Journal of Cancer Therapy
Vol.3 No.6(2012), Article ID:25412,6 pages DOI:10.4236/jct.2012.36138
Collision Anaplastic Large Cell Lymphoma (T-Cell/Histiocyte-Rich) and Diffuse Large B Cell Lymphoma: A Pathologic and Clinical Evaluation
![]()
1Department of Pathology, Division of Hematopathology, The Ohio State University Medical Center, Columbus, USA; 2The Department of Medicine, Division of Hematology, The Ohio State University Medical Center, Columbus, USA.
Email: *Weiqiang.zhao@osumc.edu
Received September 14th, 2012; revised October 13th, 2012; accepted October 22nd, 2012
Keywords: Collision Lymphoma; ALCL; DLBCL
ABSTRACT
A collision tumor of T and B-cell lymphomas occur rarely. In this article we report a collision tumor of anaplastic large cell lymphoma and diffuse large B cell lymphoma in a 46 years old female. The tumor showed predominantly neoplasmtic anaplastic large cell lymphoma (ALCL) component expressing CD30 and ALK with smaller areas of CD20+ diffuse large B cell lymphoma component. Polymerase chain reaction for T-cell receptor beta and IgH (VDJ) gene rearrangements detected a clonal T cell and a clonal B cell population. The patient developed CSF involvement approximately 3 months after treatment. CSF analysis at this time showed only monoclonal T cells, probably due to clearing of the B cell component by the chemotherapy. To the best of our knowledge this is the first case report of a composite tumor of ALCL (lymphohistiocytic variant) and DLBCL. This case raises issues related to the classification of these composite lymphomas and the treatment on initial presentation and during relapse.
1. Introduction
Collision tumors are rare disease entities in which two histologically distinct tumor types occur simultaneously at the same anatomic site. Collision tumors have been described between chronic lymphocytic leukemia (CLL) and invasive ductal carcinoma of the breast [1], T-cell lymphoma and squamous cell carcinoma of the lung [2], Hodgkin’s disease and metastatic breast or cervical cancer [3,4], Non-Hodgkin lymphoma and Kaposi sarcoma [5], mucosa-associated lymphoid tissue (MALT) lymphoma with gastric adenocarcinoma [6-8] or breast invasive ductal carcinoma [9], among other combinations. Collision lymphomas, especially those between T and B-cell lymphomas occur rarely. In a literature review by Abou-Elella and Nifong, there were only 46 published reports of such lymphomas between 1985 and 2004. Of these, T-large granular lymphocytic leukemia was the most frequently identified T-cell component, and diffuse large B-cell lymphoma (DLBCL) was the most frequent B-cell component [10]. In another case report, a composite ALCL and Hodgkin lymphoma developed in the setting of CLL [11]. The underlying pathogenesis of composite lymphomas remains unknown. Current hypotheses include cytogenetic alteration of a pluripotent cell prior to differentiating into Band T-lymphoid lineages, independent clones arising separately but simultaneously into Band T-cell tumors, immune dysregulation leading to the emergence of mixed neoplastic clones, EBV infection or reactivation and subsequent clonal expansion of immortalized EBV-infected B-cell clones, chemotherapy for one tumor resulting in the emergence of another, or coincidental existence of more than one lymphoma at the time of diagnosis [12]. Here we describe a patient with composite ALK-1 (+) primary systemic ALCL and DLBCL.
2. Clinical History
A 46-year-old Caucasian female presented with a 4-week history of lymphadenopathy in cervical, left supraclavicular, right axillary, right hilar, retroperitoneal, right external iliac chain and right inguinal areas, and associated symptoms with fever, night sweats, and weight loss. Her past medical history was notable for iron deficiency anemia, obesity, gastric bypass surgery, and cholecystectomy. Her family history was significant for breast cancer, thyroid cancer, and malignant melanoma in her siblings. Physical examination showed palpable lymph nodes in the right cervical and bilateralinguinal regions. CT imaging showed lymphadenopathy in the lower retroperitoneum, right external iliac chain, and splenomegaly. A PET scan showed increased metabolic activity in these lymph node regions, with SUV ranging from 1.9 to 19.9. Laboratory investigations showed WBC count of 3.6 K/mL, hemoglobin of 9.2 g/dL and platelet count of 187 K/mL. Her initial lactose dehydrogenase was 543 U/L and peaked at 775 U/L. The patient initially underwent a bronchoscopy at an outside institution, with the intent to biopsy one of her mediastinal lymph nodes; however, this procedure was aborted due to hypoxia and hypotension. After transferring to our hospital, she underwent an excisional biopsy of a left supraclavicular lymph node.
