Journal of Cancer Therapy
Vol. 3 No. 1 (2012) , Article ID: 17226 , 12 pages DOI:10.4236/jct.2012.31011
In Vitro Investigation of DNA Damage Induced by the DNA Cross-Linking Agents Oxaliplatin and Satraplatin in Lymphocytes of Colorectal Cancer Patients
![]()
Division of Biomedical Sciences, University of Bradford, Bradford, UK.
Email: d.anderson1@bradford.ac.uk
Received October 12th, 2011; revised November 15th, 2011; accepted December 5th, 2011
Keywords: DNA Interstrand Crosslinks; Oxaliplatin; Satraplatin; γH2AX Foci Formation
ABSTRACT
Exposure to toxic chemicals, especially chemotherapeutic drugs, may induce several DNA lesions, including DNA interstrand crosslinks. These crosslinks are considered toxic lesions to the dividing cells since they can induce mutations, chromosomal rearrangements, and cell death. Many DNA interstrand crosslinks lesions can be generated by platinum-based chemotherapeutic agents. Satraplatin is a novel orally administered platinum-based chemotherapeutic agent. In the present study, we investigated DNA interstrand crosslinks lesions induced by oxaliplatin and satraplatin in lymphocytes obtained from colorectal cancer patients and healthy volunteers. Satraplatin demonstrated an increase in interstrand crosslinks in a dose-dependent manner in the Comet assay (p < 0.001). In addition, satraplatin and oxaliplatin increased significantly the number of sister chromatid exchanges up to 8.5-fold and 5.1-fold (p < 0.001) respectively, when treated with 2 µM concentration in comparison to untreated colorectal cancer cells. Further, the γH2AX foci formation was investigated by an immunofluorescence assay with oxaliplatin and satraplatin. The γH2AX foci formation rate was increased by approximately 9-fold when lymphocytes were treated with 2 μM oxaliplatin. Satraplatin was found to significantly induce the number of γH2AX foci by 8.5-fold and 11-fold with both 0.2 μM and 2.0 μM, respectively, compared to the control volunteers that may indicate the repair system in cancer cells experiences a loss of ability to cope with the repair of DSBs. In conclusion, oxaliplatin and satraplatin effectively induced DNA interstrand crosslinks in lymphocytes obtained from colorectal cancer patients and healthy volunteers in vitro. Here, to the best of our knowledge we report for the first time evidence of DNA double strand breaks formation as a possible molecular mechanism of action for satraplatin.
1. Introduction
Exposure to toxic chemicals, especially chemotherapeutic drugs, may induce DNA damage, and consequently, many chemically induced modifications may result to the DNA. These modifications on the DNA can produce several lesions, including DNA interstrand crosslinks. Interstrand crosslinks are considered toxic lesions to the dividing cells because of their capability of inducing mutations, chromosomal rearrangements, and cell death [1,2]. Many chemotherapeutic agents can initially generate covalent adducts at DNA bases on both strands, eventually resulting in interstrand crosslinks. The formation of DNA adducts causes distortion in the DNA structure, including unwinding and bending, and obstructs DNA replication and transcription, thereby resulting in an enhancement in cell death (apoptosis) in vitro and cell-cycle arrest in the G2 phase [3]. Interstrand crosslinking (ICL) based anticancer agents such as cisplatin, carboplatin, and mitomycin C (MMC) are used generally in the clinic and novel ICL cancer anticancer therapeutics continue to be developed [4,5]. The discovery and development of platinum complexes is one of the great success stories in the advancement of cancer chemotherapy because of the pronounced activity of cisplatin in the treatment of testicular and ovarian cancers [6]. However, treatment with cisplatin is limited due to its severe side effects such as nephrotoxicity and peripheral neurotoxicity [7,8]. Highly toxic DNA interstrand crosslink lesions are generated by platinumbased chemotherapeutic agents [9]. Further, the risk of secondary malignancies is high from treatment with ICLinducing agents treatment [10] possibly due to mutagenic effects of the lesions in normal cells.
In an attempt to reduce the problematic side effects of cisplatin [11], many platinum-class derivatives were developed including oxaliplatin and satraplatin. The latter was introduced in the year 1993 [12] and it is still awaiting its approval from the FDA. Satraplatin unlike other approved platinum-based compounds (cisplatin, carboplatin, and oxaliplatin) is administrated orally, whereas the others are administrated intravenously [13]. This compound was considered a probable chemotherapeutic agent because it demonstrated a greater extent of cell killing in cisplatin-resistant cancers [14]. Satraplatin also demonstrated clinical activity against various mammalian cancers including prostate [15,16], ovarian [12], cervical, and lung [17] cancers.
Colorectal cancer is currently ranked as the third most common cancer following breast and lung cancers. In the United Kingdom, there is an average of 106 new diagnoses of colorectal cancer delivered on a daily basis [18]. However, the basic and molecular pharmacological effects of satraplatin are still unknown in all types of cancer including colorectal cancer (CRC) [19].The damage caused to DNA by satraplatin is repaired by a mammalian nucleotide excision repair pathway that is similar to repairs done after damage by cisplatin and oxaliplatin [20]. However, satraplatin-induced adducts are not known to be recognized by DNA mismatch repair proteins unlike those from cisplatin and carboplatin [21]. Some research studies have suggested that satraplatin induced adducts are unable to bind to the high mobility group 1 protein, which is capable of recognizing DNA damage due to cisplatin, and therefore trans-lesion replication is inhibited [22].
Many of the compounds used as chemotherapeutic agents exert their action in cancer cells by inducing the formation of DNA double-strand breaks. DNA double-strand breaks (DSBs) represent the most hazardous type of DNA damage since a single DSB is capable of causing cell death or disturbing the genomic integrity of the cell [23]. DSBs are difficult to repair and extremely harmful [24]. DSBs are more toxic in fast growing cells; hence, agents that cause DSB formation find application in cancer treatment. DSBs generally form when two single-strand breaks (SSBs) occur in close proximity, or when a SSB or certain other lesions are encountered during DNA-replication [23]. Mediated by DSBs, DNA lesions develop as a result of recombination between different loci [25]. DSBs are processed by a number of DNA repair pathways depending partly on the phases of the cell cycle. One of the earliest DSB repair responses is phosphorylation of the Ser-139 residue within the terminal SQEY motif of the histone H2AX [26]. The phosphorylated form of H2AX, referred to as gammaH2AX (γH2AX), is produced by a reaction catalysed by the phosphatidyl-inositol 3-kinase (PI3K) family of proteins, ATM, DNA-protein kinase catalytic subunit, and ATM and RAD3-related (ATR). The protein kinases, ATM and ATR are key DNA damage response (DDR)-signalling constituents of mammalian cells [27]. γH2AX forms discrete foci consisting of an accumulation of repair proteins in large domains of chromatin around the site of the DSB that are easily visualized by immunofluorescence [28,29], providing a measure of the number of DSBs within a cell [30]. The H2AX-interacting proteins include NBS1, 53BP1 and MDC1 [31]. The observation of intense staining of γH2AX in S phase cells and faint staining in G1 cells in some studies [32] suggest that γH2AX formation possibly occurs to safeguard cell cycle checkpoints that protect the cell from genomic instability [28].
The main aim of the present study was to measure the interstrand crosslinks formation induced by satraplatin against oxaliplatin. Also to investigate the induction of DNA double strand breaks in lymphocytes obtained from colorectal cancer patients and healthy individuals in response to satraplatin and oxaliplatin treatments.
