Advances in Chemical Engineering and Science
Vol. 2 No. 3 (2012) , Article ID: 20841 , 6 pages DOI:10.4236/aces.2012.23047
Dimerization of 1-Phenyl-1H-tetrazole-5-thiol over Metalloporphyrin Catalysts
1College of Light Industry & Chemical Engineering, Guangdong University of Technology, Guangzhou, China
2Fire Brigade of Chaoyang District, Chinese Armed Police Force (CAPF), Shantou, China
Email: *yanyan600716@hotmail.com, *gych@gdut.edu.cn
Received April 2, 2012; revised April 26, 2012; accepted May 7, 2012
Keywords: Disulfane; Dimerizatoin; Catalytic Conversion; Metalloporphyrin Catalyst
ABSTRACT
In an alkaline methanol solution, dimerization of 1-phenyl-1H-tetrazole-5-thiol (HL) was carried out over metalloporphyrin catalysts under mild conditions. The dimer product, 1,2-bis(1-phenyl-1H-tetrazol-5-yl) disulfane (L-L), was characterized by determinations of infrared (IR), HPLC, NMR and elementary analysis respectively. In situ UV-Vis spectroscopic analysis and cyclic voltammetric (CV) determinations suggested that the active intermediate for L-L formation is an axially ligated complex, RS-MnIIITHPP, which decomposes into a MnIITHPP molecule and a stable radical (·SR) for coupling to form the disulfane. Meanwhile MnIITHPP molecule can be oxidized easily to form MnIIITHPP species again by oxygen from the air for using in next catalytic circle.
1. Introduction
Metalloporphyrins have been extensively studied because their excellently catalytic functions in organic preparations [1-4]. Meanwhile their catalytic activities were dramatically enhanced by halogen substitutions in the conjugated porphyrin planes [5-8]. Recently, porphyrin molecules were anchored on the surfaces of silica and polymer for easy separation from the catalytic systems [9,10]. Furthermore, cobalt porphyrin molecules supported on pyrolysed carbon exhibited high activity in catalytic conversion from hydrogen sulfides to S8 circle molecules [11]. Catalytic conversion of organic substrates provides wide research fields in biomimetic oxidations. Oxidation of 2-substituted quinolines over metalloporphyrin catalysts showed that biomimetic oxidation can replace the biological approach, thus allowing access to large quantities of metabolites [12]. A series of ruthenium porphyrin catalysts promoted the oxidations of organic substrates such as styrenes, cycloalkenes, a,b-unsaturated ketones, steroids, benzylic hydrocarbons and arenas [13]. N4 macrocyclic complex catalysts for the electrochemical oxidation of thiols exhibited high efficiency, and their activities were adjusted dramatically by tuning the redox properties of the macrocyclic molecular electrodes [14]. Disulfane analogues are important biochemicals, which are generally produced in the alkali cleavage of insulins [15]. For instance, hydropersulfides have been implicated as important intermediates in the cell-killing action of the anticancer natural products leinamycin and varacin. It has been suggested that disulfanes mediate to convert molecular oxygen to reactive oxygen species under physicologically relevant conditions [16]. In 4-thiouridine’s biosynthesis, evidences proved that the biological sulfur was transferred via persulfide groups [17]. In the biomimic preparations of [Fe4S4] clusters, RSH compounds were partially converted into RSSR disulfanes [18-20], and these evidences were beneficial for exploring the forming mechanism of [Fe4S4] clusters. As far as we know, there was no disulfane’s preparation over metalloporphyrin catalysts has been reported. Above research works encouraged us to investigate the catalytic preparation of 1, 2-bis(1-phenyl-1H-tetrazol-5-yl) disulfane (L-L) over metalloporphyrin catalysts. Furthermore in situ UV-Vis and CV determinations supplied more kinetic information toward better understanding of the catalytic mechanism in L-L preparation.
