with ascorbic acid and progesterone receptor antagonists show promise [10,11]. Gene therapy and stem cell transfer may be the most encouraging forms of future treatment modalities for CMT. Chronic pain, both nociceptive and neuropathic, can be problematic and may require multimodal treatment with NSAIDS, opioids, tricyclic antidepressants, and anticonvulsants such as gabapentin.
4. Anesthetic Considerations
Although speculative and limited, concerns about anesthetizing patients with CMT primarily revolve around the use of neuromuscular blocking agents. While some have proposed a theoretical increased risk of developing hyperkalemia with the use of depolarizing muscle relaxants, a review of 161 surgical procedures in 86 patients with CMT found no increased risk associated with the use of succinylcholine [12]. Data is less clear on the topic of non-depolarizing agents. A small case series using nondepolarizing muscle relaxants found that mivacurium had similar activity in CMT patients when compared to children without neuromuscular disease [13]. However, both normal and prolonged neuromuscular blockade has been reported with vecuronium [14,15].
Concerns also exist around the use of regional or neuraxial anesthesia in the setting of functional denervation. In fact, many sources cite pre-existing neurologic disease as a relative contraindication for neuraxial or regional anesthesia. This stance reflects a primary concern for exacerbating the pre-existing condition or causing further neurologic impairment. Brock et al. have demonstrated the successful use of combined spinalepidural for labor and cesarean delivery in patients with CMT [16]. In a case series of 3 CMT1 patients undergoing upper extremity surgery, regional techniques were used with success [17]. The authors noted that the localization of the nerves with nerve stimulator was suboptimal, but was successful with ultrasound guidance. Bosenberg reported effective post-operative analgesia with no detrimental effects following lower extremity surgery using both single shot sciatic nerve blocks, as well as continuous blockade with a sciatic peripheral nerve catheter, placed with the aid of a nerve stimulator [18].
5. Conclusions
With appropriate diagnosis and adequate resources, patients with CMT can be successfully managed with a conservative approach utilizing occupational therapy and orthopedic devices. Unfortunately, many patients with CMT are left undiagnosed and untreated (especially in resource-poor settings) leading to severe disability and deformation necessitating surgical correction.
With a relative paucity of data addressing the safety and efficacy of current anesthetic modalities in this cohort, concerns with general, neuraxial, and regional anesthesia remain. With that said, the authors find the use of neuraxial blockade in combination with ultrasound guided peripheral nerve block to be a safe and effective method for providing anesthesia and post-operative analgesia in this setting. While this technique allowed avoidance of potential electrolyte disturbance or indeterminate duration of action associated with neuromuscular blocking agents, we were more importantly able to avoid airway instrumentation in the setting of a potentially difficult airway. Through the use of these combined regional anesthetic techniques we were able to conduct our anesthetic with minimal peri-operative sedation. Additionally, our patient required only minimal opiates throughout her hospital course. Overall, we find the benefits of this technique to outweigh the potential risk profile associated with general anesthesia in this setting.
REFERENCES
- R. D. Miller, “Miller’s Anesthesia,” 7th Edition, Churchill Livingstone, Orlando, 2009.
- A. E. Emery, “Population Frequencies of Inherited Neuromuscular Diseases—A World Survey,” Neuromuscular Disorders, Vol. 1, No. 1, 1991, pp. 19-29. doi:10.1016/0960-8966(91)90039-U
- D. Kedlaya, “Physical Medicine and Rehabilitation for Charcot-Marie-Tooth Disease,” 2012. http://emedicine.medscape.com/article/315260
- B. Bissonnette, I. Luginbuehl, B. Marciniak, B. Dalens, “Syndromes: Rapid Recognition and Perioperative Implications,” McGraw-Hill, New York, 2006, pp. 153-155.