2. Material and Methods
2.1. Chemicals
Oxaliplatin (Cat No. O9512), Mitomycin C (Cat No. M0 503), 5-bromo-2-deoxy-uridine (BrdU) (B-9285), and fetal bovine serum (FBS) (Cat No. F7524) were purchased from Sigma-Aldrich (Gillingham, UK). Satraplatin (Cat No. 129580-63-8) was purchased from LGM Pharma. RPMI 1640 medium (Cat No.72400-013), phytohaemagglutinin (PHA) (Cat No.10576-015) and penicillin-streptomycin solution (Cat No.15140-122) were obtained from Invitrogen Ltd, UK. Antirabbit primary antibody γH2AX (Phospho139) (Cat No. ab2893), Chromeo™ 488 goat anti-rabbit IgG (Cat No. ab60314) were obtained from Abcam, UK. All other chemical reagents were obtained from VWR International laboratory supplies in the UK. Both satraplatin and oxaliplatin were dissolved and diluted in dimethylsulfoxide (DMSO).
2.2. Methods
2.2.1. Blood Samples and Lymphocyte Isolation
Ethical approval (Reference no.: 04Q1202/15) for colorectal cancer patients was obtained from Bradford Royal Infirmary Hospital local Ethics Committee. Additionally, ethical approval (Reference no.: 0405/8) was granted by the University of Bradford Research Ethics sub-committee for healthy individuals. Peripheral blood was obtained after informed consent from twenty colorectal cancer patients and twenty healthy non-smoking volunteers. Blood was collected and labelled in a heparinised anticoagulant tube, then transferred to a Falcon tube and diluted 50:50 with saline (0.9% NaCl). Then, 6 ml of the diluted blood was carefully layered on top of 3 ml Lymphprep (Axis-shield, Norway) without disturbing the Lymphoprep layer followed by centrifugation at 1900 rpm (800× g) for 20 minutes. The lymphocyte layer was transferred and washed in 10ml of saline followed by centrifugation at 1500 rpm (500× g) for 15 minutes. The supernatant was discarded and the pellet was resuspended in 90% foetal bovine serum. Subsequently, the suspension was stored in 1 ml cryovials containing 10% DMSO at –20˚C overnight; the next day the cryovials were transferred to –80˚C for storage until use.
2.2.2. Lymphocyte Treatment Prior to the Comet Assay and Cell Viability
The lymphocytes were thawed in a 37˚C water bath then washed twice in PBS. The pellet was resuspended in PBS in Eppendorf tubes and supplemented with different concentrations of oxaliplatin or satraplatin. The volumes of oxaliplatin and satraplatin stock solution were always kept at 1% of the final solution volume. Lymphocytes were incubated with different oxaliplatin or satraplatin end concentrations (0.02, 0.2, 2.0, 20 and 200 µM) for 1 hour at 37˚C. After incubation, the cells were centrifuged for 5 minutes at 4000 rpm (1330× g) and washed twice with PBS. After the initial treatment with oxaliplatin or satraplatin, the cells were similarly treated for 30 minutes at 37˚C with 50 µM H2O2 also in a 1000 µl final volume. The negative control was prepared without addition of oxaliplatin or satraplatin while H2O2 the positive control was prepared by incubating the lymphocytes with 50 µM H2O2 only without any addition of oxaliplatin or satraplatin.
The viability of the treated cells was measured after the initial one-hour incubation with oxaliplatin or satraplatin by using Trypan blue exclusion according to Henderson et al. [33]. Treated cells were mixed 50:50 with 0.4% Trypan blue, and then 100 cells were scored for viability using a phase-contrast microscope. Any concentration providing cell viability below 75% was excluded.
2.2.3. The Comet Assay and Data Analysis
The Comet assay was carried out as previously described [34]. In brief, treated cells were washed twice with PBS and the supernatant was discarded except for 100 µl in which the cell pellet was resuspended. The cells were then mixed with 100 µl 1% low melting point (LMP) agarose and 100 µl of this cell suspension were transferred onto each of two glass slides pre-coated with 1% normal melting point (NMP) agarose. The slides were placed on ice for 5 minutes then 100 µl 0.5% LMP agarose were added to the slides as a third agarose layer. The slides were placed on ice again for 5 minutes to allow the agarose to set. The slides were placed overnight at 4˚C in lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM Trizma base, 1% Triton X-100 and 10% DMSO at pH 10). The next day, the slides were transferred to an electrophoresis tray and submerged for 30 minutes in alkaline electrophoresis buffer (300 mM NaOH and 1 mM EDTA, pH >13) at 4˚C for DNA unwinding. The electrophoresis was carried out at 25 Volts and 300 mA (0.75 V/cm) for 30 minutes. Afterwards, the slides were washed three times for 5 minutes each in neutralising buffer (0.4 M Tris, pH 7.5), and the DNA was stained with 60 µl of ethidium bromide (20 µg/ml). The slides were evaluated by using an image analysis system (Komet 4, Kinetic Imaging, Liverpool) using a CCD camera attached to a fluorescence microscope with a final magnification of 200×. A total of 100 cells were evaluated per specimen and dose, 50 cells per duplicate slide. The Olive tail moment (OTM) was calculated as the product of the percentage DNA in the comet tail and the distance between the centres of gravity for DNA in the head and tail [35] and the data were presented as mean ±S.E. Oxaliplatin or satraplatin crosslinks were concluded as a decrease in percentage of the OTM compared to that of hydrogen peroxide (H2O2) [36].
2.2.4. Cell Culture
Under sterile conditions, 500 µl of whole blood was cultured in 25 cm2 culture flasks containing 4.5 ml RPMI 1640 medium with Glutamax-I, 20% foetal bovine serum (FBS) and 1% penicillin-streptomycin solution. PHA was added to reach a 2.5% final concentration in order to stimulate T-lymphocyte proliferation [37]. For the SCE assay, 40 µl of 1.25 mM BrdU were additionally added to each culture. The cultures were incubated at 37˚C in the presence of 5% CO2 in air for 48 hours. Oxaliplatin or satraplatin were added to the cultures at final concentrations of 0.02, 0.2, and 2 µM after 24 hours of incubation time [38]. The cells were arrested in metaphase stage after 45 hours by adding colcemid at a final concentration of 0.15 µg/ml. The negative control was cultured without the addition of oxaliplatin or satraplatin. Mitomycin C (final concentration of 0.4 µM) was used as a positive control. Each experiment was done in duplicate with both controls included [39].
2.2.5. Sister Chromatid Exchange
The assay carried out as previously described [40].The culture medium was transferred into Falcon tubes and centrifuged at 1000 rpm (330× g) for 10 minutes. The cells were incubated in a hypotonic solution (75 mM KCl) for 15 minutes at 37˚C followed by a further centrifugation step. The cells were incubated three times in fixative (Carnoy’s solution = 1:3 acetic acid/methanol), each time followed by a centrifugation step. The cells were kept at 4˚C overnight in fixative. Slides with metaphase spreads were prepared by dropping 2 equally distant spots each of 20 µl cell suspension onto clean glass slides. Slides were left to “age” for one week at 37˚C. For SCE detection, the slides were placed in 1µg/ml bisbenzimide for 25 minutes. After a short wash in water, slides were placed under a UV A/B lamp for 90 minutes covered with PBS buffer. Next, the slides were transferred in 2 × SSC at 60˚C for 30 minutes, followed by staining in 5% Giemsa in phosphate buffer for 5 minutes. Slides were left to dry before mounting with DPX. Two slides were prepared per culture and a total of 50 metaphases were evaluated per treatment. All slides were blindly coded prior to scoring using a bright-field light microscope with 1000× magnification.
The proliferation rate index (PRI) was determined according to the formula: PRI = (M1 + 2M2 + 3M3)/N; where M1, M2 and M3 reflect the first, second and the third mitosis respectively while N represents the total cells scored.