2. Experimental
Metalloporphyrin catalysts were synthesized according to literature method [21-23]. As a typical run for catalytic reaction, 0.15 g of 1-phenyl-1H-tetrazole-5-thiol (HL) and 1.5 mg of THPPMnCl were dissolved in 20 ml of alkali methanol at 25˚C, followed by stirring for 2 h, and a yellow product was deposited. This reaction is described in Scheme 1. After filtration, washing with water and drying, the product was characterized by determinations of melting point (144˚C - 146˚C), 1H-NMR (doublets at 6.65 - 6.72 ppm and triplets at 7.10 - 7.25 ppm with integration areas of 1.2180 and 1.7257), IR (522.4 cm–1 assigned to nS-S) and elementary analysis [Found. (Calc. for C14H10N8S2%): C 47.61 (47.44), H 2.79 (2.84), N 31.58 (31.62), S 18.72 (18.60)]. The quantitative analyses were performed by HPLC and UV-Vis techniques. The electrochemical equipment was made from combination of a model JSH-1 potentiostat, a model DCG-2 multipleprogramm-function designator and a Type 3088 function recorder. Before electrochemical determinations, the solvent dimethyl sulfoxide (DMSO) was in turn dried by 5A molecular sieves, refluxed with CaH2, distilled in vacuum and sealed for later use. Purification of Ar: The gas passed in a row through two gas-washing bottles containing KMnO4 and K2Cr2O7-H2SO4 solution, three columns of active copper granules (180˚C); 5A molecular sieves and silica gel. After these processes the purity of Ar reached 99.99%.
3. Results and Discussion
3.1. Activity and Selectivity of Metalloporphyrin Catalysts
Table 1 presents catalytic activities in dimerization of HL under various conditions. Notably, the Mn-porphyrin catalysts are very active for this reaction, and only oxidative coupling products are obtained. For example, THPPMnCl exhibits the conversion at 57.6% with the selectivity for L-L at 100%. Apparently, high activity and selectivity in oxidative coupling of HL is potentially important for industrial applications in preparing L-L and its intermediated compounds. Furthermore, it is interesting to note that various Mn-porphyrin catalysts exhibit quite different activities (Table 1, Runs 1, 17-21). For example, TMOPPMnCl gives rise to the L-L yield at 48.2%, and TNPPMnCl supplies the L-L yield only at 5.6%, which is low as the yield over simple manganese salts. The activity order over these Mn-porphyrin catalysts is as follows: THPPMnCl > TMOPPMnCl > TAPPMnI5 > TCPPMnCl > TPPMnCl > TNPPFeCl. The Mn-porphyrin catalysts with electron-donating groups, such as -OH, -NH2, and -OCH3, and with large solubility in gives high
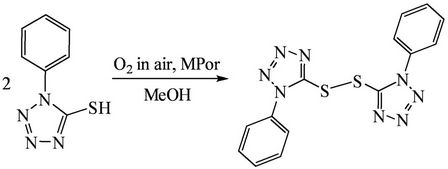
Scheme 1. The dimerization of HL over metalloporphyrin catalyst in the air.
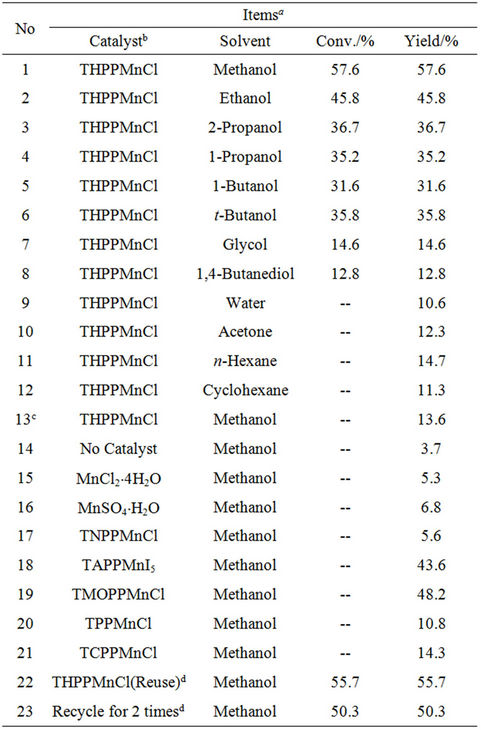
Table 1. Catalytic activities in oxidative coupling of HL (1- phenyl-1H-tetrazole-5-thiol) by air under various conditions over Mn-porphyrin and Mn-salt catalysts in the solution with NaOH (0.1 mol/L).
conversions, while those catalysts with electron-withdrawing groups of -NO2, -COOH, and even -H, or with poor solubility in alkali methanol, give low conversions. Possibly, the catalytic activities could be related to both stability and low redox potential of the intermediate of L-MnPor.
