Neuroscience & Medicine
Vol.2 No.3(2011), Article ID:7413,29 pages DOI:10.4236/nm.2011.23033
The Functions of the Amyloid Precursor Protein Gene and Its Derivative Peptides: II Experimental Evidence and Clinical Studies*
![]()
Neurodegenerative Disorders Research Pty Ltd., Subiaco, Australia.
Email: #publications@panegyres.com.au
Received November 30th, 2010; revised April 20th, 2011; accepted July 25th, 2011.
Keywords: Amyloid Precursor Protein Gene
ABSTRACT
In vitro studies suggest Amyloid Precursor Protein Gene (APP) is involved in interaction with the extracellular matrix, neurite growth, adhesion, development, synaptic function, platelet function, and interaction with GTP binding proteins. In vivo experiments show a role in embryonic development, response to cerebral excitotoxicity and gliosis, response to brain injury including ischaemia, hypothalamic function, locomotor function, learning and memory. In vitro observations indicate Aβ has a role in amyloid formation, excitotoxic neuronal injury, tachykinin interaction, endothelial vasoconstrictor response, calcium and oxidative stress, free radical interaction, cell membrane fluidity, apoptosis, astrocyte stimulation, and microglial interaction. Other studies suggest important roles for Aβ oligomers in synaptic function and as an antimicrobial peptide. In vivo investigations show involvement in memory function, the blood brain barrier, and tachykinin response to cerebral injury.
1. Functions of APP
1.1. In Vitro Studies: APP
Secretion of Amyloid Precursor Protein Gene (APP) involves the action of multiple cleavages [1] and in physiological conditions the production of non-amyloidogenic products from the action of integral membranebound metallopeptidases (secretases) [2].
APP is soluble and has soluble derivatives in CSF and other biological fluids [3,4]. Biochemical studies suggest that APP is proteolytically cleaved by a putative enzyme termed α-secretase, this cleavage occurring at position 16/17 of the Aβ region [5]. The generated N-terminal fragment is soluble, termed sAPPα, and the C-terminal fragment remains in the membrane. APP may be reinternalized [6] and its proteolysis diverted towards endosomes and liposomes where a β-secretase acts at the Nterminus of Aβ to generate the sAPPβ fragment containing the Aβ domain [7,8]. The Aβ sequence is liberated from the membrane by the action of γ-secretase which acts at the C-terminus of Aβ [9].
APP is first synthesized as a membrane-bound protein carrying asparagine-linked sugar chains. It is then processed into higher molecular mass forms by the additions of sulphate, phosphate and sialic acid residues—adding about 20 kDa in molecular weight [10]. The chondroitin sulfate proteoglycan form of APP is known as APPICAN or L-APP and is an alternatively spliced form of APP lacking exon 15. This results in the formation of AspXaa-Ser-Gly consensus sequence site for chondroitin sulfate chain attachment to serine 619 of L-APP which is 16 amino acids upstream of the Aβ sequence; this enhances the binding of APP to the extracellular matrix [11].
Embryonic cortical neurons are believed to have two pools of APP: one within the axons and cell bodies which is mobilized after paraformaldehyde exposure and elevation of the extracellular Ca2+ concentration [12]. This APP is on the cell membrane and interacts with the cytoskeleton through the C-terminal. The other pool, which is probably endosomal, is not mobilized by these treatments. The C-terminal fragment of APP has been shown to form non-selective ion channels or pores in membranes of Xenopus oocytes, a property which is modulated by metal ions including Ca2+, Zn2+, Fe2+/Fe3+ and Al3+ [13].
