Health
Vol. 4 No. 10 (2012) , Article ID: 24076 , 4 pages DOI:10.4236/health.2012.410130
Cancerous multi-drug resistance is reduced by Leptomycin B treatment in CCRF-CEM/Taxol cells
![]()
1Institute of Aviation Medicine, Beijing, China
2The Fourth Military Medical University, Xi’an, China
3Beijing Institute of Pharmacology and Toxicology, Beijing, China; *Corresponding Author: GuanYB@Hotmail.com
Received 16 August 2012; revised 13 September 2012; accepted 25 September 2012
Keywords: Leptomycin B; CCRF-CEM; Multi-Drug Resistance; CRM1; Paclitaxel
ABSTRACT
Objectives: Multi-drug resistance (MDR) to chemotherapy remains a major obstacle to overcome in the successful treatment of patients with cancers. It was recently discovered that Leptomycin B (LMB) reduces the paclitaxel-induced MDR in CCRF-CEM/Taxol cells. However, the mechanism remains unclear. Here we sought to explore the mechanism of LMB to reduce the MDR induced by paclitaxel. Results: LMB has remarkable cytotoxic effects in both sensitive CCRF-CEM and resistant CCRF-CEM/Taxol cell lines. The paclitaxel-induced MDR was reduced by 0.013 μm of LMB. Lower concentration of LMB regulated cell cycle progress, in situ expressions of P-gp, MRP1, and LRP, expression of CRM1, and localization of P-gp and CRM1 in CCRF-CEM/ Taxol cells. Study Design: Cytotoxicity of LMB on cancerous cell lines was determined by MTT assay. Cell cycle progress and in situ expressions of P-gp, MRP1, and LRP were analyzed by flow cytometry. Expression of CRM1 in the cells was examined by Western blot. And co-localization between P-gp and CRM1 was determined by laser confocal microscopy. Conclusion: The paclitaxel-induced MDR of CCRF CEM/Taxol cells was reduced by lower concentration of LMB. The mechanisms might be related to decreasing in situ expression of drug transporter proteins, promoting cell cycle progress, and altering colocalization between P-gp and CRM1 in the resistant cells.
1. INTRODUCTION
Acquisition of multi-drug resistance (MDR) is the leading cause of treatment failure in cancer therapy. MDR is, in large part, a story of drug transporters [1]. Multidrug transporters extrude antitumor agents from the cells and promote MDR phenotypes in cancer cells, which are composed of primary active transporters and secondary active transporters. The primary transporters are represented by the ATP-binding cassette (ABC) transporter super-family members energized by ATP hydrolysis, such as P-glycoprotein (P-gp), Multi-drug Resistance-related Protein 1 (MRP1), ect. The second class consists of secondary active transporters, including the small MDR superfamily, the multidrug and toxic compound extrusion family, the resistance-nodulation-cell division family, and the major facilitator superfamily [2]. In addition, intracellular nuclear export of either tumor suppressive agents or drug targets (proteins, mRNAs, or DNAs) result in drug resistance due to overexpression of nucleo-cytoplasmic transporters, including human major vault proteins, represented by Lung Resistance-related Protein (LRP) [3], and nuclear export signal (NES) mediated nuclear export proteins, represented by Chromosomal region maintenance 1 (CRM1) [4,5].
Acquired drug resistance can manifest in many ways: for example, cell signaling pathways can arrest cell cycle and delay apoptosis [6]; ATP binding cassette transporter drug efflux pumps can bind to and extrude chemotherapeutic drugs from cancer cells [2]; intracellular nuclear export of either antitumor agents or drug targets can result in drug resistance [7]. In addition, the tumor microenvironment has been reported to be very hypoxic due to inadequate blood supply and subsequent oxygen diffusion. Hypoxic tumor cells are resistant to both radiotherapy and chemotherapy [8]. Since multi-drug resistant cancer cells present a direct resistance to chemotherapeutic drugs, exhibiting increased IC50 concentration to the drugs. Reversal effect of drug resistance was usually evaluated using decreasing IC50 values of drug resistant cancer cells [9].
Leptomycin B (LMB) is an unsaturated; branchedchain fatty acid originated from Streptomyces sp., and was firstly discovered as an antifungal agent. Recently, some studies addressed that it can serve as a specific inhibitor of the substrates containing Nuclear Export Signal sequence (NESs) and block the molecules to export from nucleus, such as HIV-1 Rev protein and Revdependent mRNA [10,11], transcription factor E2F4 [12], Extra-cellular signal-Regulated Kinase 3 (ERK3) [13], p53 [14], Inhibitors of Apoptosis Proteins (IAPs) [15], and the Mad family protein Mad4 [16]. The suggested blocking mechanism has been identified as that LMB inhibits the cellular target CRM1 protein to bind with its substrates containing NESs and blocks the CRM1-dependent nuclear export of these molecules, which leads the substrates inactivation [17]. As a specific inhibitor, LMB covalently binds to the individual binding-domain sulphydryl group of Cys-529 in CRM1 via it’s α, β-unsaturated δ-lactone [11]. More recently, some studies reported that LMB exhibits antitumor activities by inhibiing nuclear export of topoisomerase Ⅱα in human myeloma cells, which enhances the cancerous cells sensitive to the topoisomerase Ⅱα-targeted anti-cancer drugs, such as etoposide (VP-16) [4,5]. Some other studies suggested that LMB also causes cell cycle G1 arrest [13] and induces apoptosis by sequestrating NES-containing p53 or BCR-ABL in the nucleus [14,18]. It was recently discovered by our studies for LMB to reduce paclitaxedinduced MDR of CCRF-CEM/Taxol cells. However, the mechanism remains unclear.
Purpose of the present study was to explore the reversal effect of LMB on MDR induced by paclitaxel. By means of investigating the cytotoxicity of LMB on cancerous cell lines and examining the cellular events cell cycle progress, in situ expression of drug transporter protein P-gp, MRP1, and LRP, expression of CRM1, and colocalization between P-gp and CRM1 to explore the mechanisms of LMB for the effects. Results from the current study demonstrated that LMB reduces the cancerous MDR induced by paclitaxel and enhances the sensitivity of CCRF-CEM/Taxol cells to chemotherapeutic drugs.