2.2.6. Immunofluorescence Detection
Lymphocyte Treatment Separated lymphocytes were incubated with different concentrations (0.02, 0.2 and 2 µM) of oxaliplatin or satraplatin as a final concentration for 1h at 37˚C in Eppendorf ® tubes containing 79% of RPMI 1640 media with Glutamax-I, 20% foetal bovine serum (FBS) and 1% penicillin-streptomycin solution. Next the cells were centrifuged for 5 minutes at 4000 rpm (1330× g) and the cells were washed twice with PBS then cells were resuspended in RPMI 1640 media and incubated at 37˚C for 16 hours [41].
The washing steps with PBS were repeated twice then the cells were resuspended in 100 µl PBS and dropped on clean slides. Once the slides dried the cells were fixed in 4% formaldehyde solution for 15 minutes at room temperature (RT). The cells were washed with PBS for 5 minutes three times followed by incubation in permeabilising solution (0.1% Triton X-100 in PBS) for 10 minutes at RT. The unspecific binding sites were blocked for one hour in antibody blocking buffer (1% BSA in PBT). The cells were incubated with the anti-rabbit primary antibody γH2AX (Phospho139) (Abcam, 2893) in antibody blocking buffer (1:100) for 45 minutes at RT then the cells were incubated with Chromeo™ 488 goat antirabbit IgG (Abcam, 60314) in 1% BSA/PBT (1:250) for 45 minutes in the dark after washing twice with PBS. Afterwards, the cells were washed three times with PBS, and the nuclear DNA was stained with DAPI (5 mg/ml). ProLong® Gold antifade reagent was used as mounting media then the slides were covered by cover slips and left to dry on a flat surface in the dark. Untreated cells were used as the negative control without the addition of oxaliplatin or satraplatin. Ethyl methanesulfonate (EMS) (final concentration of 1.5 mM) was used as a positive control [42]. Each experiment was done in duplicate with both controls included A total of 100 cells was analysed by a fluorescence microscope connected with a CCD camera (Nikon Digital Sight DS-SMC, Surrey, UK) with 1000× magnification. Two slides were prepared per treatment and all of them were coded prior to analysis.
2.2.7. Statistical Analysis
Statistical analysis was carried out by using the statistical software SPSS 13.0. The data were presented as mean ±SE and the data normality was assessed by using normal probability plots. To analyse the significance between the different treatment concentrations, the data were analysed by one-way ANOVA followed by Dunnett’s post hoc test for significant differences. The level of significance was considered at p < 0.05.
3. Results
3.1. DNA Crosslinks Formation Induced by Oxaliplatin and Satraplatin
To qualitatively measure the DNA crosslinking action of oxaliplatin and satraplatin in vitro, the treatment of lymphocytes for the alkaline Comet assay was modified. This modification allowed the detection of the formation of DNA crosslinks via the pseudo-reduction in DNA damage induced by H2O2. Colorectal cancer patient lymphocytes were treated with increasing concentrations of either oxaliplatin or satraplatin (0.02 µM - 200 µM). Despite an increase in the oxaliplatin and satraplatin concentrations, our data showed that in vitro treatment of lymphocytes resulted in a dose-dependent decrease of induced DNA damage, measured with the Comet parameter Olive tail moment (OTM) (Figure 1(a)). Oxaliplatin concentrations of 0.02, 2, and 200 µM resulted in approximately 44, 66, and 70% decreased OTM, respecttively, compared to the H2O2 control. The data were statistically significant with p < 0.01.
The OTM reduction induced by satraplatin was 49, 67, and 75% when the lymphocytes from colorectal cancer patients were treated correspondingly with 0.02, 2, and 200 µM. The OTM reduction was statistically significant with satraplatin concentrations relative to control treatment with H2O2 (p < 0.01).
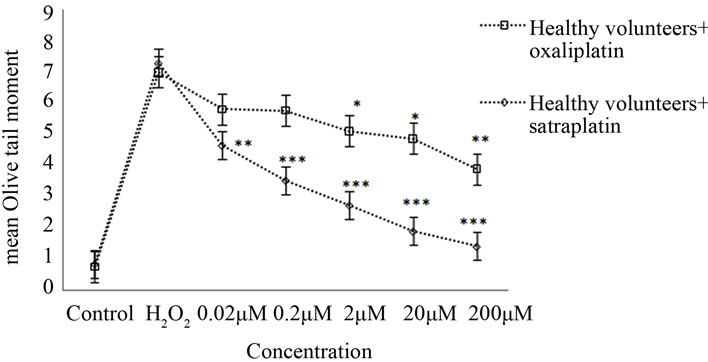
In healthy volunteers, oxaliplatin treatments of 0.2, 2, 20, and 200 µM resulted in an apparent reduction of OTM by approximately 18%, 25% (p < 0.05), 30% (p < 0.05), and 44% (p < 0.01), respectively, when compared to the positive control (50 µM H2O2) (Figure 1(b)).
Healthy lymphocytes treated with various satraplatin concentrations produced a statistically significant reduction in mean OTM with p < 0.01. Further, the rate of
 (a)
(a) (b)
(b)
Figure 1. Cross-links induced by satraplatin and oxaliplatin. (a) Lymphocytes from colorectal cancer patients, untreated (control) or treated with different concentrations of satraplatin or oxaliplatin followed by treatment with 50 µM H2O2; (b) Lymphocytes from healthy control individuals, untreated (control) or treated with different concentrations of satraplatin or oxaliplatin. The data are presented as the mean ± SE of Olive tail moments (OTM). Data normality was assessed using normal probability plots. The data were analysed by one-way ANOVA followed by Dunnett’s post hoc test for significant differences compared to the positive control, 50 µM H2O2 (*p < 0.05, **p < 0.01, ***p < 0.001).
decrease was inversely proportional to the concentration of satraplatin used. Thus, while the least reduction in mean OTM of ~30% was observed with 0.02 μM satraplatin, the maximum reduction of ~75% was noted in the case of healthy lymphocytes incubated with 200 μM satraplatin (Figure 1(b)).
3.2. Sister Chromatid Exchange (SCE) Induction by Oxaliplatin and Satraplatin
The DNA damage induced by oxaliplatin and satraplatin was investigated with the sister chromatid exchange (SCE) assay. SCE data for colorectal cancer patients and healthy control lymphocytes incubated with oxaliplatin or satraplatin for 48 hours are summarised in Table 1. The number of SCEs formed per metaphase was significantly increased in colorectal cancer lymphocytes from the 6.5-fold (p < 0.01) to 8.5-fold (p < 0.001) when treated with 0.2 µM and 2 µM satraplatin correspondingly in comparison to untreated colorectal cancer cell (control). In addition, the SCEs frequencies induced by satraplatin increased approximately 1.7-fold in case of both oxaliplatin treatments (0.2 µM and 2 µM).
The proliferative rate index (PRI) decreased from the untreated colorectal cancer cell (control) value of 2.51 ± 0.37 to 1.82 ± 0.22 and 1.49 ± 0.83 in the presence of 2 μM of either oxaliplatin or satraplatin, respectively.
Incubation of lymphocytes obtained from healthy volunteers with concentration of 0.2 µM either of oxaliplatin or satraplatin significantly increased the mean number of exchanges to approximately 17 and 30 SCE per metaphase, respectively, compared to 2 SCE per metaphase in untreated lymphocytes. The data indicated that when compared to the negative control the induced SCE frequency significantly increased by 7-fold (p < 0.01) and 12-fold (p < 0.001) in the treatment of 2 µM of oxaliplatin and satraplatin, correspondingly (Table 1).

Table 1. Sister Chromatid Exchange (SCE). The mean of the number of SCE ± SE was measured between healthy controls and colorectal cancer patients’ cells treated with oxaliplatin and satraplatin. The results were normally distributed. Therefore, the data were analysed by one-way ANOVA followed by Dunnett’s post hoc test for significant differences compared to the untreated control as appropriate for each group (*p < 0.01,**p< 0.01, ***p < 0.001).