APP isoforms with and without KPI domains stimulate adhesion of PC12 cells to tissue culture dishes [14]. These effects were enhanced by adding NGF and basic fibroblast growth factor (bFGF). NGF induced neuronal differentiation is accompanied by differential splicing of APP in PC12 cells [15]. The neurite promoting properties of APP on PC12 cells was present at low concentrations (10–10 M) and with APP in both soluble and membraneassociated forms [16]. APP increased neurite length and branching but had no effect on the number of neurites. At high doses APP was toxic [17]. Addition of APP antibodies reduced the effect of NGF, suggesting an interaction between these two substances. The effect of APP on neurite outgrowth may be mediated through an interaction with heparin sulfate proteoglycans and NGF [18]. The effect of NGF on the APP probably involves neutrophin binding to the p75 neurotrophin receptor [19]. Toxicity of APP at high concentrations was confirmed in a murine embryonal carcinoma line transfected with human APP cDNA [20]. When differentiation into postmitotic neurons was induced by retinoic acid the cells possessed large amounts of APP and died. APP may be a substrate for caspase-3 in motor neurons [21].
Transfection of APP into a cell line that does not have APP revealed greater adherence, increased neurite outgrowth, and more rapid growth in low serum [22]. These cells were protected from the toxic effects of glutamate and Aβ. Substratum bound APP stimulated neurite outgrowth in cultures of chick sympathetic ganglia and mouse hippocampal neurons and was dependent on the presence of a substrate-bound proteoglycan similar to laminin [23]. A 14 kDa N-terminal fragment of APP bound to heparin, pointing to an interaction between APP and the extracellular matrix.
CHO cells transfected with APP cDNA stimulated neurite outgrowth and cell adhesion in hippocampal neurons after co-culture [24]. These effects were greater with the KPI+ isoforms APP 770 and 751, than with APP 695. Amino acids 361 - 648 mediated this effect. This suggested that two extracytoplasmic domains promoted neurite outgrowth: one common to all isoforms, and the other localized to KPI+ domain. In a clonal neuronal cell line the RERMS sequence of APP (independent of KPI+) encouraged neurite induction, cell surface binding, and inositol polyphosphate stimulation [25]. These effects were shared with Aβ if it contained this sequence. An antagonist containing the sequence RMSQ successfully inhibited these effects, pointing to a possible endogenous antagonist.
With immunoelectron microscopy APP was found to be incorporated into the extracellular matrix between preand post-synaptic cells [26]. The secretory N-terminal of APP (sAPPα) binds to a saturable, specific receptor and binding protein which is probably a component of cell surface microdomains [27]. The presence of APP in extracellular microparticles was demonstrated using confocal immuno-fluorescence microscopy [28]. All APP isoforms had high affinity interactions with heparin sulfate proteoglycan [29]. The APP-collagen interaction is probably mediated through a heparin bridge mechanism [30]. KPI+ isoforms had the weakest affinity, possibly related to steric or conformational effects. Specific binding of APP to the IKVAV site of laminin A has been demonstrated [31]. Laminin, heparin sulfate proteoglycan, and other extracellular matrix glycoproteins are important in advancing neurite outgrowth.
The binding of vascular heparin sulfate proteoglycan to APP may be mediated by the N-terminal region of the Aβ sequence and may influence its proteolysis and promote fibril formation [32,33]. The heparin binding neuritotropic site is probably distinct [34]. The extracellular matrix probably influences the synthesis of APP in microglial cells and influences their migration, target recognition and binding [35]. APP binding to heparin is probably increased by zinc [36]. The heparin binding domains are found in the alpha-helical regions of APP [37]. There may be four distinct heparin binding domains suggesting that APP may interact with more than one glycosaminoglycan [38]. Treatment of PC12 cells with APP antisense oligonucleotides reduced neuritic processes in the presence of laminin, confirming an interaction between APP and molecules of the extracellular matrix [31]. In neuronal cultures APP colocalizes with integrins on the cellular surface in a discontinuous manner, further supporting a role for APP in cellular adhesion [39].
All APP isoforms have been detected in mouse oocytes, preimplantation embryos, and in all postimplantation stages—strongly supporting a wider role for APP in development [40]. Induction of neuronal differentiation with retinoic acid in a murine embryonal carcinoma cell line (P19) was accompanied by the production of APP 695 [41]; this was also seen in hippocampal cultures. Another study with P19 cells showed that differentiation to neuronal phenotype with retinoic acid was accompanied by the expression of all the isoforms of APP [42]. APP 695 was the most abundant in the early stages, with APP 751 and 770 increasing in amount with greater age of the cultures.