2. MATERIALS AND METHODS
2.1. Antibodies
Mouse primary monoclonal antibodies to P-gp, MRP1, and LRP were purchased from Sigma-Aldrich (St. Louis, USA). Rabbit primary polyclonal antibody to CRM1 and rabbit polyclonal antibody to β-actin (HRP) were from Abcam (Cambridge, UK). Mouse monoclonal antibody iso-type matched reagents and goat anti-mouse IgG (Fc specific) antibody conjugated with FITC were from Sigma-Aldrich. Goat anti-mouse IgG (H + L) Alexa Fluor® 488 and goat anti-rabbit IgG (H + L) Alexa Fluor® 555 were from Molecular probes (Eugene, USA). Horse-radish peroxidase (HRP) conjugated goat anti-rabbit IgG (H + L) was from SouthernBiotech (Birmingham, USA).
2.2. Cell Culture
Multi-drug resistant CCRF-CEM/Taxol cells induced by paclitaxel and its parental sensitive CCRF-CEM (Acute Lymphoblastic Leukemia, T-cell, Human) cells were maintained in RPMI-1640 media supplemented with 10% (v/v) fetal bovine serum (FBS, from Biochrom, Berlin). The cancer cells were cultured in a humidified 5% CO2 /95% air incubator at 37˚C [19]. In order to sustain the drug resistance, CCRF-CEM/Taxol cells were kept in exposure to an indicated dosage of paclitaxel during the experiments.
2.3. Cytotoxicity Assay
Exponentially growing CCRF-CEM/Taxel and CCRFCEM cells were harvested and seeded in 24-well plates at a density of 3 to 6 × 104 cells/500μl/well in complete medium. The first column of wells was exposed to DMSO as negative control. The other 5 columns of wells were exposed to different concentration of LMB or chemotherapeutic drugs as testing groups. The antiproliferative effect of the drugs was evaluated after 48 hrs by MTT Assay. All the studies were conducted in triplicate for each sample concentration.
Drug resistance of CCRF-CEM/Taxol cells was evaluated using the resistance fold calculated by the ratio of IC50 values of resistant cells versus that of its parental sensitive CCRF-CEM cells.
For examination of the effects of LMB on the paclitaxel-induced MDR of CCRF-CEM/Taxol cells, the cells were exposed to different concentration of chemotherapeutic drugs simultaneously with a lower concentration of LMB. The effects were evaluated using decreasing the resistance folds of CCRF-CEM/Taxol cells to different chemotherapeutic drugs.
2.4. Flow Cytometry Analysis
To quantitatively assess in situ expression of the drug transporter proteins in CCRF-CEM and CCRF-CEM/Taxol cells, the cells were harvested and fixed with 2% formaldehyde in PBS at room temperature for 10 min, washed with PBS (containing saponin:BSA:sodium azide = 0.04%:0.1%:0.01%) twice, then were incubated with mouse primary monoclonal antibodies to P-gp, MRP1 and LRP, respectively. Washed with PBS (containing 0.04% saponin) twice, the cells were incubated with goat anti-mouse IgG (FITC) and washed with PBS in presence of saponin twice and in absence of saponin once prior to be fixed in 1% paraformaldehyde. To account for nonspecific background fluorescence, cells were incubated with murine IgG1 antibody as control and backgro-0 und fluorescence intensity was subtracted from specific signals [20,21]. The flow cytometry analysis was performed on a FACSCalibur with CellQuest Pro v5.2.1. (Becton Dickinson). All the studies were conducted in triplicate.
For assessment of P-gp expression on the membrane of CCRF-CEM and CCRF-CEM/Taxol cells, the cells were processed according to the method described above just in absent of saponin.
For cell cycle analysis, CCRF-CEM/Taxol cells were treated with LMB as testing groups paralleled with untreated CCRF-CEM/Taxol cells as positive control and with CCRF-CEM cells as negative control, respectively. Cell cycle analyses were carried out after the cells were synchronized and cultured for 48 h. The cell populations were fixed in 70% of ice-cold ethyl alcohol for 3 h, treated with 5 u/μl of RNase H solution at 37˚C for 30 min, stained with 50 μg/ml of propidium iodide for 15 min, and then subjected to flow cytometry with a FACScan flow cytometer (Becton Dickinson). 10,000 events were collected from each sample. Data acquisition was carried out using Cell-Quest software and cell cycle distribution calculated using the ModFit LT 3.0 (Verity Software House, ME, USA) analysis software [22].
2.5. Western Blot Analysis
To explore the expression of CRM1 affected by lower concentration of LMB in CCRF-CEM/Taxol cells, the cells were treated with 0.013 and 0.015 μm of LMB and compared to CCRF-CEM cells as negative control and CCRF-CEM/Taxol cells as positive control. The whole cell lysates were prepared from both CCRF-CEM and CCRF-CEM/Taxol cell line cancerous cells. The protein concentration of lysates was determined with 2-D Quant Kit (Amersham Biosciences), and 30 μg proteins were applied to a 10% SDS-polyacrylamide gel and electrophoresed for 90 min at 100 V. The protein was then transferred to nitrocellulose membrane (Millipore). After blocking with 5% of skimmed milk in PBST (PBS-0.2% Tween 20) overnight at 4˚C, the membranes were incubated together with rabbit primary antibodies to CRM1 for 3 hrs at 37˚C. HRP-conjugated goat anti-rabbit IgG (HRP) served as the secondary antibody. The membranes were briefly incubated with enhanced ECL detection reagent (SuperECL Plus, Applygen Technolgies Inc.) to visualize the protein and exposed to an X-ray film. β-actin served as the internal control. Density of plot on lanes was quantified using Image J software (version 1.33).
2.6. Confocal Microscopy Analysis
For confocal analysis to visualize the effect of LMB on intracellular localization of CRM1 and P-gp in CCRFCEM/Taxol cells, the cells and negative CCRF-CEM cells were grown in phenol red-free RPMI-1640 media for 48 hrs and fixed with 70% ethanol for 15 min at room temperature, permeated with 0.04% saponin for 5 mins, then reacted with mouse monoclonal primary antibody to P-gp and rabbit polyclonal primary antibody to CRM1 for 2 hrs at 37˚C. The cells were stained by goat antimouse IgG conjugated with Alexa Fluor 488 (green, Molecular Probes) and goat anti-rabbit IgG conjugated with Alexa Fluro 555 (red, Molecular Probes), then were mounted with DAPI (blue) to detect nucleus. Confocal microscopy was performed on a Zeiss LSM 510 META system (60 × oil-immersion objectives) equipped with a 150 Mw Omnichrome Ar-Kr laser exciting at 488 nm and 555 nm. Images were captured using Carl Zeiss Laser Scanning Microscope LSM 510 Release (Version 4.2).