3.2. Influences of the Reaction Media
Moreover, it is also observed that NaOH is a necessary agent for the catalytic conversion (Table 1, Runs 1 and 13). Without sodium hydroxide, only 13.6% of the product could be prepared. Possibly, this catalytic reaction could be promoted by the alkali media to help the mercapto ionization in HL molecules, and therefore the produced L– anions were easy to coordinate the center metals to form the active intermediates of RS−-Mn(III)- porphyrin.
It is worth noting that the solvents strongly influence the catalytic activities over THPPMnCl catalyst (Table 1, Runs 1-12). Notably, by using both the solvents with strong polarity such as H2O, glycol, and 1,4-butanediol and with weak polarity such as n-hexane and acetone, THPPMnCl is catalytically inactive for oxidative coupling of HL. However, the use of the solvents with medium polarity including methanol, ethanol, 1-propanol, 1-butanol, tert-butanol, and 2-propanol shows high activities for the reaction (conversions at 31.6% - 57.6%). Particularly, methanol gives the highest conversion at 57.6%. Obviously, the selection of solvents with suitable polarity is important for the reaction.
3.3. Reuse of Active Catalyst
More importantly, a recycle of THPPMnCl catalyst also shows high activity (Table 1, Runs 22 and 23). For example, the reuse of the catalyst exhibits the conversion at 55.7%, and the recycled catalyst for 2 times gives the activity at 50.3%. However, the recycled catalyst for 5 times gives the catalytic activity at 11.6%, which is reasonably assigned to the loss of the catalyst during the separation of the catalyst with the product. Because the catalyst for the first run is only 1.5 mg, and the separation of the catalyst with the product for 5 times resulted in the loss of the catalyst significantly. If the catalyst used in the reaction are 10 mg for the first run, the recycled catalyst for 5 times still gives the catalytic activity at near 45%. Obviously, the catalyst losing should not be ignored in a system with larger amount of the catalyst.
3.4. In Situ UV-Vis Determinations
The in situ UV-Vis spectra for the catalytic conversion systems under various conditions were given in Figure 1. Generally, MnIIITHPP exhibited a Soret band at 472 nm. However, after mixing with HL under flowing nitrogen, the band at 472 nm reduced, and a new Soret band from MnIITHPP species appeared gradually at lmax = 423 nm. In contrast, without HL, the Soret band from MnIIITHPP species kept changeless. These results suggest that MnIII- THPP species can be easily reduced by HL to form MnIITHPP species. After exposing the mixture to the air
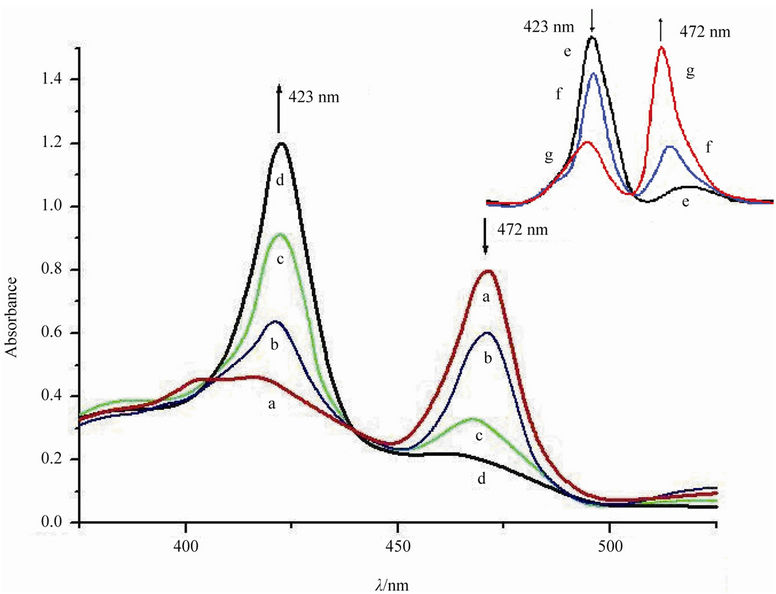
Figure 1. UV-Vis spectra of a mixture of MnIIITHPPCl (1 ´ 10–3 mol/dm3), HL (0.1 mol/dm3), NaOH (0.1 mol/dm3), and methanol treated by nitrogen for (a) 0 min; (b) 20 min; (c) 50 min; (d) 80 min, after d, exposure air for (e) 0 min; (f) 10 min; (g) 20 min. For removing the dissolved oxygen, a mixture of MnIIITHPPCl, NaOH, and methanol treated by nitrogen for 20 min. All experiments were done at room temperature (25˚C) in 20 mL methanol solution containing 0.1 mol/L of NaOH.