- J. Burns, P. Bray, L. A. Cross, K. N. North, M. M. Ryan and R. A. Ouvrier, “Hand Involvement in Children with Charcot-Marie-Tooth Disease Type 1A,” Neuromuscular Disorders, Vol. 18, No. 12, 2008, pp. 970-973. doi:10.1016/j.nmd.2008.08.004
- E. M. Yiu, N. Geevasinga, G. A. Nicholson, E. R. Fagan, M. M. Ryan and R. A. Ouvrier, “A Retrospective Review of X-Linked Charcot-Marie-Tooth Disease in Childhood,” Neurology. Vol. 76, No. 5, 2011, pp. 461-466. doi:10.1212/WNL.0b013e31820a0ceb
- B. G. Kousseff, T. A. Hadro, D. L. Treiber, T. Wollner and C. Morris, “Charcot-Marie-Tooth Disease with Sensorineural Hearing Loss—An Autosomal Dominant Trait,” Birth Defects Original Article Series, Vol. 18, No. 3B, 1982, pp. 223-228.
- T. Stojkovic, P. Latour, A. Vandenberghe, J. F. Hurtevent and P. Vermersch, “Sensorineural Deafness in X-Linked Charcot-Marie-Tooth Disease with Connexin 32 Mutation (R142Q),” Neurology, Vol. 52, No. 4, 1999, pp. 1010- 1014. doi:10.1212/WNL.52.5.1010
- T. Sevilla, et al., “Vocal Cord Paresis and Diaphragmatic Dysfunction Are Severe and Frequent Symptoms of GDAP1-Associated Neuropathy,” Brain, Vol. 131, No. 11, 2008, pp. 3051-3061. doi:10.1093/brain/awn228
- J. Burns, R. A. Ouvrier, E. M. Yiu and M. M. Ryan, “Extended Treatment of Childhood Charcot-Marie-Tooth Disease with High-Dose Ascorbic Acid,” Journal of the Peripheral Nervous System, Vol. 16, No. 3, 2011, pp. 272- 274. doi:10.1111/j.1529-8027.2011.00348.x
- J. Bradbury, “Antiprogesterone Hope for Inherited Neuropathy,” The Lancet Neurology, Vol. 3, No. 1, 2004, p. 6. doi:10.1016/S1474-4422(03)00636-7
- J. F. Antognini, “Anaesthesia for Charcot-Marie-Tooth Disease: A Review of 86 Cases,” Canadian Journal of Anesthesia, Vol. 39, No. 4, 1992, pp. 398-400. doi:10.1007/BF03009054
- H. J. Schmitt, S. Wick and T. Münster, “Onset and Duration of Mivacurium-Induced Neuromuscular Blockade in Children with Charcot-Marie-Tooth Disease. A Case Series with Five Children,” Paediatric Anaesthesia, Vol. 16, No. 2, 2006, pp. 182-187. doi:10.1111/j.1460-9592.2005.01627.x
- A. S. Baraka, “Vecuronium Neuromuscular Block in a Patient with Charcot-Marie-Tooth Syndrome,” Anesthesia & Analgesia, Vol. 84, No. 4, 1997, pp. 927-928.
- D. Pogson, J. Telfer and S. Wimbush, “Prolonged Vecuronium Neuromuscular Blockade Associated with Charcot Marie Tooth Neuropathy,” British Journal of Anaesthesia, Vol. 85, No. 6, 2000, pp. 914-917. doi:10.1093/bja/85.6.914
- M. Brock, C. Guinn and M. Jones, “Anesthetic Management of an Obstetric Patient with Charcot-Marie-Tooth Disease: A Case Study,” American Association of Nurse Anesthetists, Vol. 77, No. 5, 2009, pp. 335-337.
- S. Dhir, S. Balasubramanian and D. Ross, “UltrasoundGuided Peripheral Regional Blockade in Patients with Charcot-Marie-Tooth Disease: A Review of Three Cases,” Canadian Journal of Anesthesia, Vol. 55, No. 8, 2008, pp. 515-520.
- A. Bosenberg and K. Larkin, “Anaesthesia and CharcotMarie-Tooth Disease,” Southern African Journal of Anaesthesia & Analgesia, Vol. 12, No. 4, 2006, pp. 131-133.