2.7. Statistic Analyses
Statistical analysis in this study were carried out with OriginPro 8 SR0 (v. 8.0724) software. The cell fraction of cell cycle progression and in situ expression of the drug transporter proteins were evaluated by 2-way ANOVA followed by Student’s t-test. Differences with P values of less than 0.05 were considered to be statistically significant.
3. RESULTS
3.1. Cytotoxic Effects of LMB
To determine the cytotoxicity of LMB, CCRF-CEM/ Taxol and CCRF-CEM cells were treated with different concentration of LMB. The antiproliferative effect of LMB was observed after treating the cells with LMB for 48 hrs (Figure 1(A)). LMB reduced the proliferation of CCRF-CEM and CCRF-CEM/Taxol cells in a concentration-dependent manner. The remarkable effects were observed at the concentration range from 0.015 to 8.1 μm in CCRF-CEM cells and 0.15 to 39.38 μm in CCRF-CEM/ Taxol cells. IC50 values of CCRF-CEM/Taxol cell lines are 2.38 μm and that of CCRF-CEM cells is 1.36 μm. The results demonstrated that CCRF-CEM cells are susceptible to LMB more than CCRF-CEM/Taxol cells. 0.013 μm is 0.0054-fold of the IC50 concentration of CCRF-CEM/Taxol cells and 0.01-fold of CCRF-CEM cells, respectively. It can not significantly impact the viability of CCRF-CEM/Taxol cells. Thus the reversing MDR effects of LMB were explored at this concentration in the subsequent studies.
3.2. Effects of LMB on Paclitaxel-Induce MDR
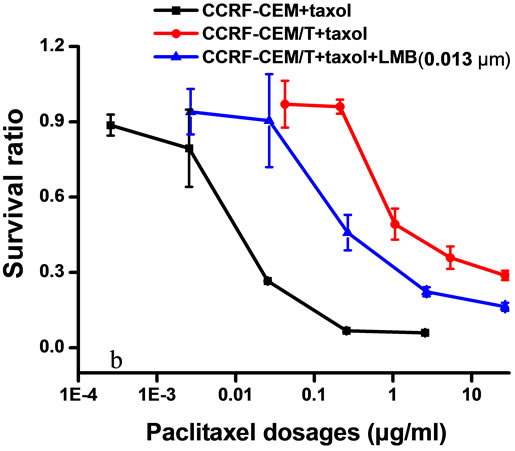
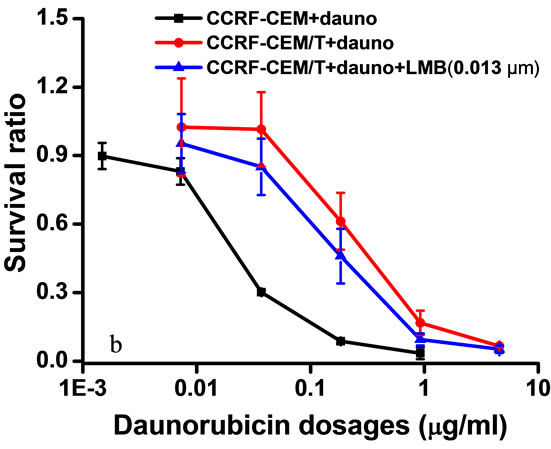
CCRF-CEM/Taxol cells are resistant to paclitaxel, vinblastin, and daunorubicin simultaneously. Treatment with 0.013 μm of LMB for 48 hrs decreased the drug resistance of CCRF-CEM/Taxel cells to paclitaxel from 250.5- to 125.1-fold (at 50.06%) (Figure 1(B,b), to vinblastin from 2.03- to 1.83-fold (at 9.8%) (Figure 1(C,a), and to daunorubincin from 8.93- to 7.93-fold (at 11.2%) (Figure 1(c,b). However, it was observed that treatment with different concentration (0.0065, 0.013, 0.065, and 0.13 μm) of LMB result in no significant impact on the IC50 values of CCRF-CEM cells to paclitaxel (Figure 1(B, a)). The results suggested that LMB only enhances susceptibility of CCRF-CEM/Taxol cells to chemotherapeutic drugs, but it does not affect CCRF-CEM cells.
 (A)
(A)
 (B)
(B)
 (C)
(C)
Figure 1. Cytotoxicity assays of CCRF-CEM/Taxol and CCRF-CEM cells. Cells were exposed to different concentration of LMB (0.063 - 39.38 μm and 0.013 - 8.1 μm, respectively) for 48 hrs (A). Different concentration of LMB does not significantly impact the IC50 values of CCRF-CEM cells to paclitaxel (B, a). 0.013 μm of LMB rendered CCRF-CEM/Taxol cells with reduced MDR to Paclitaxel (B, b). Daunorubicin (C, a) and Vinblastin (C, b). Results are expressed as the average of three separate repeats. Points, means; bars, SD.
3.3. Effects of LMB on Cell Cycle Progression
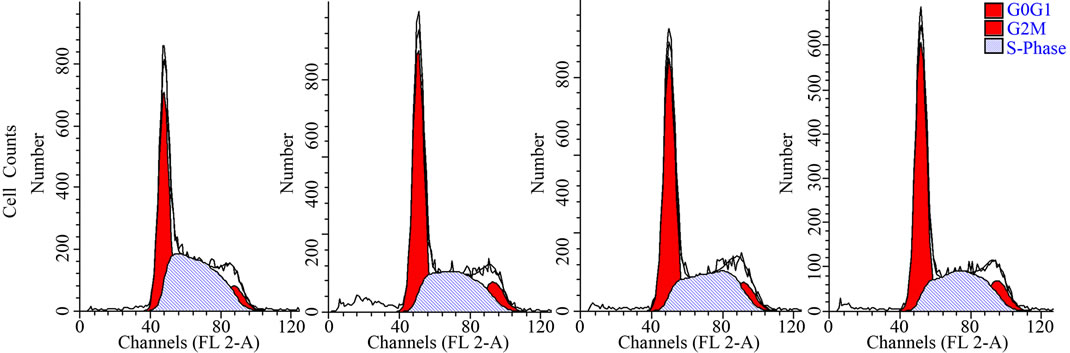
To examine the effects of LMB on cell cycle progresssion, flow cytometric analysis was performed. Compared to CCRF-CEM cells, the amount of G0/G1 and G2/M phase cells of CCRF-CEM/Taxol are larger, and the amount of S phase and apoptosis cells are lesser (Figure 2). CCRF-CEM/Taxol cells showed a pronounced G0/G1 phase arrest and released more cells into G2/M phase with showing a significant decrease in apoptotic cells (P < 0.05). Treatment with 0.013 μm and 0.065 μm of LMB significantly reduced the amount of G0/G1 and G2/M phase cells and significantly enhanced the amount of S phase and apoptosis cells of CCRF-CEM/T/axol (P < 0.05), wich made the cell cycle progression of CCRFCEM/Taxol cells similar to that of CCRF-CEM cells.