Immunofluorescence, laser confocal microscopy, and immunoelectron microscopy showed co-localization of APP and synaptophysin—a 39 kDa synaptic vesicle protein [43]. This co-localization was demonstrated in human and rat brain, and at neuromuscular junctions. Over expression of APP in neuronal and other cell lives has been shown to enhance toxicity and change its processing by augmenting C-terminal fragments [44]. The Cterminal fragment of APP may decrease the frequency of spontaneous postsynaptic currents through an effect on K channels via a cGMP pathway, and the N-terminal fragment may increase impulse evoked postsynaptic currents involving protein kinase [45]. APP was also found in the vesicular elements of neuronal perikarya, dendrites and axons. The triplet lysine residue of APP (Lys 724 - Lys 725 - Lys 726), which is located in the carboxyl region after the transmembrane domain, may anchor APP in the cell membrane and influence the action of secretases [46]. Using immunodetection in rat peripheral nerve preparations, it was shown that APP is synthesized in the soma and is exported by fast axonal transport like neuropeptides and enzymes [47]. APP is found in the plasma membrane and intracellular membrane residues [48].
APP is released by human platelets [49] and inhibits coagulation factor X1α [50] Calcium ionophores, phosphatase inhibitors, anti mu-chain or anti CD3 antibodies in T cells, and phorbol esters in monocytes were able to induce surface membrane APP suggesting a role as a receptor in the haemopoietic and immune system [51]. Endothelial cells have been shown to be vulnerable to oxidative injury if there is over expression of APP [52]. Heat stress increases APP on endothelial cells and endothelial cells enzymes cleave APP on platelets [53,54]. APP KPI+ isoforms are identical to protease nexin II and inhibit tissue plasminogen activators, urokinase and thrombin [55,56]. A new member of the protein X11 family, X11L2, has been shown to bind APP in its intracellular domain further supporting the concept of an interaction of APP with coagulation factors [57]. Protease inhibitors may stimulate neurite extension and may be produced by glial cells [55,58,59].
Aurintricarboxylic acid (ATA), a metabolic inhibitor, decreased APP mRNA in cultured neurons [60]. In the same study interleukin 1 (IL-1) stimulated APP mRNA. The ATA-induced reduction in APP was accompanied by loss of neuronal phenotype, reduced adhesion, and neurite retraction. Cultures of sympathetic and cortical neurons grown initially with NGF showed a reduction in APP 695; KPI+ isoforms were increased when NGF was withdrawn [61]. Heat-shock, IL-1, acetylcholine, and TGF β1 have all been shown to induce APP mRNA in neuronal and other cells [62-66]. In human glioma cells IL-1β decreased the expression of APP [67]. Interleukin- 1 may alter processing and secretion of APP to augment sAPP and decrease Aβ [68]. There may also be a cellular selectivity in the influence of cytokines on APP with 1L-6 augmenting APP mRNA in neuronal but not glial cell lives [69]. The translation of APP may be determined by 1L-1α and 1L-1β operating upon a 90 nucleotide sequence in the untranslated regions (S-UTR of APP); this translational enhancer maps +55 to +144 nucleotides from the 5’ cap site and is homologous to regulatory elements found in ferritin genes [70].
APP may interact and form protein—protein complexes with the GTP binding protein G(o) [71], the enzyme glyceraldehyde-3-phosphate dehydrogenase [72]; and APOE [73]. The APOEε4 isoform was more potent than the APOEε3 isoform in compromising the neuroprotective properties of sAPP. The binding of APP to APOE is probably through its N-terminal involving regions 1 - 207 of APP and 1 - 191 of APOE and is independent of the Aβ sequence [74]. APOE may therefore have a chaperone function by which the metabolism and function of APP may be modulated. The isoforms of APP may form complexes with themselves and other proteins [75]. The neuronal adapter proteins FE65 and DISABLED bind to the C-terminal of APP where they may serve as molecular scaffolds for the assembly of protein complexes [76]. Heterotrimetric protein complexes involving X11α, mLin2 and mLin7 may be important in the localization of proteins in cells and the trafficking of APP in neurons [77,78].