3. Materials and Methods
Immunohistochemistry was performed on 4-micro section of paraffin-embedded tissue (FFPE) on a positively charged slide. After deparaffinization, the tissue was rehydrated through xylenes and graded ethanol solutions to water. All slides were quenched for 5 minutes in a 3% hydrogen peroxide solution in water to block for endogenous peroxidase. Antigen retrieval was performed by a heat method in which the specimens were placed in a citric acid solution (pH 6.1) for 25 minutes at 94˚C and cooled for 15 minutes using a vegetable steamer. Slides were then placed on a DakoAutostainer System (Dako Tucson, AZ), for use with immunohistochemistry. The first antibody, CD20 (L26, Dako, 1:400), or CD30 (Ber-H2, Dako, 1:120), was incubated for 30 minutes at room temperature. The detection system for the primary antibody was Envision Plus HRP from Dako. For double staining, serum-free protein block (Dako) was added followed by anti-ALK (SP8, Abcam, 1:100) and incubated for 30 minutes at room temperature. The detection system for ALK was Mach 4 alkaline phosphatase (Biocare Medical) and Vulcan Fast Red was used to develop ALK producing a bright Fuchsia precipitate so that the two primary antibodies could be easily differentiated. Slides were then counterstained in Richard Allen hematoxylin, dehydrated through graded ethanol solutions and cover slipped.
Chromogenic In-Situ Hybridization (CISH) was performed to detect EBV infection using Ventana Medical System’s INFORM EBER (Epstein-Barr Early RNA) Probe Reagent on the Ventana Benchmark XT Slide Staining System (Ventana Medical Systems). Briefly, the FFPE sections were cut, placed on positively charged slides, and dried in a 60 degree oven for an hour prior to deparaffinization. Once slides had been oven-dried, they were then loaded onto the XT, where they were subsequently deparaffinized, rehydrated, and treated with Protease 3 (Ventana) at 37˚C for 4 minutes. Slides were rinsed. Approximately 100mL of INFORM EBER probe, a DNP-labeled oligonucleotide probe in a formamidebased diluent, was then applied to each slide, denatured at 85˚C for 12 minutes, and then hybridized at 47˚C for one hour. A stringency wash (2× SSC) was performed at 65˚C for 4 minutes, followed by 3 additional stringency washes at room temperature, 4 minutes each. Detection was performed using Ventana Medical Systems Blue plus Detection Kit. This kit consists of a primary rabbit anti-DNP reagent, which detects the DNP labeled probes bound to the target sequence. This is followed by an amplification reagent (mouse anti rabbit antibody) and the binding of a biotinylated secondary antibody (goat antimouse IgG). Streptavidin conjugated alkaline phosphatase is then utilized as a chromogenic enzyme (BCIP/ NBT), which generates an intense blue color reaction at the target site. Slides are then counterstained for 4 minutes using Ventana Medical Systems Red Stain II. Once staining is completed, the slides are removed from the XT automated stainer, briefly washed in warm, soapy water, and rinsed in ddH2O. Slides are then dehydrated through graded alcohols, acetone, and cleared in 3 changes of xylene. They are then mounted with a Xylene based mounting media (Micromount, Surgipath).
Fluorescent in Situ Hybridization (FISH) was performed to detect the translocation of ALK gene on (2p23) using ALK dual color break apart probe (Vysis®, Abbott Laboratories, Abbott Park, IL) on tissue sections at 2-µm as described previously [13]. The slides were visualized on a fluorescent microscope (Olympus BX51; Olympus, Tokyo, Japan) equipped with a digital image analysis system. The presence of ALK translocation was defined by the presence of a normal intact signal (yellow) and separated green and red signals apart from each other at a distance of at least 3 signal widths.