3.4. Effects of LMB on Expression of CRM1
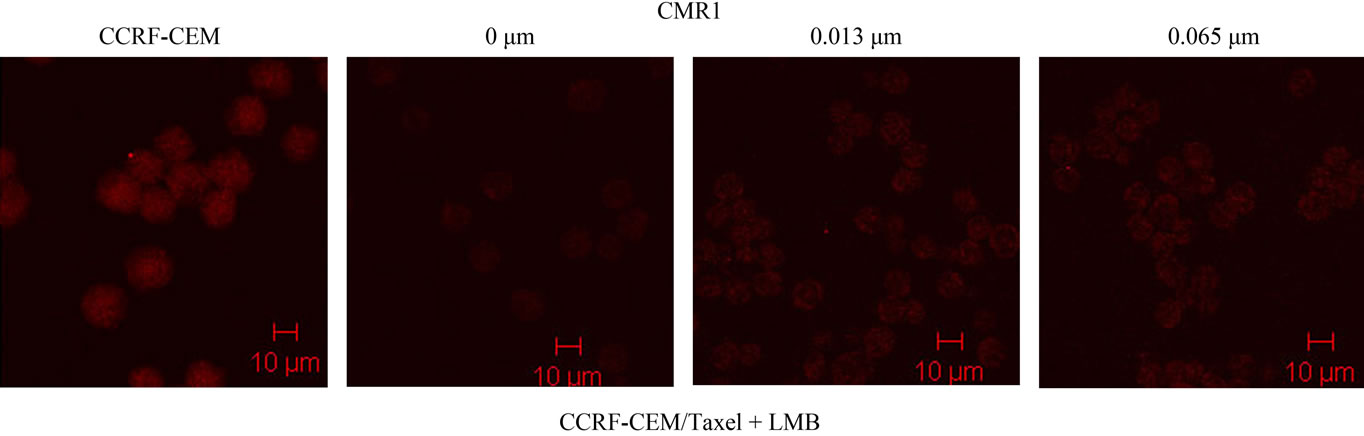
The effect of LMB treatment to enhance the expression of CRM1 in CCRF-CEM/Taxol cells was determined at the protein level, using Western blotting on protein extracts from LMB treated CCRF-CEM/Taxol cells compared to CCRF-CEM cells as negative control. CRM1 in CCRF-CEM/Taxol cells was found to be expressed at lower level compared to that in CCRF-CEM cells, as demonstrated in Figure 3(c). Treatment with lower concentration of LMB clearly enhanced CRM1 expression in CCRF-CEM/Taxol cells at protein level confirming the results above by the Confocal Microscopy assay (Figure 3(a)). Although the Western blots suggested a variable degree of CRM1 expression after LMB treatment, the LMB treated CCRF-CEM/Taxol cells demonstrated higher protein levels of CRM1 compared to untreated CCRFCEM/Taxol cells (Figure 3(c)).
3.5. Effects of LMB on in Situ Expression of Drug Transporter Proteins
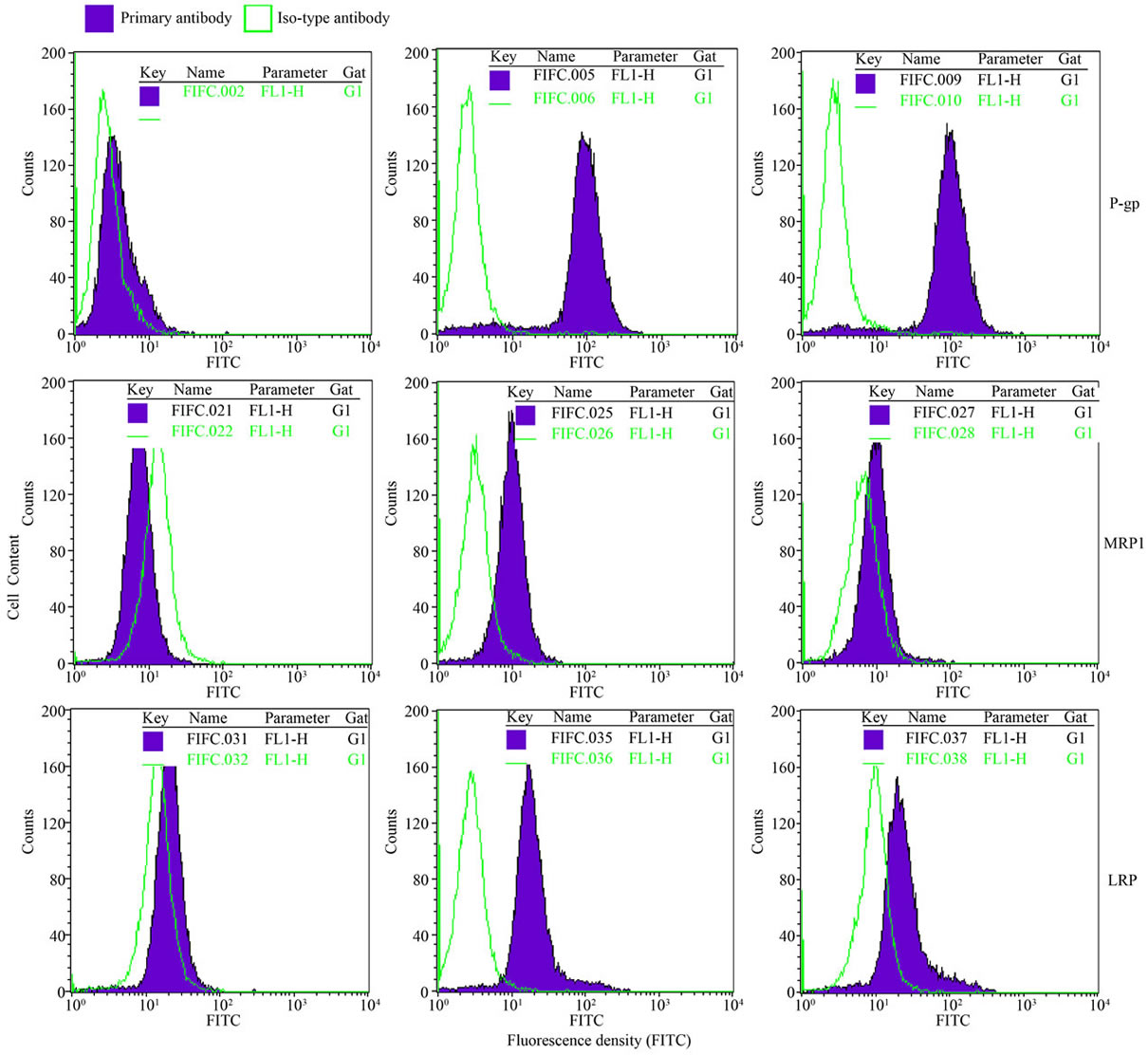
Quantitative analysis of immnofluorescence staining by flow cytometry revealed that drug transporter proteins P-gp, MRP1, and LRP were expressed in both CCRFCEM/Taxol and CCRF-CEM cells. CCRF-CEM/Taxol cells expressed high levels of the three proteins at 15.4- fold to P-gp, 3.08-fold to MRP1, and 2.19-fold to LRP compared to CCRF-CEM cells (Figure 4). All the three proteins in situ expression in CCRF-CEM/Taxol cells were decreased by 0.013 μm of LMB at 32.86% to P-gp, 34.66% to MRP1, and 35.96% to LRP compared to untreated CCRF-CEM/Taxol cells (Figure 4(b)).