3.3. Phosphorylation of Histone H2AX Induced by Oxaliplatin and Satraplatin
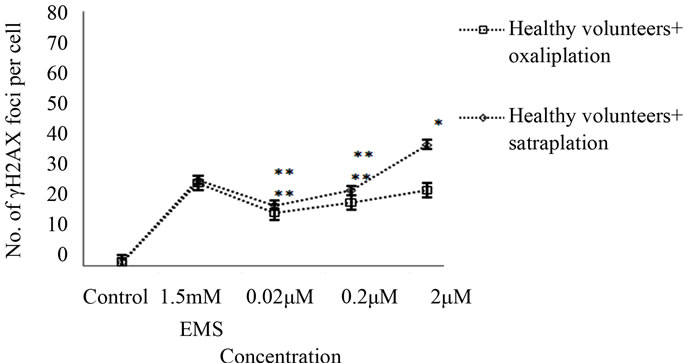
The γH2AX foci formation was investigated by the immunofluorescence assay in lymphocytes gained from colorectal cancer patients and healthy volunteers. As shown in Figure 2, an increased number of γH2AX foci formations were observed after treatment of the lymphocytes with oxaliplatin or satraplatin. In colorectal cancer cells, treatment with 0.02 μM oxaliplatin was enough to generate more of γH2AX foci than in untreated cells by 4.5- fold. Additionally, foci formation rate was increased by approximately 7-fold when lymphocytes treated with 0.2 μM and by 9-fold with 2 μM oxaliplatin (Figures 3(a) and (b)). In comparison to the positive EMS-treated cells 2 μM oxaliplatin increased the number of γH2AX foci observed per cell nearly by 29%. Foci yielded per cell were significantly enhanced with the highest concentration of oxaliplatin used with p < 0.001 and p < 0.01 in all other cases.
After satraplatin treatments in cancer cells, the incidence of the foci formation was increased with the increased concentrations of satraplatin (Figure 3(a)). The γH2AX foci induction values indicated a good correlation (r = 0.86) with the concentrations of 0.02 µM, 0.2 µM and 2 µM satraplatin. Compared to the number of foci generated by control, both 0.2 μM and 2.0 μM satraplatin registered significant increases in the number of γH2AX foci by 8.5-fold and 11-fold, respectively. The induction of γH2AX foci was statistically significant (p < 0.01) with 0.02 μM satraplatin, and p < 0.001 in both of the concentrations of satraplatin 0.2 μM and 2.0 μM.
Treatment of healthy cells with the 0.02 µM, 0.2 µM, and 2 µM concentrations of oxaliplatin produced a higher induction of foci by 17%, 20%, and 24%, respectively, compared to untreated healthy cells (Figure 3(b)).
The formation of γH2AX foci increased from 2% in untreated healthy cells to 38% when treated with 2 µM satraplatin. As indicated, the treatment of healthy cells with satraplatin was observed to induce a significantly (p < 0.01) higher γH2AX foci formation in healthy cells as against those healthy cells that received no treatment.
4. Discussion
Satraplatin (JM-216), or bis (acetato) ammine dichloro (cyclohexylamine) platinum (IV), is a novel platinum (IV) compound that is administrated orally unlike all platinum (II) class of compounds. Upon administration of satraplatin, a number of metabolites (JM118, JM383, JM518, JM559 and JM149) are formed [43]. Satraplatin and other platinum-based compounds exhibit anti-tumour activity via the formation of DNA crosslinks in vitro [44] which in turn results in specific cellular responses, namely, apoptosis and inhibition of transcription leading to arrest
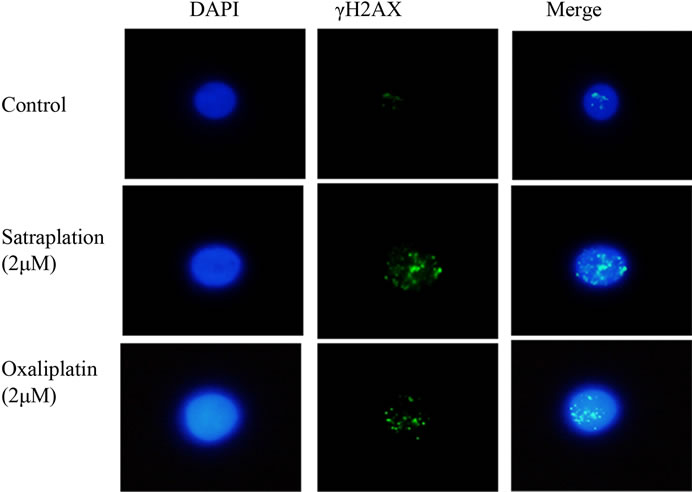
Figure 2. γH2AX phosphorylation in lymphocytes from colorectal cancer patients presented in untreated cells (control) and treated cells with 2 µM either of satraplatin or oxaliplatin. DNA is counterstained with DAPI and the nuclei size may vary due to treatment. The image is analysed with Nikon Digital Sight DS-SMC, Surrey, UK with 1000× magnification.
 (a)
(a)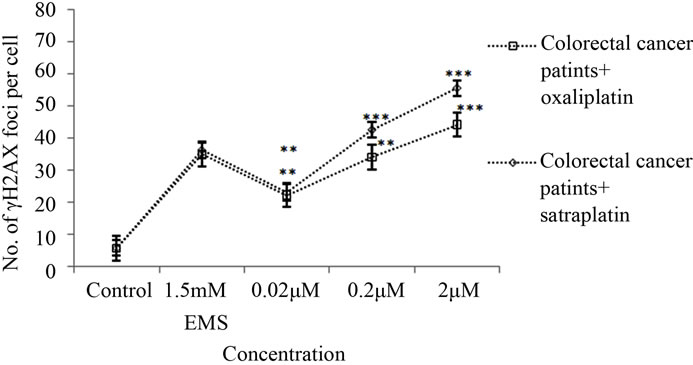 (b)
(b)
Figure 3. γH2AX foci induction by satraplatin and oxaliplatin in lymphocytes from (a) colorectal cancer patient or (b) healthy controls after treatment with different concentrations of satraplatin or oxaliplatin. Data were analysed by one-way ANOVA followed by Dunnett’s post hoc test for significant differences compared to the untreated control for each group (*p < 0.05, **p < 0.01, ***p < 0.001).
of cell replication [45-47]. It has been amply demonstrated that the primary mechanism of cell death induced by platinum compounds is through the formation of DNA adducts [45,48,49]. The present study investigates the effect of oxaliplatin versus satraplatin in terms of interstrand crosslinking action, SCE frequencies in S phase and induction of DNA double-strand breaks detected by γH2AX foci formation in lymphocytes from colorectal cancer patients and healthy individuals in vitro.
Peripheral blood lymphocytes appear to be the cells of choice due to the fact that in a variety of cancers, these cells demonstrate basal DNA damage [50] Many chemoand radiotherapeutic agents have consequently been stu0 died to establish increased baseline DNA damage and diminished repair efficiency following exposure in cancer patients [51]. These appear to be the best choice of surrogate human cells due to the fact that the collection of lymphocytes is sufficiently non-invasive in comparison to collection of other target tissues.