within enough time, the band at 472 nm appeared again, while the band at 423 nm reduced gradually. These results suggest that MnIITHPP species was gradually oxidized by oxygen from the air, and MnIIITHPP species was obtained again.
3.5. Electrochemistry
The electrolyte is a dimethyl sulfoxide (DMSO) solution containing 2.0 ´ 10–3 mol/L MnIIITPPCl and 0.1 mol/L (But)4NClO4 (tetrabutylammonium perchloride, TBAP) in which the O2 is removed by flowing argon. Under argon protection and at scan rate of 200 mV/s, the cyclic voltammetric experiments are carried out, and the cyclic voltammograms (CV) are seen in Figure 2. The CV of MnTPPCl (Figure 2(a)) presents three pair of current peaks around –0.72 (omitted), –1.77 and –2.28 V, reflecting the redox processes of Mn(VI)TPP-Mn(III)TPP, Mn(III)TPP-Mn(II)TPP, and the redox process of porphyrin ring (Mn(II)TPP-Mn(II)TPP–) respectively. After HL adding, the pa around –1.77 V disappears, and a new pc appears around 1.90 V (Figure 2(b)). The new pc must be caused by the oxidation of a new species containing MnTPP and HL, an active species in L-L forming process.
3.6. Possible Mechanism in HL’s Dimerzation
Based on the in situ UV-Vis and electrochemical determinations, a possible mechanism for catalytic dimerization of HL was described as Scheme 2. Step 1 axial coordination: After dehydrogenation of HSR, the produced –SR anion coordinated with Mn(III)TPP molecules at the axial position. Step 2 chemical reduction and hemolytic cleavage: The electron in –SR was transferred to the central metal and Mn(III)TPP species was reduced to Mn- (II)TPP species, and the cathodic current peak from Mn- (III)TPP to Mn(II)TPP species around –1.77 V (Figure 2(a)) therefore was not observed (Figure 2(b)). This is a typical chemical-electrical (CE) process. Meanwhile RSMn(II)TPP was quickly homolyzed to form Mn(II)TPP molecule and ·SR radical. Owing to their conjugating
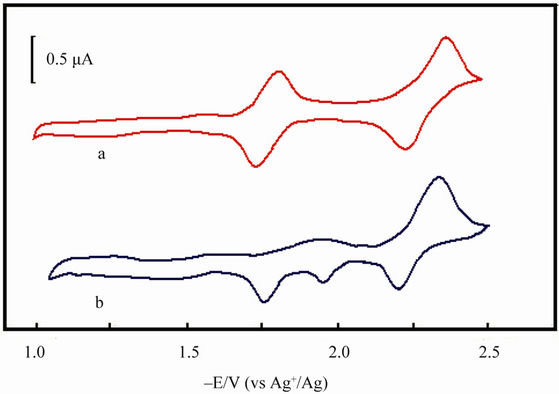
Figure 2. The cyclic voltammetric curves of Mn(III)THPPCl (1 ´ 10–3 mol/dm3) in DMSO with HL (0.1 mol/dm3) (b) and without HL (a). Supporting electrolyte: (But)4NClO4 (0.1 mol/L); scan rate: 200 mV/s; reference electrode: luggin capillary, Ag/AgNO3 in MeCN (0.1 mol/L).