TCR-beta and IgH (VDJ) gene rearrangement were performed as described previously (Christian et al., 2010) on the FFPE tissue sections. To detect the clonality present in the CSF, the cell pellet was obtained after spinning down at 400 rpm on a table centrifuge at room temperature. The genomic DNA was extracted from the cell pellet using QiAamp DNA Mini Kit (Qiagen) according to the manufacturer’s procedure. Ten (10) ng of prepared genomic DNA was applied to a 25-mL reaction mixture containing HotStarTaq Master Mix (Qiagen) and forward and reverse primer mixtures (Invivoscribe Technologies, San Diego, CA), and PCR-based clonality assay for TCR-beta or IgH (VDJ) gene rearrangement was performed as described previously [13].
4. Results
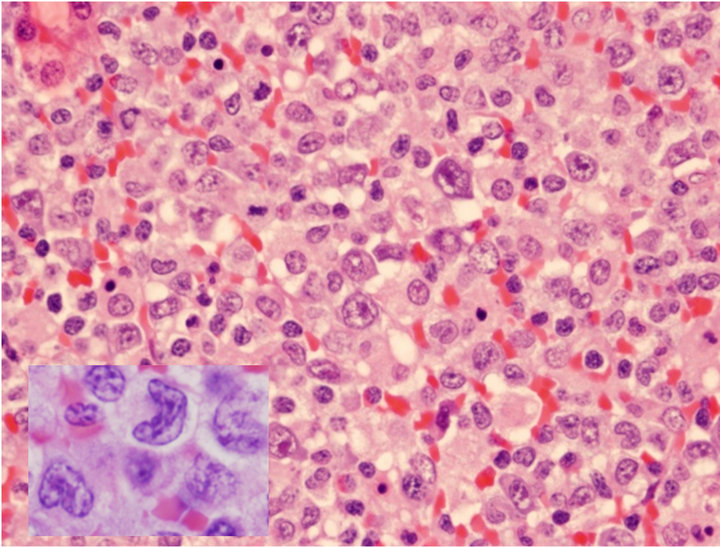
The H&E of the lymph node biopsy shows the effaced nodal structures by heterogenous populations of cells including numerous histiocytes, small lymphocytes, and anaplastic large cells (Figure 1(a)). The anaplastic large lymphoid cells had abundant cytoplasm, horseshoe (“hallmark”) nuclei with prominent nucleoli (Figure 1(a)). They infiltrated confluently along the vessels, or colonized the residual germinal centers, forming motheaten appearances. In the same lymph node there were simultaneously infiltrated by a monotonous population of large atypical lymphoid cells with relatively round nuclei without anaplastic morphology. Immunophenotype by flow cytometry demonstrated an aberrant T cell population expressing CD2+, CD3+, CD7+, and CD8+ with loss of CD4 and CD5. The B-lymphocytes were polyclonal. The differential diagnosis of ALCL versus peripheral T-cell lymphoma (PTCL) was considered.
Immunohistochemical stains were performed for CD3, CD20, CD15, CD30, and ALK. There were numerous histiocytes highlighted by CD56 and reactive T-lymphocytes highlighted by CD3 stains. The anaplastic large cells were positive for CD30+ (Figure 1(b)) and ALK (Figure 1(c)) but negative for CD15 or CD3, which supported the diagnosis of ALCL. Results of FISH demonstrated the presence of translocated ALK gene in the ALCL cells (Figure 1(d)).
CD20 stain, however, highlighted numerous monotonous large lymphoma cells (Figure 2(a) and insert), highly suspicious for a collisional B-cell lymphoma. Molecular studies by the polymerase chain reaction (PCR) for T-cell receptor-beta (TCRb) and IgH (VDJ) gene rearrangements detected both as clonal in the specimen, supporting the diagnosis of composite lymphoma (upper and lower panels, Figure 2(b), respectively).