Using a modified treatment for the alkaline Comet assay the induction of crosslinks by satraplatin versus oxaliplatin was investigated. Hence, lymphocytes were pre-treated in vitro with different concentrations of satraplatin or oxaliplatin (0.02 µM - 200 µM) to induce crosslinks followed by an exposure to 50 µM H2O2 which reduced alkali labile sites as well as strongly induced single-and double-strand breaks in the DNA. The reduced detection of these breaks due to crosslinked DNA served to identify the action of a cross-linking agent using the Comet assay. Our results showed a dose-dependent decrease in OTM up to 75% (p < 0.01) for the highest concentration of satraplatin; i.e. a decrease of the migration of DNA fragments and open ends in the electrophoretic field due to an increased amount of crosslinks (Figures 1(a) and (b)). However, a direct relation between satraplatin-induced DNA crosslinks and satraplatin concentrations has also been detected. This could be explained by the fact that satraplatin is capable of forming intrastrand and interstrand crosslinks between the adjacent purine bases, which is similar to how cisplatin binds to the DNA [52]. Oxaliplatin forms intrastrand crosslinks between two adjacent guanine residues and between guanine-adenosine mainly at the reactive N7- position. Oxaliplatin also forms interstrand crosslinks, which represent 10% of the total number of adducts [53]. The mode of action of many chemotherapeutic drugs of importance in cancer treatment is through production of DNA interstrand crosslinks detectible by the comet assay in cells exposed to the drug [54]. It has recently been demonstrated that oxaliplatin-induced crosslinks were detected by the alkaline Comet assay using a H460 tumour cell line and patients’ lymphocytes. This suggests that the Comet assay provides the means for predicting patient response to chemotherapy with platinum compounds [45]. Similar studies indicated that the alkaline Comet assay is widely applicable for detecting DNA cross-linking action induced by a variety of DNA cross-linking agents [36, 55].
To determine whether the DNA damage induced by satraplatin and oxaliplatin has an effect on chromosomes during S phase, the SCE assay was employed. Here, to our knowledge, we report for the first time that treatment of lymphocytes from colorectal cancer patients with 2 µM satraplatin was sufficient to induce a significant 8.5- fold increase (p < 0.001) in the number of sister chromatid exchanges (Table 1). On an equimolar basis, the data indicated that SCE frequencies induced by satraplatin was a 1.7-fold more than oxaliplatin induced SCE in both colorectal cancer cells and healthy cells when treated with 0.2 µM and 2 µM. It is long known, that Satraplatin was found 2 - 7 times more potent than cisplatin for lung cancer cell lines [17]. Another study showed that JM 118 one of satraplatin metabolites was up to 16 times more potent against prostate cancer cell lines than its parent satraplatin [15,16]. Our data from colorectal cancer cells indicated that the proliferation rate index (PRI) decreased significantly up to 1.49 ± 0.83 (p < 0.01) and 1.82 ± 0.22 (p < 0.001) when treated with satraplatin and oxaliplatin in the highest concentration. Interstrand crosslink repair would necessitate both nucleotide excision and recombinational repair pathways [56]. During replication, the decreased DNA synthesis is partially responsible in the chemotherapeutic effect of drugs which form bulky adducts on the DNA. The postreplication repair pathway in mammalian cells is one possible way that allows cells to overcome replication blocks caused by these adducts [57].
Adduct lesions induced by satraplatin and oxaliplatin may bypass the replication process successfully after producing a replication fork collapse. DNA replication continues even in the presence of DNA adducts leading to DNA translesion synthesis and to miscoding errors in the newly synthesised DNA [58]. Platinum-adducts may be successfully bypassed by translesional DNA polymerases [59] and several translesional DNA polymerases are capable of even bypassing platinum-adducts [22,60, 61]. DNA polymerase β is able to synthesise DNA with greater efficiency in the presence of oxaliplatin adducts than those of cisplatin [57]. The efficiency and fidelity of such a bypass of these induced adducts by these translesional polymerases is likely to have an effect on mutagenicity.
In order to investigate DNA double-strand break induction induced by satraplatin, immunofluorescence detection of γH2AX foci formation was used. γH2AX is a sensitive marker of DNA double-strand breaks produced by chemotherapeutics that cause double-strand breaks [26]. In the present study, treatment of lymphocytes from colorectal cancer patients with different concentrations of oxaliplatin and satraplatin caused significant DNA double-strand breaks as observed from the data on γH2AX foci in individual cells (Figures 3(a) and (b)). Our data indicated that the damage caused by satraplatin followed a dose-related linear increase in the number of γH2AX foci produced per cell. Furthermore, the γH2AX foci formation varied linearly with the oxaliplatin dose employed. To the best of our knowledge, these results represent for the first time that γH2AX foci formation was detected in lymphocytes either from colorectal cancer or from healthy individuals after satraplatin treatments. The measurement of γH2AX is closely related to the number of DNA strand breaks, and level of cell death [62]. Therefore, based on the results obtained in the present study it can be inferred that satraplatin causes more DNA doublestrand breaks and, perhaps, cell death as compared to equimolar oxaliplatin. The reason for this could be the higher interstrand crosslinks formation and cellular accumulation exhibited by satraplatin as compared with oxaliplatin. Satraplatin potentially produces DNA adducts that are more resistant to DNA nucleotide excision repair enzymes [63]. A recent study reported a fourfold higher accumulation of satraplatin than did oxaliplatin with CRC cell lines [19].
Following exposure to genotoxic agents, DNA replication is generally blocked through inhibition of replication initiation and through inhibition of DNA chain elongation when high doses of genotoxicants are used [64]. Many ICL agents can also obstruct DNA replication fork progression in dividing cells, resulting in the formation of DSBs [41]. It is believed that the activity of endonucleases at the sites of stalled replication forks causes DSBs [65]. The blockage of DNA replication results in the formation of γH2AX foci at sites of stalled replication forks [66]. Several proteins support the replication fork in its action of correction of replication errors or DNA strand breakages so that the cell cycle can continue. The stalled replication forks can resume DNA synthesis after the replication block has been removed, that is, after DNA damage has been resolved [67]. It is yet to be confirmed whether the generation of γH2AX at the sites of stalled replication forks is triggered by the stalled replication forks themselves [66] or occurs as a result of DSB formation [68]. Time course experiments conducted on a Chinese hamster ovary cell line exposed to ionising radiation and alkylating agents showed a decrease in the γH2AX foci number at sites of replication forks that undergo repair; however, unrepaired DSBs are likely to cause chromosomal aberrations if a cell undergoes mitosis, subsequently leading to cell death [69].
The results of this study showing significantly higher levels of γH2AX foci in lymphocytes of colorectal cancer patients in relation to lymphocytes from healthy donors would indicate that the repair system in cancer cells experiences a loss of ability to cope with the repair of DSBs when treated with either oxaliplatin or satraplatin. Persistence of γH2AX foci after exposure to interstrand crosslinking agents such as Pt-based compounds cisplatin, oxaliplatin, and satraplatin, could reflect a defective homologous repair system that is unable to repair replication-associated DSBs [41].
The clinical diagnostic uses of γH2AX are based on the fact that γH2AX levels essentially reflect endogenous genomic instability in tissues and, as such, serve to indicate pre-cancerous lesions [70]. It may be more relevant to measure DSBs for anticancer agents whose efficacy is dependent on the patient’s metabolism. A patient’s sensitivity to ICL cancer anticancer therapeutics could be evaluated by γH2AX measurements done in peripheral blood lymphocytes or skin samples at different times during or after treatment [70]. Following radiotherapy, in vivo measurements of γH2AX in white blood cells showed a very good linear correlation between the number of γH2AX foci per lymphocytes and radiation dose [71]. While the level of DSBs as measured by the surrogate γH2AX in blood cells may provide information on the effect of anticancer therapy, the response of tumour cells to the same treatment could be dependent on several factors including an altered gene expression and the proportion of S-phase cells [70].
In conclusion, satraplatin effectively induced DNA interstrand crosslinks in lymphocytes obtained from colorectal cancer patients and healthy volunteers in vitro. Phosphorylation of H2AX is a process that is crucial for the recognition and repair of DNA double strand breaks. The measurement of γH2AX is closely related to the number of DNA strand breaks. An important clinical application of γH2AX measurements is to determine the levels DSBs generated by chemotherapeutic compounds as a marker of treatment efficacy. Further, γH2AX measurements would be useful to determine the sensitivity or resistance to DNA damaging anticancer agents.