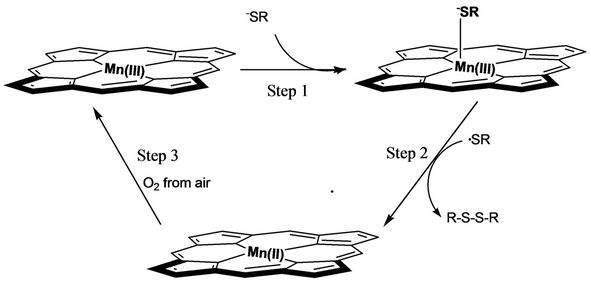
Scheme 2. The proposed mechanism for the catalytic dimerization of HL based on in situ UV-Vis and CV determination.
configurations, the ·SR radicals were stable enough to collide with each other, and as a result the coupling disulfane products were generated. By the way, in the CV of Mn(III)TPP with HL in DMSO system, the anodic current peaks (ipa) from RS-Mn(II)TPP and Mn(II)TPP to Mn(III)TPP were clearly monitored (Figure 2(b)). Step 3 catalyst recovery: Actually, in this catalysis the deactive Mn(II)TPP was oxidized by O2 from air to form the active species of Mn(III)TPP. The in situ UV-Vis determinations also indicated the interconversion between Mn- (III)TPP and Mn(II)TPP (Figure 1).
4. Conclusion
In conclusion, an organic compound of 1,2-bis(1-phenyl-1H-tetrazol-5-yl) disulfane (L-L) has been synthesized by catalytic dimerization of 1-phenyl-1H-tetrazole-5-thiol (HL) over metalloporphyrin catalysts in alkaline methanol solutions. Experimental results indicate that Mn-porphyrin catalysts with electron-donating groups and with enough solubility in alkaline solvents give high conversions of HL. Meanwhile, a possible mechanism for catalytic dimerization of HL was proposed based on in situ UV-Vis and electrochemical determinations. This catalytic dimerization is useful in research on sweetening of fuel oil [24] and biodegradation [25].
5. Acknowledgements
This work is financially supported by The National Scientific Foundation of China (No. 20771073) and the 211 Project of Guangdong Province (3rd), China.
REFERENCES
- Y. Yan, E. H. Kang, K. E. Yang, S. L. Tong, C. G. Fang, S. J. Liu and F. S. Xiao, “High Activity in Selective Catalytic Oxidation of Naphthol to 2-Hydroxy-1,4-naphthoquinone by Molecular Oxygen under Air Pressure over Recycled Iron Porphyrin Catalysts,” Catalysis Communications, Vol. 5, No. 8, 2001, pp. 387-390. doi:10.1016/j.catcom.2004.05.003
- T. Chen, E. H. Kang, G. P. Tan, S. J. Liu, S. D. Zheng, K. E. Yang, S. L. Tong, C. G. Fang, F. S. Xiao and Y. Yan, “Highly Selective Catalytic Preparation of Bis(4-Oxobenzo- 2-cyclohexen-1-yl) Amine from 1-Naphthylamine Oxidation over Metalloporphyrin Catalysts by Molecular Oxygen under Air Pressure and by Hydrogen Peroxide,” Journal of Molecular Catalysis A: Chemical, Vol. 252, No. 1-2, 2006, pp. 56-62. doi:10.1016/j.molcata.2006.02.039
- B. Meunier, “Metalloporphyrins as Versatile Catalysts for Oxidation Reactions and Oxidative DNA Cleavage,” Chemical Reviews, Vol. 92, No. 6, 1992, pp. 1411-1456. doi:10.1021/cr00014a008
- M. L. Merlau, S.-H. Cho, S.-S. Sun, S. T. Nguyen and J. T. Hupp, “Anthracene-Induced Turnover Enhancement in the Manganese Porphyrin-Catalyzed Epoxidation of Olefins,” Inorganic Chemistry, Vol. 44, No. 15, 2005, pp. 5523-5529. doi:10.1021/ic0505596