Since ALK Anaplastic lymphoma kinase (ALK)-positive diffuse large B-cell lymphoma (DLBCL) is a rare variant of DLBCL that has been described in previous reports, we used dual antibody immunohistochemistry in a combination of CD20 and ALK or CD30 and ALK to further characterize the composite lymphoma. We applied mouse monoclonal antibodies for CD20 or CD30 as first primary antibody and detected by Envision Plus HRP (Dako) and DAB chromogens which produced a brown precipitate on the cell membranes and cytoplasm. Then ALK proteins were detected by rabbit monoclonal antibody using Mach 4 alkaline phosphatase and Vulcan Fast Red (Biocare Medical). Using this strategy, two populations of neoplastic cells, CD30+ were ALK positive (CD30+/ALK+) (Figure 2(c)) and CD20+ were ALK negative (CD20+/ALK−) (Figure 2(d)), were easily distinguished and these results confirmed that DLBCL
 (a)
(a) (b)
(b)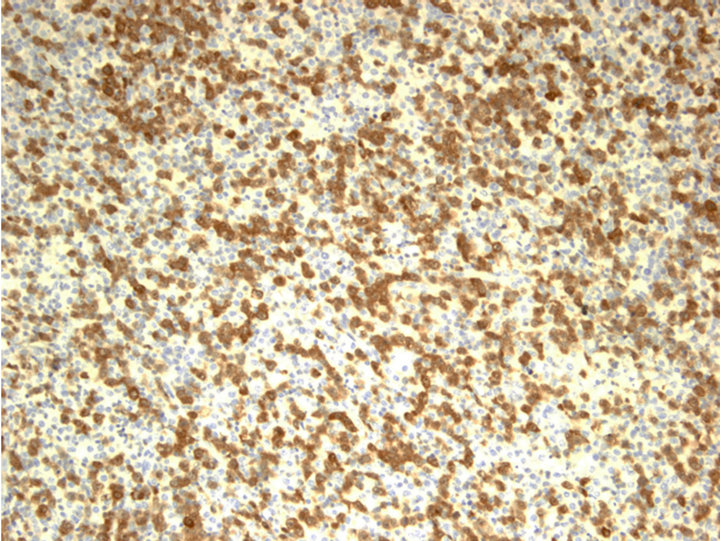 (c)
(c) (d)
(d)
Figure 1. Characterization of ALCL components. (a) Large atypical cells are present in a background of small lymphocytes and numerous histiocytes (H&E stain, original 400×). Some of the large atypical cells show hallmark cells with features of “horseshoe” nuclei (Insert: 500×); (b) Large neoplastic cells were positive for CD30 with membranous and Golgi staining (IHC, anti-CD30, 400×); (c) ALCL cells reactive to antibody for ALK-1 (20×); (d) FISH with dual color break apart ALK probe shows splitting orange and green signals indicating ALK gene break apart due to the translocation (1000×).