REFERENCES
- M. L. G. Dronkert and R. Kanaar, “Repair of DNA Interstrand Cross-Links,” Mutation Research/DNA Repair, Vol. 486, No. 4, 2001, pp. 217-247. doi:10.1016/S0921-8777(01)00092-1
- D. M. Noll, T. M. Mason and P. S. Miller, “Formation and Repair of Interstrand Cross-Links in DNA,” Chemical Reviews, Vol. 106, No. 2, 2005, pp. 277-301. doi:10.1021/cr040478b
- C. F. O'Neill, B. Koberle, J. R. W. Masters and L. R. Kelland, “Gene-Specific Repair of Pt/DNA Lesions and Induction of Apoptosis by the Oral Platinum Drug JM216 in Three Human Ovarian Carcinoma Cell Lines Sensitive and Resistant to Cisplatin,” The British Journal of Cancer, Vol. 81, No. 8, 1999, pp. 1294-1303. doi:10.1038/sj/bjc/6694381
- L. Kelland, “The resurgence of platinum-based cancer chemotherapy,” Nature Reviews Cancer, Vol. 7, No. 8, 2007, pp. 573-584. doi:10.1038/nrc2167
- C. Martin, T. Ellis, C. J. McGurk, T. C. Jenkins, J. A. Hartley, M. J. Waring and D. E. Thurston, “Sequence-Selective interaction of the minor-groove interstrand cross-linking agent SJG-136 with naked and cellular DNA: Footprinting and enzyme inhibition studies,” Biochemistry, Vol. 44, No. 11, 2005, pp. 4135-4147. doi:10.1021/bi0479813
- E. Reed, “ERCC1 Measurements in Clinical Oncology,” The New England Journal of Medicine, Vol. 355, No. 10, 2006, pp. 1054-1055. doi:10.1056/NEJMe068162
- E. Donzelli, M. Carfi, M. Miloso, A. Strada, S. Galbiati, M. Bayssas, G. Griffon-Etienne, G. Cavaletti, M. G. Petruccioli and G. Tredici, “Neurotoxicity of platinum compounds: comparison of the effects of cisplatin and oxaliplatin on the human neuroblastoma cell line SH-SY5Y,” Journal of Neuro-Oncology, Vol. 67, No. 1-2, 2007, pp. 65-73. doi:10.1023/B:NEON.0000021787.70029.ce
- A. Stachurska, M. Dudkowska, A. Czopek, M. ManteuffelCymborowska and B. Grzelakowska-Sztabert, “Cisplatin Up-Regulates the in Vivo Biosynthesis and Degradation of Renal Polyamines and c-Myc Expression,” Biochimica et Biophysica Acta (BBA)—Molecular Basis of Disease, Vol. 1689, No. 3, 2004, pp. 259-266.
- M. Raschle, P. Knipsheer, M. Enoiu, T. Angelov, J. C. Sun, J. D. Griffith, T. E. Ellenberger, O. D. Scharer and J. C. Walter, “Mechanism of Replication-Coupled DNA Interstrand Crosslink Repair,” Cell, Vol. 134, No. 6, 2008, pp. 969-980. doi:10.1016/j.cell.2008.08.030
- X. Wang, C. A. Peterson, H. Y. Zheng, R. S. Nairn, R. J. Legerski and L. Li, “Involvement of Nucleotide Excision Repair in a Recombination-Independent and Error-Prone Pathway of DNA Interstrand Cross-Link Repair,” Molecular and Cellular Biology, Vol. 21, No. 3, 2001, pp. 713-720. doi:10.1128/MCB.21.3.713-720.2001
- D. M. Kweekel, H. Gelderblom and H. J. Guchelaar, “Pharmacology of oxaliplatin and the use of pharmacogenomics to individualize therapy,” Cancer Treatment Reviews, Vol. 31, No. 2, 2005, pp. 90-105. doi:10.1016/j.ctrv.2004.12.006
- L. R. Kelland, G. Abel, M. J. McKeage, M. Jones, P. M. Goddard, M. Valenti, B. A. Murrer and K. R. Harrap, “Preclinical Antitumor Evaluation of Bis-acetato-amminedichloro-cyclohexylamine Platinum (iv)—An Orally Active Platinum Drug,” Cancer Research, Vol. 53, No. 11, 1993, pp. 2581-2586.
- H. Choy, C. Park and M. Yao, “Current status and future prospects for satraplatin, an oral platinum analogue,” Clinical Cancer Research, Vol. 14, No. 6, 2008, pp. 1633-1638. doi:10.1158/1078-0432.CCR-07-2176
- L. Martelli, F. Di Mario, E. Ragazzi, P. Apostoli, R. Leone, P. Perego and G. Fumagalli, “Different Accumulation of cisplatin, oxaliplatin and JM216 in sensitive and cisplatin-resistant human cervical tumour cells,” Biochemical Pharmacology, Vol. 72, No.6 , 2006, pp. 693-700. doi:10.1016/j.bcp.2006.06.008
- K. Wosikowski, L. Lamphere, G. Unteregger, V. Jung, F. Kaplan, J. P. Xu, B. Rattel and M. Caligiuri, “Preclinical antitumor activity of the oral platinum analog satraplatin,” Cancer Chemotherapy and Pharmacology, Vol. 60, No. 4, 2007, pp. 589-600. doi:10.1007/s00280-007-0502-z
- K. Wosikowski, F. Obermayr and B. Rattel, “Efficacy of satraplatin, an oral platinum analogue in human breast cancer models: synergistic activity with capecitabine,” Breast Cancer Research and Treatment, Vol. 100, 2006, pp. S74-S74.
- P. R. Twentyman, K. A. Wright, P. Mistry, L. R. Kelland and B. A. Murrer, “sensitivity to Novel Platinum Compounds of Panels of Human Lung-Cancer Cell-Lines with Acquired and Inherent Resistance to Cisplatin,” Cancer Research, Vol. 52, 1992, pp. 5674-5680.
- NIC Registry, “Cancer Incidence and Mortality,” 2010.
- M. Kalimutho, A. Minutolo, S. Grelli, A. Formosa, G. Sancesario, A. Valentini, G. Federici and S. Bernardini, “Satraplatin (JM-216) mediates G2/M cell cycle arrest and potentiates apoptosis via multiple death pathways in colorectal cancer cells thus overcoming platinum chemoresistance,” Cancer Chemotherapy and Pharmacology, Vol. 67, No. 6, 2011, pp. 1299-1312. doi:10.1007/s00280-010-1428-4
- J. T. Reardon, A. Vaisman, S. G. Chaney and A. Sancar, “Efficient Nucleotide Excision Repair of Cisplatin, Oxaliplatin, and Bis-aceto-ammine-dichloro-cyclohexylamineplatinum (IV) (JM216) Platinum Intrastrand DNA Diadducts,” Cancer Research, Vol. 59, No. 16, 1999, pp. 3968- 3971.
- D. Fink, S. Nebel, S. Aebi, H. Zheng, B. Cenni, A. Nehme, R. D. Christen and S. B. Howell, “The role of DNA mismatch repair in platinum drug resistance,” Cancer Research, Vol. 56, No. 21, 1996, pp. 4881-4886.
- A. Vaisman, S. E. Lim, S. M. Patrick, W. C. Copeland, D. C. Hinkle, J. J. Turchi and S. G. Chaney, “Effect of DNA polymerases and high mobility group protein 1 on the carrier ligand specificity for translesion synthesis past platinum-DNA adducts,” Biochemistry, Vol. 38, No. 34, 1999, pp. 11026-11039. doi:10.1021/bi9909187
- S. P. Jackson and J. Bartek, “The DNA-damage Response in human biology and disease,” Nature, Vol. 461, No. 7267, 2009, pp. 1071-1078. doi:10.1038/nature08467
- K. K. Khanna and S. P. Jackson, “DNA Double-Strand Breaks: Signaling, Repair and the Cancer Connection,” Nature Genetics, Vol. 27, No. 3, 2001, pp. 247-254. doi:10.1038/85798
- N. Kongruttanachok, C. Phuangphairoj, A. Thongnak, W. Ponyeam, P. Rattanatanyong, W. Pornthanakasem and A. Mutirangura, “Research Replication Independent DNA Double-Strand Break Retention May Prevent Genomic Instability,” Molecular Cancer, Vol. 9, No. 1, 2010, p. 70.