- J. Rosenthal, T. D. Luckett, J. M. Hodgkiss and D. G. Nocera, “Photocatalytic Oxidation of Hydrocarbons by a Bis-iron(III)-μ-oxo Pacman Porphyrin Using O2 and Visible Light,” Journal of the American Chemical Society, Vol. 128, No. 20, 2006, pp. 6546-6547. doi:10.1021/ja058731s
- N. A. Stephenson and A. T. Bell, “Mechanistic Study of Iron(III) [Tetrakis(pentafluorophenyl)Porphyrin Triflate (F20TPP)Fe(OTf) Catalyzed Cyclooctene Epoxidation by Hydrogen Peroxide,” Inorganic Chemistry, Vol. 46, No. 6, 2007, pp. 2278-2285. doi:10.1021/ic060757c
- C.-M. Che, J. L. Zhang, R. Zhang, J. S. Huang, T. S. Lai, W. M. Tsui, X. G. Zhou, Zh. Y. Zhou, N. Y. Zhu and C. K. Chang, “Hydrocarbon Oxidation by β-Halogenated Dioxoruthenium(VI) Porphyrin Complexes: Effect of Reduction Potential (RuVI/V) and C-H Bond-Dissociation Energy on Rate Constants,” Chemistry—A European Journal, Vol. 11, No. 23, 2005, pp. 7040-7053. doi:10.1002/chem.200500814
- N. A. Stephenson and A. T. Bell, “A Study of the Mechanism and Kinetics of Cyclooctene Epoxidation Catalyzed by Iron(III) Tetrakispentafluorophenyl Porphyrin,” Journal of the American Chemical Society, Vol. 127, No. 24, 2005, pp. 8635-8643. doi:10.1021/ja043380n
- A. D. Q. Ferreira, F. S. Vinhado and Y. Iamamoto, “Characterization of Mn(III)porphyrin Immobilized on Modified Silica Surfaces by EXAFS Spectroscopy: A Promising Tool for Analysis of Supported Metalloporphyrin Catalysts,” Journal of Molecular Catalysis A: Chemical, Vol. 243, No. 1-2, 2006, pp. 111-119. doi:S1381116905005121
- M. Moghadam, S. Tangestaninejad, M. H. Habibi and V. Mirkhani, “A Convenient Preparation of Polymer-Supported Manganese Porphyrin and Its Use as Hydrocarbon Monooxygenation Catalyst,” Journal of Molecular Catalysis A: Chemical, Vol. 217, No. 1-2, 2004, pp. 9-12. doi:10.1016/j.molcata.2004.03.001
- A. Goifman, J. Gun, V. Gitis, A. Kamyshny Jr., O. Lev, J. Donner, H. Börnick and E. Worch, “Pyrolysed Carbon Supported Cobalt Porphyrin: A Potent Catalyst for Oxidation of Hydrogen Sulfide,” Applied Catalysis B: Environmental, Vol. 54, No. 4, 2004, pp. 225-235. doi:S0926337304004242
- B. Akagah, A. T. Lormier, A. Fournet and B. Figadère, “Oxidation of Antiparasitic 2-Substituted Quinolines Using Metalloporphyrin Catalysts: Scale-Up of a Biomimetic Reaction for Metabolite Production of Drug Candidates,” Organic & Biomolecular Chemistry, Vol. 6, No. 24, 2008, pp. 4494-4497. doi:10.1039/b815963g
- C.-M. Che and J.-S. Huang, “Metalloporphyrin-Based Oxidation Systems: From Biomimetic Reactions to Application in Organic Synthesis,” Chemical Communications, No. 27, 2009, pp. 3996-4015. doi:10.1039/b901221d
- F. Bedioui, S. Griveau, T. Nyokong, A. J. Appleby, C. A. Caro, M. Gulppi, G. Ochoa and J. H. Zagal, “Tuning the Redox Properties of Metalloporphyrinand MetallophthaLocyanine-Based Molecular Electrodes for the Highest Electrocatalytic Activity in the Oxidation of Thiols,” Physical Chemistry Chemical Physics, Vol. 9, No. 26, 2007, pp. 3383-3396. doi:10.1039/B618767F