 (a)
(a)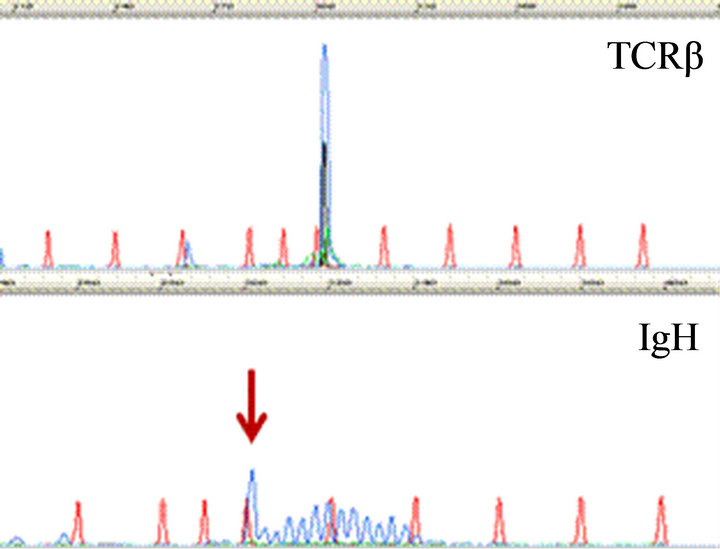 (b)
(b) (c)
(c)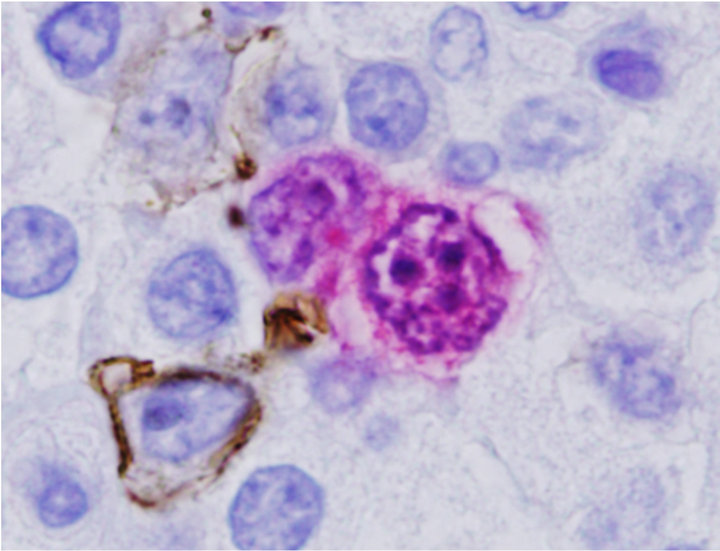 (d)
(d)
Figure 2. Characterization of Collision with DLBCL. (a) Diffuse large B-cell lymphoma cells were highlighted by anti-CD20 (2×) with inset showing CD20+ cells at 400×; (b) Molecular study for TCR-beta (upper panel) and IgH (lower panel, arrow) gene rearrangements showed clonal peaks present in the specimen, indicating 2 distinct clonal populations of T and B-lymphocyte, respectively; (c) CD30 (Brown) and ALK-1 (Red) dual IHC show the population of ALK+ ALCL lymphoma cells (1500×); (d) CD20 and ALK-1 dual IHC show the population of DLBCL cells reacted only to anti-CD20 (brown) but not to anti-ALK (red).
are negative forALK [14-16]. The lymphoma cells are negative for Epstein Barr Virus encoded RNA (EBER) by ISH.
A bone marrow biopsy and aspirate showed no evidence of either lymphoma in the marrow. The patient was initiated with CHOP and then switched to 6 cycles of rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP). While getting CHOP, the patient was found to have CNS involvement 2.5 months later by the T-cell component only as evidenced by rearranged TCR-beta gene while IgH gene rearrangement was negative. The patient received intrathecal methotrexate therapy for that. Disease reevaluation with a PET scan performed 5 months later showed complete remission. One month later, the patient developed skin nodules, pruritic skin rash as well as lymphadenopathy of left cervical area and left axilla but systemic review was unremarkable. A skin biopsy as well as cervical lymph node biopsy was performed and consistent with ALCL component. The patient received salvage chemotherapy with ICE and planed for stem cell transplantation. She developed fever, respiratory failure, and septic shock even with broad spectrum antibiotic treatment. The patient passed away 9 month later due to cardiac arrest.
5. Discussion
A composite lymphoma of B and T cells is a rare occurrence. To our knowledge, a composite lymphoma composed of ALCL (lymphocytic-histiocytic rich variant) and ALK-negative DLBCL has not been described in literature. Our case is unique in several aspects. Firstly, it is a lymphohistiocytic rich of ALCL, a rare variant of ALCL in which there are diffuse infiltration of histiocytes and small reactive T-lymphocytes. Flow cytometric analysis might reveal the immunophenotypic abnormal T-cell population, such as loss of CD3, CD5 and CD4. ALK+ ALCL cells were clearly co-expressing CD30. The ALCL cells were clonal showing rearranged TCRbeta genes, presence of ALK+ translocations and these cells expressed CD30. The second DLBCL population is small and was not detectable by flow cytometry. However, the clonality of rearranged IgH gene demonstrated DLBCL were clonal in nature and expressed CD20. It might be difficult to differentiate if DLBCL expresses ALK or not, but dual color and dual antibody IHC solved this problem as shown in this case. In this case, the DLBCL cells didn’t express ALK. Therefore, there is no clonal relation between ALCL and DLBCL in this collision tumor.