- E. P. Rogakou, D. R. Pilch, A. H. Orr, V. S. Ivanova and W. M. Bonner, “DNA Double-Stranded Breaks Induce Histone H2AX Phosphorylation on Serine 139,” Journal of Biological Chemistry, Vol. 273, No. 10, 1998, pp. 5858- 5868. doi:10.1074/jbc.273.10.5858
- Y. Shiloh, “ATM and related protein kinases: Safeuarding Genome Integrity,” Nature Reviews Cancer, Vol. 3, No. 3, 2003, pp. 155-168. doi:10.1038/nrc1011
- I. H. Ismail and M. J. Hendzel, “The Gamma-H2A.X: Is it just a surrogate marker of double-strand breaks or much more?” Environmental and Molecular Mutagenesis, Vol. 49, No. 1, 2008, pp. 73-82. doi:10.1002/em.20358
- R. S. Vasireddy, M. M. Tang, L.-J. Mah, G. T. Georgiadis, A. El-Osta and T. C. Karagiannis,” Evaluation of the spatial distribution of gammaH2AX following ionizing radiation,” Journal of Visualized Experiments, No. 47, 2010, pp. 4-7.
- I. H. Ismail, T. I. Wadhra and O. Hammarsten, “An optimized method for detecting gamma-H2AX in blood cells reveals a significant interindividual variation in the gamma-H2AX response among humans,” Nucleic Acids Research, Vol. 35, No. 2, 2007, p. e36.
- M. Stucki, J. A. Clapperton, D. Mohammad, M. B. Yaffe, S. J. Smerdon and S. P. Jackson, “MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks,” Cell, Vol. 123, No. 7, 2005, pp. 1213-1226. doi:10.1016/j.cell.2005.09.038
- T. M. Marti, E. Hefner, L. Feeney, V. Natale and J. E. Cleaver, “H2AX phosphorylation within the G(1) Phase after UV Irradiation Depends on Nucleotide Excision Repair and Not DNA Double-Strand Breaks,” Proceedings of the National Academy of Sciences of USA, Vol. 103, No. 26, 2000, pp. 9891-9896. doi:10.1073/pnas.0603779103
- L. Henderson, A. Wolfreys, J. Fedyk, G. Bourner and S. Windebank, “The ability of the Comet assay to discriminate between genotoxins and cytotoxins,” Mutagenesis, Vol. 13, No. 1, 1998, pp. 89-94. doi:10.1093/mutage/13.1.89
- R. R. Tice, E. Agurell, D. Anderson, B. Burlinson, A. Hartmann, H. Kobayashi, Y. Miyamae, E. Rojas, J. C. Ryu and Y. F. Sasaki, “Single Cell Gel/Comet Assay: Guidelines for in Vitro and in Vivo Genetic Toxicology Testing,” Environmental and Molecular Mutagenesis, Vol. 35, No. 3, 2000, pp. 206-221. doi:10.1002/(SICI)1098-2280(2000)35:3<206::AID-EM8>3.0.CO;2-J
- P. L. Olive, P. J. Johnston, J. P. Banath and R. E. Durand, “The Comet Assay: A New Method to Examine Heterogeneity Associated with Solid Tumors,” Nature Medicine, Vol. 4, No. 1, 1998, pp. 103-105. doi:10.1038/nm0198-103
- M. Volpato, J. Seargent, P. M. Loadman and R. M. Phillips, “Formation of DNA Interstrand Cross-Links as a Marker of Mitomycin C Bioreductive Activation and Chemo Sensitivity,” European Journal of Cancer, Vol. 41, No. 9, 2005, pp. 1331-1338. doi:10.1016/j.ejca.2005.03.014
- A. Baumgartner, T. E. Schmid, E. Cemeli and D. Anderson, “Parallel evaluation of doxorubicin-induced genetic damage in human lymphocytes and sperm using the comet assay and spectral karyotyping,” Mutagenesis, Vol. 19, No. 4, 2004, pp. 313-318. doi:10.1093/mutage/geh032
- D. Meynard, V. Le Morvan, J. Bonnet and J. Robert, “Functional analysis of the gene expression profiles of colorectal cancer cell lines in relation to oxaliplatin and cisplatin cytotoxicity,” Oncology Reports, Vol. 17, No. 5, 2007, pp. 1213-1221.
- M. Fenech, “Cytokinesis-Block Micronucleus Cytome Assay,” Nature Protocols, Vol. 2, 2007, pp. 1084-1104. doi:10.1038/nprot.2007.77
- S. Wolff and P. Perry, “Differential Giemsa Staining of Sister Chromatids and Study of Sister Chromatid Exchanges without Autoradiography,” Chromosoma, Vol. 48, No. 4, 1974, pp. 341-353. doi:0.1007/BF00290991
- P. H. Clingen, J. Y. H. Wu, J. Miller, N. Mistry, F. Chin, P. Wynne, K. M. Prise and J. A. Hartley, “Histone H2AX phosphorylation as a molecular pharmacological marker for DNA interstrand crosslink cancer chemotherapy,” Biochemical Pharmacology, Vol. 76, No. 1, 2008, pp. 19-27. doi:10.1016/j.bcp.2008.03.025
- T. Nikolova, M. Ensminger, M. Löbrich and B. Kaina, “Homologous recombination protects mammalian cells from replication-associated DNA double-strand breaks arising in response to methyl methanesulfonate,” DNA Repair, Vol. 9, No. 10, 2010, pp. 1050-1063. doi:10.1016/j.dnarep.2010.07.005
- F. I. Raynaud, P. Mistry, A. Donaghue, G. K. Poon, L. R. Kelland, C. F. J. Barnard, B. A. Murrer and K. R. Harrap, “Biotransformation of the Platinum Drug JM216 Following Oral Administration to Cancer Patients,” Cancer Chemotherapy and Pharmacology, Vol. 38, No. 2, 1996, pp. 155-162. doi:10.1007/s002800050464
- K. J. Mellish, C. F. J. Barnard, B. A. Murrer and L. R. Kelland, “dna-Binding Properties of Novel cisand trans Platinum-Based Anticancer Agents in 2 Human Ovarian-Carcinoma Cell-Lines,” International Journal of Cancer, Vol. 62, No. 6, 1995, pp. 717-723. doi:10.1002/ijc.2910620612
- G. M. Almeida, T. L. Duarte, W. P. Steward and G. D. D. Jones, “Detection of Oxaliplatin-Induced DNACrosslinks in Vitro and in Cancer Patients Using the Alkaline Comet Assay,” DNA Repair, Vol. 5, No. 2, 2006, pp. 219-225. doi:10.1016/j.dnarep.2005.09.010
- R. C. Todd and S. J. Lippard, “Inhibition of Transcription by Platinum Antitumor Compounds,” Metallomics, Vol. 1, No. 4, 2009, pp. 280-291. doi:10.1039/b907567d
- V. Brabec, “DNA Modifications by Antitumor Platinum and Ruthenium Compounds: Their recognition and repair,” In: Progress in Nucleic Acid Research and Molecular Biology, Academic Press, Cambridge, 2002, pp. 1-68.