- D. Cavallini, G. Federici, E. Barboni and M. Marcucci, “Formation of Persulfide Groups in Alkaline Treated Insulin,” FEBS Letters, Vol. 10, No. 2, 1970, pp. 125-128. doi:10.1016/0014-5793(70)80432-X
- T. Chatterji, K. Keerthi and K. S. Gates, “Generation of Reactive Oxygen Species by a Persulfide (BnSSH),” Bioorganic & Medicinal Chemistry Letters, Vol. 15, No. 17, 2005, pp. 3921-3924. doi:10.1016/j.bmcl.2005.05.110
- C. M. Wright, P. M. Palenchar and E. G. Mueller, “A Paradigm for Biological Sulfur Transfers via Persulfide Groups: A Persulfide-Disulfide-Thiol Cycle in 4-Thiouridine Biosynthesis,” Chemical Communications, Vol. 22, 2002, pp. 2708-2709. doi:10.1039/B208626C
- R. H. Holm, P. Kennepohl and I. E. Solomon, “Structural and Functional Aspects of Metal Sites in Biology,” Chemical Reviews, Vol. 96, No. 7, 1996, pp. 2239-2314. doi:10.1021/cr9500390
- C. Y. Zhou, J. W. Raebiger, B. M. Segal and R. H. Holm, “The Influence of Net Charge on the Redox Potentials of Fe4S4 Cubane-Type Clusters in Aprotic Solvents,” Inorganic Chimica Acta, Vol. 300-302, 2000, pp. 892-902. doi:10.1016/S0020-1693(99)00593-9
- C. Zhou, L. Cai and R. H. Holm, “Synthesis of a [Fe4S4]- S-Ferriheme Bridged Assembly Containing an Isobacteriochlorin Component: A Further Analogue of the Active Site of Sulfite Reductase,” Inorganic Chemistry, Vol. 35, No. 10, 1996, pp. 2767-2772. doi:10.1021/ic951493p
- A. D. Adler, F. R. Longo, J. D. Finarelli, J. Goldmacher, J. Assour and L. Korakoff, “A Simplified Synthesis for Meso-Tetraphenylporphine,” Journal of Organic Chemistry, Vol. 32, No. 2, 1967, pp. 476-478. doi:10.1021/jo01288a053
- A. D. Adler, F. R. Longo, F. Kampas and J. Kim, “On the Preparation of Metalloporphyrins,” Journal of Inorganic and Nuclear Chemistry, Vol. 32, No. 7, 1970, pp. 2443- 2445. doi:10.1016/0022-1902(70)80535-8
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney, and A. M. Marguerettaz, “Rothemund and Adler-Longo Reactions Revisited: Synthesis of Tetraphenylporphyrins under Equilibrium Conditions,” Journal of Organic Chemistry, Vol. 52, No. 5, 1987, pp. 827-836. doi:10.1021/jo00381a022
- D.-H. Xia, Y.-X. Su and J.-L. Qian, “Study on the Oxidation Mechanism of Mixed Thiols in Light Oil Sweetening. 3. Apparent Kinetics of the Catalytic Cooxidation of Mixed Thiols in Gas-Liquid-Solid Systems,” Vol. 38, No. 4, 1999, pp. 1291-1294. doi:10.1021/ie980541l
- R. C. van Leerdam, M. Bonilla-Salinas, F. A. M. de Bok, H. Bruning, P. N. L. Lens, A. J. M. Stams and A. J. H. Janssen, “Anaerobic Methanethiol Degradation and Methanogenic Community Analysis in an Alkaline (pH 10) Biological Process for Liquefied Petroleum Gas Desulfurization,” Biotechnology and Bioengineering, Vol. 101, No. 4, 2008, pp. 691-701. doi: 10.1002/bit.21933
NOTES
*Corresponding authors.