 (a)
(a) (b)
(b)
Figure 2. Cell cycle analysis of CCRF-CEM/Taxel cells treated with LMB. 0.013 μm and 0.065 μm of LMB interferes cell cycle arrest in G0/G1 phase and releases cells into S-phase, enhanced cancerous cells apoptosis in CCRF-CEM/Taxol cells, which was demonstrated by Flow cytometry assay (a) and Cell fraction-phases histogram (b). Columns, means; bars, SD; *P < 0.05 (t test), compared to CCRFCEM cells; #P < 0.05 (t test), compared to CCRF-CEM/Taxol cells.
 (a)
(a) (b)
(b) (c)
(c)
Figure 3. Expression of CRM1 in CCRF-CEM/Taxol cells. The protein level of CRM1 in LMB treated CCRF-CEM/Taxol cells compared to controls were analyzed using confocal microscopy assay (a) and western blots assay (b). Densitometry analysis was shown by Histogram (c). Scale bars, 10 μm.
3.6. Effects of LMB on Localization of P-gp and CRM1
To further clarify the effect of LMB on P-gp, the expression and distribution of P-gp in CCRF-CEM/Taxol cells were determined following 48 and 96 h of LMB exposure. Flow cytometry analysis revealed that CCRFCEM/Taxol cells expressed significantly higher level of P-gp at total cellular amount and on membrane compared to CCRF-CEM cells (Figure 5(b)). The surface P-gp was decreased in LMB treated CCRF-CEM/Taxol cells compared to the untreated CCRF-CEM/Taxol cells (Figure 5(b)). The results were confirmed by the Confocal microscopy analysis (Figure 5(a)).
 (a)
(a)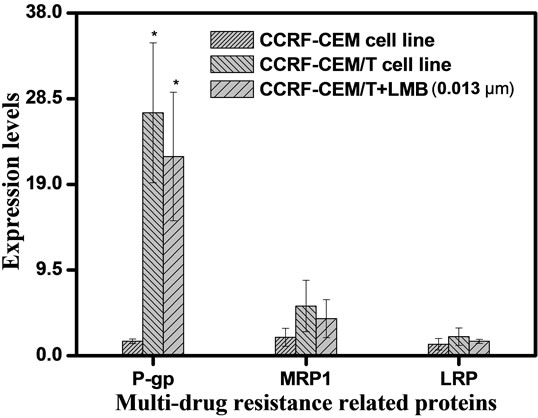 (b)
(b)
Figure 4. In situ expression of drug transporter protein P-gp, MRP1, and LRP in CCRF-CEM/taxol cells. CCRF-CEM/ Taxol cells expressed the three proteins at higher level compared to CCRF-CEM cells. Decreased levels of the proteins induced by 0.013 μm of LMB were demonstrated using Flow cytometry assay (a) and Statistic histogram. The experiments were performed in triplicate (b). Filled profiles, fluorescence density of primary mAbs; open profiles, fluorescence density of iso-type matched monoclonal antibodies; Columns, means; bars, SD; *P < 0.05 (t test), compared to CCRF-CEM cells.
 (a)
(a)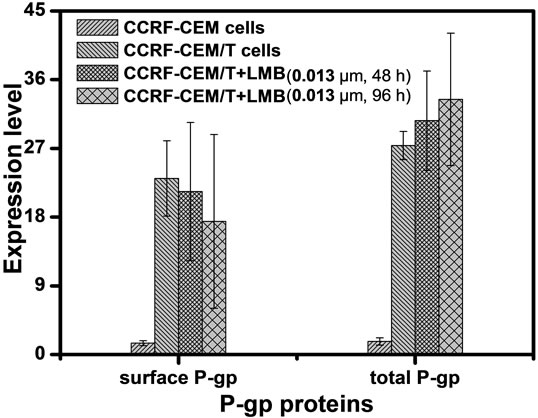 (b)
(b)
Figure 5. Localization and in situ expression of P-gp in CCRF-CEM/Taxol cells. Lower concentraton of LMB decreases the surface P-gp level and increases the cytoplasmic P-gp level in CCRF-CEM/Taxol cells in a time-dependent manner, compared to untreated CCRF-CEM/Taxol cells control. The results were demonstrated by Laser confocal microscopy assay (a) and Flow cytometry assay (b). Scale bar, 10 μm; Filled profiles, fluorescence density of primary mAb; open profiles, fluorescence density of iso-type matched monoclonal antibodies to primary mAbs. Scale bars, 10 μm.
Colocalization studies between P-gp and CRM1 for intracellular compartments were performed to investigate whether sequestration plays a role in any of the drug resistance phenotypes. The stronger colocalization was observed between P-gp and CRM1 in cytoplasm of CCRFCEM cells (Figure 6(d)). No significant overlap was observed between the two proteins in CCRF-CEM/Taxol cells, indicating that the localization of CRM1 in cytoplasm of the cells was reduced by Paclitaxel treatment. Although 0.013 and 0.015 μm of LMB enhanced the expression of CRM1, co-localization between P-gp and CRM1 in cytoplasm of CCRF-CEM/Taxol cells is still no significant (Figures 6(h), (l), (p)).