- S. S. Hah, R. A. Sumbad, R. W. de Vere White, K. W. Turteltaub and P. T. Henderson, “Characterization of Oxaliplatin-DNA Adduct Formation in DNA and Differentiation of Cancer Cell Drug Sensitivity at Microdose Concentrations,” Chemical Research in Toxicology, Vol. 20, No. 12, 2007, pp. 1745-1751. doi:10.1021/tx700376a
- G. Boysen, B. F. Pachkowski, J. Nakamura and J. A. Swenberg, “The formation and biological significance of N7-guanine adducts,” Genetic Toxicology and Environmental Mutagenesis, Vol. 678, No. 2, 2009, pp. 76-94. doi:10.1016/j.mrgentox.2009.05.006
- F. Faust, F. Kassie, S. Knasmüller, R. H. Boedecker, M. Mann and V. Mersch-Sundermann, “The use of the alkaline comet assay with lymphocytes in human biomonitoring studies,” Mutation Research/Reviews in Mutation Research, Vol. 566, No. 3, 2004, pp. 209-229. doi:10.1016/j.mrrev.2003.09.007
- D. J. McKenna, S. R. McKeown and V. J. McKelvey-Martin, “Potential use of the comet assay in the clinical management of cancer,” Mutagenesis, Vol. 23, No. 3, 2008, pp. 183-190. doi:10.1093/mutage/gem054
- J. F. Hartwig and S. J. Lippard, “DNA-binding properties of [Pt(NH3)(C6H11nh2)cl2], a metabolite of an orally active platinum anticancer drug,” Journal of American Chemical Society, Vol. 114, No. 14, 1992, pp. 5646- 5654.
- E. D. Scheeff, J. M. Briggs and S. B. Howell, “Molecular modeling of the intrastrand guanine-guanine DNA adducts produced by cisplatin and oxaliplatin,” Molecular Pharmacology, Vol. 56, No. 3, 1999, pp. 633-643.
- S. D. Webley, R. J. Francis, R. B. Pedley, S. K. Sharma, R. H. J. Begent, J. A. Hartley and D. Hochhauser, “Measurement of the critical DNA lesions produced by antibody-directed enzyme prodrug therapy (ADEPT) in vitro, in vivo and in clinical material,” British Journal of Cancer, Vol. 84, No. 12, 2001, pp. 1671-1676. doi:10.1054/bjoc.2001.1843
- E. Cemeli, E. Mirkova, G. Chiuchiarelli, E. Alexandrova and D. Anderson, “Investigation on the mechanisms of genotoxicity of butadiene, styrene and their combination in human lymphocytes using the Comet assay,” Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, Vol. 664, No. 1-2, 2009, pp. 69-76. doi:10.1016/j.mrfmmm.2009.02.010
- G. Chipitsyna, D. Slonina, K. Siddiqui, F. Peruzzi, T. Skorski, K. Reiss, B. E. Sawaya, K. Khalili and S. Amini, “HIV-1 Tat Increases Cell Survival in Response to Cisplatin by Stimulating Rad51 Gene Expression,” Oncogene, Vol. 23, No. 15, 2004, pp. 2664-2671.
- A. Vaisman and S. G. Chaney, “The efficiency and fidelity of translesion synthesis past cisplatin and oxaliplatin GpG adducts by human DNA polymerase beta,” The Journal of Biological Chemistry, Vol. 275, No. 17, 2000, pp. 13017-13025. doi:10.1074/jbc.275.17.13017
- S. G. Chaney, S. L. Campbell, E. Bassett and Y. B. Wu, “Recognition and processing of cisplatinand oxaliplatin-DNA adducts,” Critical Reviews in Oncology/Hematology, Vol. 53, No. 1, 2005, pp. 3-11. doi:10.1016/j.critrevonc.2004.08.008
- A. Vaisman, M. W. Warren and S. G. Chaney, “The effect of DNA structure on the catalytic efficiency and fidelity of human DNA polymerase beta on templates with platinum-DNA adducts,” The Journal of Biological Chemistry, Vol. 276, 2001, pp. 18999-19005. doi:10.1074/jbc.M007805200
- [61] E. Bassett, A. Vaisman, K. A. Tropea, C. M. McCall, C. Masutani, F. Hanaoka and S. G. Chaney, “Frameshifts and deletions during in Vitro translesion synthesis past Pt-DNA adducts by DNA polymerases β and η,” DNA Repair, Vol. 1, No. 12, 2002, pp. 1003-1016. doi:10.1016/S1568-7864(02)00150-7
- [62] J. M. Havener, S. A. N. McElhinny, E. Bassett, M. Gauger, D. A. Ramsden and S. G. Chaney, “Translesion synthesis past platinum DNA adducts by human DNA polymerase μ,” Biochemistry, Vol. 42, No. 6, 2003, pp. 1777- 1788. doi:10.1021/bi0270079
- [63] J. P. Banath and P. L. Olive, “Expression of Phosphorylated Histone H2AX as a Surrogate of Cell Killing by Drugs That Create DNA Double-Strand Breaks,” Cancer Research, Vol. 63, No. 15, 2003, pp. 4347-4350.
- [64] R. Sánchez-Olea, M. R. Calera and A. Degterev, “Molecular pathways involved in cell death after chemically induced DNA damage,” In: A. Luch, Ed., Molecular, Clinical and Environmental Toxicology, Birkhäuser, Basel, 2009, pp. 209-230.
- [65] W. G. McGregor, “DNA repair, DNA replication, and UV mutagenesis,” Journal of Investigative Dermatology Symposium Proceedings, Vol. 4, 1999, pp. 1-5. doi:10.1038/sj.jidsp.5640172
- [66] B. Kaina, “The Interrelationship between sce Induction, Cell-Survival, Mutagenesis, Aberration Formation and DNA Synthesis Inhibition in V79 Cells Treated with N-methyl-N-nitrosourea or N-methyl-N'-nitro-N-nitrosoguanidine,” Mutation Research Letters, Vol. 142, No. 1-2, 1985, pp. 49-54. doi:10.1016/S0165-7992(85)80012-9
- [67] I. M. Ward and J. Chen, “Histone H2AX Is Phosphorylated in an ATR-Dependent Manner in Response to Replicational Stress,” The Journal of Biological Chemistry, Vol. 276, No. 51, 2001, pp. 47759-47762.
- [68] A. J. Osborn, S. J. Elledge and L. Zou, “Checking on the Fork: The DNA-Replication Stress-Response Pathway,” Trends in Cell Biology, Vol. 12, No. 11, 2002, pp. 509- 516. doi:10.1016/S0962-8924(02)02380-2
- [69] B. Pascucci, M. T. Russo, M. Crescenzi, M. Bignami and E. Dogliotti, “The accumulation of MMS-induced single strand breaks in G1 phase is recombinogenic in DNA polymerase β defective mammalian cells,” Nucleic Acids Research, Vol. 33, No. 1, 2005, pp. 280-288. doi:10.1093/nar/gki168
- [70] O. Staszewski, T. Nikolova and B. Kaina, “Kinetics of γH2AX focus Formation upon Treatment of Cells with UV Light and Alkylating Agents,” Environmental and Molecular Mutagenesis, Vol. 49, No. 9, 2008, pp. 734-740.
- [71] W. M. Bonner, C. E. Redon, J. S. Dickey, A. J. Nakamura, O. A. Sedelnikova, S. Solier and Y. Pommier, “γH2AX and Cancer,” Nature Reviews Cancer, Vol. 8, No. 12, 2008, pp. 957-967. doi:10.1038/nrc2523
- [72] A. Sak, S. Grehl, P. Erichsen, M. Engelhard, A. Grannaß, S. Levegrün, C. Pöttgen, M. Groneberg and M. Stuschke, “Gamma-H2AX Foci Formation in Peripheral Blood Lymphocytes of Tumor Patients after Local Radiotherapy to Different Sites of the Body: Dependence on the DoseDistribution, Irradiated Site and Time from start of treatment,” International Journal of Radiation Biology, Vol. 83, No. 10, 2007, pp. 639-652. doi:10.1080/09553000701596118