In the majority of collision T and B cell lymphomas reported in the literature, EBV could be demonstrated, indicating the role of this virus in the development of these tumors [12]. Our patient did not have any predisposing immunological condition and EBER was negative, the latter was consistent with previous reported results that EBV usually was absent in ALCL [17]. The patient did not have any prior history of chemotherapy. However, a family history of malignancy and her past chemical exposure to PFOA might suggest tumorigenesis and multiple clonal evolutions of different tumor clusters. In our patient, CSF involvement, consisting only of the T cell component, was diagnosed within 3 months of initial diagnosis. This finding further proves that there were two separate neoplastic clones. She might have cleared her CSF B cell component after intrathecal methotrexate. However, the ALCL component revolved and disseminated which conferred in an inferior prognosis.
This case raises issues related to the classification of these composite lymphomas and the treatment on initial presentation as well as during relapse. Also questions regarding age/sex predilection and chromosomal aberrations associated with these tumors are raised. Clearly more studies are needed to help in understanding the pathogenesis, treatment and prognosis of these rare tumors.
REFERENCES
- K. J. Cheung, W. Tam, E. Chuang and M. P. Osborne, “Concurrent Invasive Ductal Carcinoma and Chronic Lymphocytic Leukemia Manifesting as a Collision Tumor in Breast,” The Breast Journal, Vol. 13, No. 4, 2007, pp. 413-417.
- O. Kawashima, S. Sakata, M. Kamiyoshihara, A. Maeshima, S. Ishikawa and Y. Morishita, “Primary Pulmonary Collision Tumor Including Squamous Cell Carcinoma and T-Cell Lymphoma,” Lung Cancer, Vol. 23, No. 1, 1999, pp. 67-70.
- A. S. Allal, J. Weintraub, S. Remadi and R. Abele, “Concurrent Interfollicular Hodgkin’s Disease and Metastatic Breast Carcinoma in Lymph Nodes,” Pathology International, Vol. 46, No. 10, 1996, pp. 787-790.
- M. O. Lovell and P. T. Valente, “Unique Collision of Hodgkin Lymphoma and Adenosquamous Carcinoma in the Uterine Cervix: Synchronous Malignant Neo Plasms of the Cervix,” Journal of Lower Genital Tract Disease, Vol. 7, No. 4, 2003, pp. 307-310.
- M. Tunc, M. L. Simmons, D. H. Char and B. Herndier, “Non-Hodgkin Lymphoma and Kaposi Sarcoma in an Eyelid of a Patient with Acquired Immunodeficiency Syndrome. Multiple Viruses in Pathogenesis,” Archives of Ophthalmology, Vol. 115, No. 11, 1997, pp. 1464- 1466.
- T. Isaka, T. Nakamura and M. Tajika, “API2-MALT1 Chimeric Transcript-Positive Gastroduodenal MALT Lymphoma with Subsequent Development of Adenocarcinoma as a Collision Tumour over a Clinical Course of 7 Years,” Histopathology, Vol. 51, No. 1, 2007, pp. 119- 123.
- G. Cammarota, L. M. Larocca, D. D’Ugo, R. Persiani, R. Cianci, R. Nocente, A. Picciocchi and G. Gasbarrini, “Synchronous Gastric Adenocarcinoma and MALT Lymphoma in a Patient with H. Pylori Infection. Could the Two Neoplasms Share a Common Pathogenesis?” Hepato-Gastroenterology, Vol. 48, No. 37, 2001, pp. 104-106.
- A. C. Wotherspoon and P. G. Isaacson, “Synchronous Adenocarcinoma and Low Grade B-Cell Lymphoma of Mucosa Associated Lymphoid Tissue (MALT) of the Stomach,” Histopathology, Vol. 27, No. 4, 1995, pp. 325- 331.