4. DISCUSSION
Multi-drug resistance to chemotherapy remains a major obstacle to overcome in the successful treatment of patients with cancers. Due to the inherent side effect of many therapeutic drugs, the resistance presented by cancerous cells cannot be overcome by simply increasing doses to cancer targets. Many anticancer drugs have a steep dose-toxicity curve. Therefore, changes in the doses of drugs will likely have significant impacts on the clinical responses to drug treatments.
Leptomycin B (LMB) was originated from the actinomycetes of Streptomyces. It was reported as a cytotoxic agent inducing cell apoptosis [23]. The conclusion supported by data achieved from our studies was that 2.38 and 1.36 μm of LMB induced 50% of CCRF-CEM/Taxol and CCRF-CEM cells to death, respectively. Chemotherapeutic drug sensitive CCRF-CEM cells treated by paclitaxel became multi-drug resistant CCRF-CEM/Taxol cells, showing resistance to different chemotherapeutic drugs simultaneously. Lower concentration (0.013 μm) of LMB cannot impact the viabilities of CCRF-CEM/Taxol cells; however, it can enhance the responsibility of CCRF-CEM/Taxol cells to paclitaxel at 56.43%, daunorubicin at 11.2%, and vinblastin at 9.8% (Figures 1(C), 6(d), (e)). As the MTT results alone cannot be used as a direct evidence of reversing MDR of CCRF-CEM/Taxol cells, we determined the effects of LMB on in situ expression of three drug transporter proteins (P-gp, MRP1,
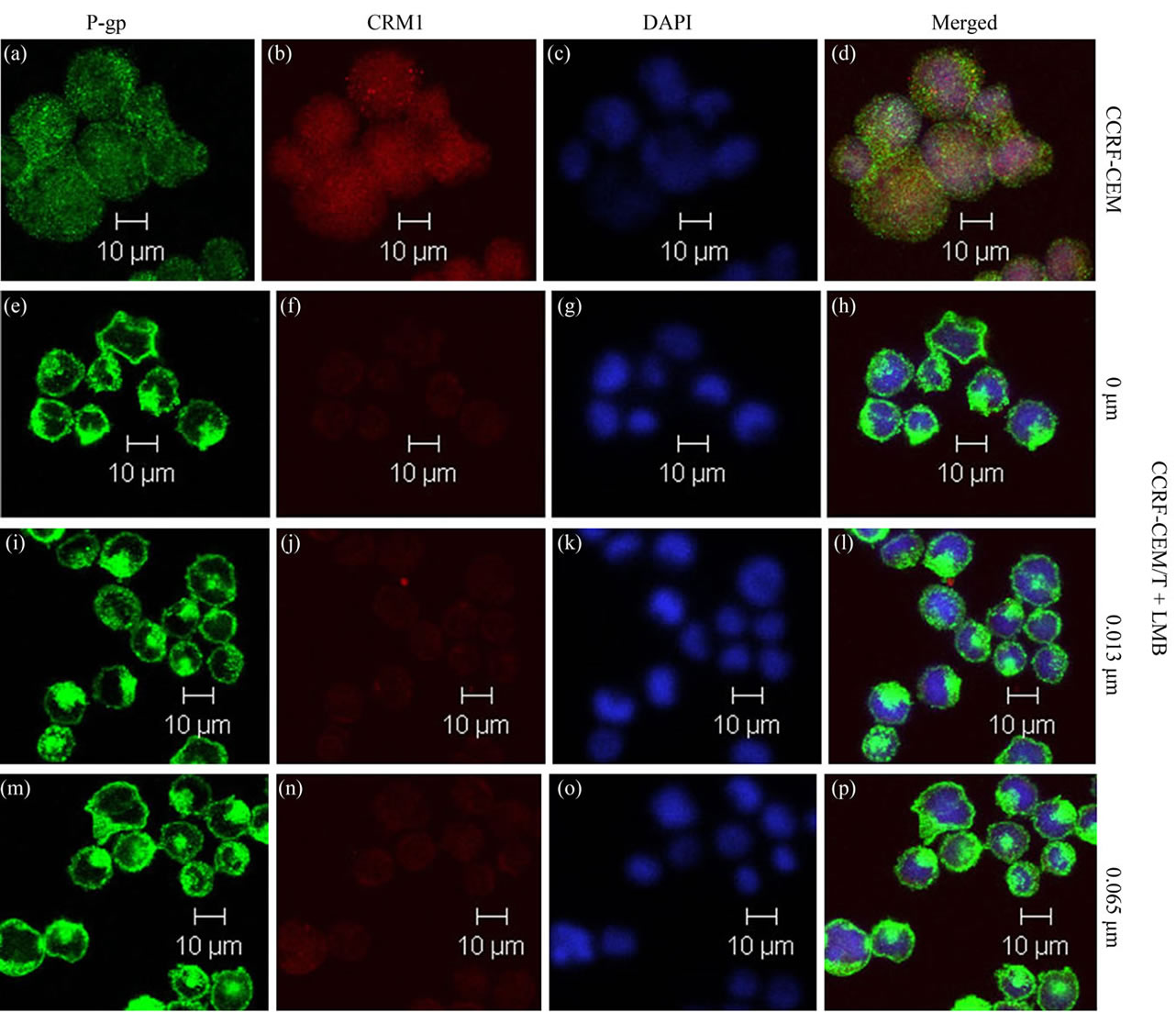
Figure 6. Colocalization of CRM1 with P-gp in CCRF-CEM/Taxol cells. Confocal analysis of the expression, distribution, and overlapping of P-gp (green; (a), (e), (i), (m)) and CRM1 (red; (b), (f), (j), (n)) is shown for 0.013 and 0.065 μm of LMB treatment in CCRF-CEM/Taxol cells. The yellowish color represents colocalization, or a very similar distribution of the two proteins. The nuclei were stained with DAPI (blue; (c), (g), (k), (o)). Imagines are representative of three independent experiments. Scale bars, 10 μm.
and LRP) and expression of nuclear export protein CRM1, cell cycle progression, colocalization between P-gp and CRM1 in the resistant cells.