- B. Susnik, J. Jordi Rowe, P. N. Redlich, C. Chitambar, C. C. Chang and B. Kampalath, “A Unique Collision Tumor in Breast: Invasive Ductal Carcinoma and Mucosa-Associated Lympoid Tissue Lymphoma,” Archives of Pathology & Laboratory Medicine, Vol. 128, No. 1, 2004, pp. 99-101.
- A. A. Abou-Elella and T. P. Nifong, “Composite EBV Negative Peripheral TCell Lymphoma and Diffuse Large B-Cell Lymphoma Involving the Ileum: A Case Report and a Systematic Review of the Literature,” Leukemia & Lymphoma, Vol. 47, No. 10, 2006, pp. 2208- 2217.
- A. van den Berg, E. Maggio, R. Rust, R. Rust, K. Kooistra, A. Diepstra and S. Poppema, “Clonal Relation in a Case of CLL, ALCL, and Hodgkin Composite Lymphoma,” Blood, Vol. 100, No. 4, 2002, pp. 1425-1429.
- Y. Xu, R. W. Mckenna, M. P. Hoang, R. H. Collins and S. H. Kroft, “Composite Angioimmunoblastic T-Cell Lymphoma and Diffuse Large B-Cell Lymphoma: A Case Report and Review of the Literature,” American Journal of Clinical Pathology, Vol. 118, No. 6, 2002, pp. 848- 854.
- B. Christian, W. Zhao, M. Hamadani, E. M. Sotomayor, W. Navarro, S. M. Devine, F. Racke and K. A. Blum, “Mantle Cell Lymphoma 12 Years after Allogeneic Bone Marrow Transplantation Occurring Simultaneously in Recipient and Donor,” Journal of Clinical Oncology, Vol. 28, No. 31, 2010, pp. e629-e632. doi:10.1200/JCO.2010.29.8992
- E. Haralambieva, K. A. Pulford, L. Lamant, S. Pileri, G. Roncador, K. C. Gatter, G. Delsol and D. Y. Mason, “Anaplastic Large-Cell Lymphomas of B-Cell Phenotype Are Anaplasticlymphoma Kinase (ALK) Negative and belong to the Spectrum of Diffuse Large B-Celllymphomas,” British Journal of Haematology, Vol. 109, No. 3, 2000, pp. 584-591. doi:10.1046/j.1365-2141.2000.02045.x
- P. Adam, T. Katzenberger, H. Seeberger, S. Gattenlöhner, J. Wolf, C. Steinlein, M. Schmid, H. K. Müller-Hermelink and G. Ott, “A Case of a Diffuse Large B-Cell Lymphoma of Plasma Blastic Type Associated with the t(2;5)(p23;q35) Chromosome Translocation,” American Journal of Surgical Pathology, Vol. 27, No. 11, 2003, pp. 1473-1476. doi:10.1097/00000478-200311000-00012
- R. D. Gascoyne, L. Lamant, J. I. Martin-Subero, V. S. Lestou, N. L. Harris, H. K. Müller-Hermelink, J. F. Seymour, L. J. Campbell, D. E. Horsman, I. Auvigne, E. Espinos, R. Siebert and G. Delsol, “ALK-Positive Diffuse Large B-Cell Lymphoma Is Associated with ClathrinALK Rearrangements: Report of 6 Cases,” Blood, Vol. 102, No. 7, 2003, pp. 2568-2573. doi:10.1182/blood-2003-03-0786
- M. Herling, G. Z. Rassidakis, D. Jones, A. Schmitt-Graeff, A. H. Sarris and L. J. Medeiros, “Absence of Epstein-Barr Virus in Anaplastic Large Cell Lymphoma: A Study of 64 Cases Classified According to World Health Organization Criteria,” Human Pathology, Vol. 35, No. 4, 2004, pp. 455-459. doi:10.1016/j.humpath.2003.10.013
NOTES
*Corresponding author.