Increasing drug efflux predominantly via ATP-driven extrusion pumps frequently of the ATP-banding cassette (ABC) superfamily may be the major of modalities of chemotherapeutic drug resistance. P-glycoprotein (P-gp) and multi-drug resistance protein 1 (MRP1) belong to the ABC superfamily members. Lung cancer resistance-related protein (LRP) is not the member of ABC transporter proteins. LRP corresponds to the human major cytoplasmic vault protein composed of 4 proteins and one small RNA molecule, which have been hypothesized to be involved in nucleo-cytoplasmic transport of drugs [3,24]. These pumps significantly decrease the intracellular concentration of a multitude of endogenous and exogenous cytotoxic agents, thereby resulting the cancer cells in MDR [25-27]. Higher the proteins expression was found to be strongly correlation with intrinsic resistance to chemotherapeutic drugs. In the present study, the three proteins expression level were detected in LMB-treated and untreated CCRF-CEM/Taxol cells. The established paclitaxel-induced CCRF-CEM/Taxol cells had higher level of the proteins than its parental CCRF-CEM cells, and its MDR to drugs induced by paclitaxel in the cell viability assay (Figure 1(B)) was more pronounced than its parental CCRF-CEM cells. 0.013 and 0.065 μm of LMB did not appear to totally reverse the paclitaxel-induced MDR of CCRF-CEM/Taxol cells since the concentrations of LMB could not decrease the in situ expression of all three drug transporter proteins to their base level in CCRF-CEM cells.
The P-gp localizing on cell membrane plays a role of drug efflux pump, which directly contributes to export of chemotherapeutic drugs from cancerous cells, which is responsible for MDR phenotype of cancerous cells, in spite of the chemotherapeutic drugs are a wide variety of structurally and functionally unrelated with cytotoxicity [28]. 0.013 μm of LMB decreased P-gp on membrane and increased P-gp in cytoplasm of CCRF-CEM/Taxol cells in a time-dependent manner (Figure 5(b)). The present work indicates that lower concentration of LMB in CCRF-CEM/Taxol cells induces a mobilization of P-gp to cytoplasm from the membrane.
Another possible mechanism for LMB-mediated MDR is the regulation of cell cycle progression. CRM1 is the major cellular target of LMB, a nuclear protein that shuttles between nucleus and cytoplasm as a nuclear-cytoplasmic transporter for a NES-mediated nuclear export pathway [11,29,30]. On the other hand, CRM1 inactivates one or more components of the pocket protein G1 arrest pathway. CRM1-dependent nuclear export regulates the G1 arrest function of E2F4 and E2F5. Over-expression of CRM1 prevents cell cycle G1 arrest by an E2F4-dependent process [12]. In the present study, expression and localization of CRM1 was detected. CCRFCEM/Taxol cells expressed lower level of CRM1 than CCRF-CEM cells (Figure 3). Using flow cytometry analysis of synchronized cells, we showed that significant blockage of S-phase entry (cell cycle G1 arrest) and lower apoptosis are observed in the paclitaxel-induced drug resistant CCRF-CEM/Taxel cells (Figure 2). After exposure to 0.013 and 0.065 μm of LMB for 48 h, G0/G1 arrest of CCRF-CEM/Taxel cells was inefficiently maintained; allowing progression of G0/G1 phase cells into S-phase, and more apoptosis was induced. We assumed that CCRF-CEM/Taxol cells expressed lower level of cytoplasmic CRM1 (Figures 3(a), 6(f), (g), (n)) might be resulted from its sequestration in nuclear to prevent single DNA double-strand from breakage in G2 phase. Further studies are required to discern the effective roles of CRM1 in the paclitaxel-induced MDR.
Colocalization between CRM1 and P-gp was observed in chemotherapeutic drug sensitive CCRF-CEM cells. No significant overlap of the two proteins occurred in paclitaxel-induced drug resistant CCRF-CEM/Taxol cells. Further studies will be carrying out to explore the mechanism of colocalization between CRM1 and P-gp in MDR phenotype.
In summary, we detected LMB cytotoxicity in cancerous cell lines and the effects of LMB on in situ expression of three and expression of one drug transporter proteins, cell cycle progression, and colocalization between P-gp and CRM1 in drug sensitive CCRF-CEM cells and paclitaxel-induced drug resistant CCRF-CEM/ Taxol cells. Our data demonstrated that LMB reduces paclitaxel-induced MDR and enhances susceptibility of drug resistant CCRF-CEM/Taxol cells to chemotherapeutic drugs. The present work indicates that LMB renders CCRF-CEM/Taxol cells with decreased MDR to paclitaxel, vinblastin and daunorubicin. We are confident that the effect of LMB is most likely related to decreasing in situ expression level of drug transporter proteins P-gp, MRP1, and LRP, regulating cell cycle progression, and altering the localization between P-gp and CRM1 proteins in CCRF-CEM/Taxol cells.
5. ACKNOWLEDGEMENTS
The study was supported by the grant from Department of Drug Safety Evaluation and Research of Beijing Institute of Pharmacology and Toxicology. We thank Dr. Chou Ting-Chao for the generous gifts of cancerous cell lines.
REFERENCES
- Higgins, C.F. (2007) Multiple molecular mechanisms for multi-drug resistance transporters. Nature, 446, 749-757. doi:10.1038/nature05630
- Lage, H. (2003) ABC-transporters: Implications on drug resistance from microorganisms to human cancers. International Journal of Antimicrobial Agents, 22, 188-199. doi:10.1016/S0924-8579(03)00203-6
- Scheffer, G.L., Wijngaard, P.L., Flens, M.J., Izquierdo, M.A., Slovak, M.L., Pinedo, H.M., et al. (1995) The drug resistance-related protein LRP is the human major vault protein. Nature Medicine, 6, 578-582. doi:10.1038/nm0695-578
- Engel, R., Valkov, N.I., Gump, J.L., Hazlehurst, L., Dalton, W.S. and Sullivan, D.M. (2004) The cytoplasmic trafficking of DNA topoisomerase IIa correlates with etoposide resistance in human myeloma cells. Experimental Cell Research, 295, 421-431. doi:10.1016/j.yexcr.2004.01.012
- Turner, J.G., Engel, R., Derderian, J.A., Jove, R. and Sullivan, D.M. (2004) Human topoisomerase IIa nuclear export is mediated by two CRM-1-dependent nuclear export signals. Journal of Cell Science, 117, 3061-3071. doi:10.1242/jcs.01147
- Longley, D.B. and Johnston, P.G. (2005) Molecular mechanisms of drug resistance. The Journal of Pathology, 205, 275-292. doi:10.1002/path.1706
- Turner, J.G., Dawson, J. and Sullivan, D.M. (2012) Nuclear export of proteins and drug resistance in cancer. Biochemical Pharmacology, 83, 1021-1032. doi:10.1016/j.bcp.2011.12.016
- Kim, J.W., Ho, W.J. and Wu, B.M. (2011) The role of the 3D environment in hypoxia-induced drug and apoptosis resistance. Anticancer Research, 31, 3237-3245.
- Zhu, J.W. and Guan, Y.B. (2010) Construction of cancerous multi-drug resistant cell model induced by paclitaxel from human leukemia CCRF-CEM cells. Chinese Journal of Pharmacology and Toxicology, 24, 134-139.
- Fischer, U., Huber, J., Boelens, W.C., Mattaj, I.W. and Lührmann, R. (1995) The HIV-1 rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell, 82, 475-483. doi:10.1016/0092-8674(95)90436-0
- Kudo, N., Matsumori, N., Taoka, H., Fujiwara, D., Schreiner, E.P., Wolff, B., et al. (1999) Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proceedings of the National Academy of Sciences, 96, 9112-9117.
- Gaubatz, S., Lees, J.A., Lindeman, G.L. and Livingston, D.M. (2001) E2F4 is exported from the nucleus in a CRM1-dependent Manner. Molecular and Cellular Biology, 21, 1384-1392. doi:10.1128/MCB.21.4.1384-1392.2001
- Julien, C., Coulombe, P. and Meloche, S. (2003) Nuclear export of ERK3 by a CRM1-dependent mechanism regulates its inhibitory action on cell cycle progression. The Journal of Biological Chemistry, 278, 42615-42624. doi:10.1074/jbc.M302724200
- Stommel, J.M., Marchenko, N.D., Jimenez, G.S., Moll, U.M., Hope, T.J. and Wahl, G.M. (1999) A leucine-rich nuclear export signal in the p53 tetramerization domain: Regulation of subcellular localization and p53 activity by NES masking. The EMBO Journal, 18, 1660-1672. doi:10.1093/emboj/18.6.1660
- Monte, M., Benetti, R., Collavin, L., Marchionni, L., Sal, G.D. and Schneider, C. (2004) hGTSE-1 expression stimulates cytoplasmic localization of p53. The Journal of Biological Chemistry, 279, 11744-11752. doi:10.1074/jbc.M311123200
- Grinberg, A.V., Hu, C.D. and Kerppola, T.K. (2004) Visualization of Myc/Max/Mad family dimers and the competition for dimerization in living cells. Molecular and Cellular Biology, 24, 4294-4308. doi:10.1128/MCB.24.10.4294-4308.2004
- Nishi, K., Yoshida, M., Fujiwara, D., Nishikawa, M., Horinouchi, S. and Beppu, T. (1994) Leptomycin B targets a regulatory cascade of crm1, a fission yeast nuclear protein, involved in control of higher order chromosome structure and gene expression. The Journal of Biological Chemistry, 269, 6320-6324.
- Vigneri, P. and Wang, J.Y. (2001) Induction of apoptosis in chronic myelogenous leukemia cells through nuclear entrapment of BCR-ABL tyrosine kinase. Nature Medicine, 7, 228-234. doi:10.1038/84683
- Shah, D., Naciri, M., Clee, P. and Mohamed, A.R. (2006) Nucleocounter: An efficient technique for the determination of cell number and viability in animal cell culture processes. Cytotechnology, 51, 39-44. doi:10.1007/s10616-006-9012-9
- Yamada, M., Ariga, T., Kawamura, N., Yamaguchi, K., Ohtsu, M., Nelson, D.L., et al. (2000) Determination of carrier status for the wiskott-aldrich syndrome by flow cytometric analysis of wiskott-aldrich syndrome protein expression in peripheral blood mononuclear cells. The Journal of Immunology, 165, 1119-1122.
- Meschini, S., Marra, M., Calcabrini, A., Monti, E., Gariboldi, M., Dolfini, E., et al. (2002) Role of the lung resistance-related protein (LRP) in the drug sensitivity of cultured tumor cells. Toxicol in Vitro, 16, 389-398. doi:10.1016/S0887-2333(02)00035-8
- Cortner, J. and Farnham, P.J. (1991) Cell cycle analysis of Krox-20, c-fos, and JE expression in proliferating NIH3T3 fibroblasts. Cell Growth & Differentiation, 2, 465-473.
- Jang, B.C., Paik, J.H. and Jeong, H.Y. (2004) Leptomycin B-induced apoptosis is mediated through caspase activetion and down-regulation of Mcl-1 and XIAP expression, but not through the generation of ROS in U937 leukemia cells. Biochemical Pharmacology, 68, 263-274. doi:10.1016/j.bcp.2004.03.007
- Laurencot, C.M., Scheffer, G.L. and Shoemaker, R.H. (1997) Incresed LRP mRNA expression is associated with the MDR phenotype in intrinsically resistant human cancer cell lines. International Journal of Cancer, 72, 1021-1026. doi:10.1002/(SICI)1097-0215(19970917)72:6<1021::AID-IJC17>3.0.CO;2-7
- Larsen, A.K., Escargueil, A.E. and Skladanowski, A. (2000) Resistance mechanisms associated with altered intracellular distribution of anticancer agents. Pharmacology & Therapeutics, 85, 217-229. doi:10.1016/S0163-7258(99)00073-X
- Krishna, R. and Mayer, L.D. (2000) Multidrug resistance (MDR) in cancer Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. European Journal of Pharmaceutical Sciences, 11, 265-283. doi:10.1016/S0928-0987(00)00114-7
- Laurencot, C.M., Scheffer, G.L., Schrper, R.J. and Shoemarker, R.H. (1997) Increased LRP mRNA expression is associated with the MDR phenotype in intrinsically resistant human cancer cell lines. International Journal of Cancer, 72, 1021-1026. doi:10.1002/(SICI)1097-0215(19970917)72:6<1021::AID-IJC17>3.0.CO;2-7
- Ferrao, P., Sincock, P., Cole, S. and Ashman, L. (2001) Intracellular P-gp contributes to functional drug efflux and resistance in acute myeloid leukaemia. Leukemia Research, 25, 395-405. doi:10.1016/S0145-2126(00)00156-9
- Stade, K., Ford, C.S., Guthrie, C. and Weis, K. (1997) Exportin 1 (Crm1p) is an essential nuclear export factor. Cell, 90, 1041-1050. doi:10.1016/S0092-8674(00)80370-0
- Jang, B.C., Ursula, M.N., Paik, J.H., Claffey, K., Yoshida, M. and Hla, T. (2003) Leptomycin B, an inhibitor of the nuclear export receptor CRM1, inhibits COX-2 expression. The Journal of Biological Chemistry, 278, 2773-2776. doi:10.1074/jbc.C200620200