The interaction of APP with FE65 may mobilize APP to the cell surface [79]. The cytosolic molecular chaperone Hsc73 binds adjacent to the post-transmembrane region of APP at the KFFEQ site of the C-terminal fragment and may be the targeting signal that determines lysosomal proteolysis [80]. APP may contain a domain which inhibits a matrix metallo-proteinase known as gelatinase A [81].
Other neurotransmitter and endocrine factors have been shown to interact with APP and may be involved in its regulation. Serotonin 5HT2α and 5HT2c receptors stimulate sAPPα (and APLP2) through a mechanism involving phospholipases and kinases [82]. In the same experiments muscarinic acetylcholine receptor agonists also stimulated sAPPα. Additional experiments have confirmed that an agent that is an M1 muscarinic receptor agonist and M2 antagonist stimulates sAPP and reduces Aβ [83]. Other data reveals that M1 and M3 muscarinic stimulation of sAPP may involve coupling with protein kinase C [84]. Thyroid hormone reduces APP transcription via a negative hormone response element [85]. Prostaglandin E2 stimulates APP mRNA via cAMP and this may be a mechanism by which brain injury and inflammation activates APP [86].
Electrical stimulation of rat hippocampal slices released N-terminal fragments of APP supporting the concept of APP’s involvement in synaptic processing [87].
1.2. In Vivo Studies: APP
APP immunoreactivity is widely distributed in the brains of mammals and mostly found in neurons [88,89]. Oligonucleotide probes to APP mRNA isoforms showed that APP 695 and 751 are the most abundant types in rodent and human brain [90,91]. APP 714 is greatest in the hippocampus, and APP 770 in ependymal cells in normal conditions. Full length APP probably accumulates at presynaptic terminals along with C-terminal fragments containing the Aβ domain and probably modulates synaptic activity in both forms [92].
With aging, APP immunoreactivity is found in smaller neurites in most regions of the rat CNS and this has been attributed to a decrease in axonal transport [93]. APP in the Purkinje cells of aging rats is located in the cell body and dendrites, and exists as a full length or truncated forms [94]. Development may influence the alternative splicing of APP such that KPI—isoforms predominate in early post natal life and correlate with maximum neurotrophic responsiveness [95].
The excitotoxin kainic acid damages the hippocampus and other brain regions after systemic and intraventricular administration [96]. Increased APP immunoreactivity has been observed several days after kainic acid treatment [97]. APP KPI+ deposition was shown immunocytochemically in astrocytes three days after kainic acid [98]. With in situ hybridization, increases in KPI+ isoforms were found after one day and were associated with reductions in KPI– isoforms in the hippocampus and other brain regions [99]. This stimulation in KPI+ correlated with changes in GFAP mRNA. Another study showed increases in KPI+ isoforms 12 hours after kainic acid in the dentate gyrus, CA1, and CA3 regions [100]. Neuroprotective agents including interleukin-1 receptor antagonists may modify the effects of excitotoxicity on APP expression [101,102]. Inflammatory processes may also influence the expression of APP [103].
APP KPI+ isoforms contribute to the plasticity of hippocampal sclerosis where their expression correlates with neuronal loss and gliosis in experimental temporal lobe epilepsy [104]. The changes in APP are preceded by induction of the immediate early genes c-fos and c-jun suggesting that these molecules are a component of the cascade of events leading to changes in APP [104,105]. There is tissue culture evidence for a direct role for c-jun in the regulation of APP [106].
Striatal injections of quinolinic acid in rats reduced APP KPI—in neurons, and stimulated APP KPI+ in glial cells [107]. Chemical lesions of the nucleus basalis of Meynert, adrenergic bundle, and dorsal raphe nucleus enhanced total APP expression in the cerebral cortex [108]. Ibotenic acid and colchicine have also been shown to stimulate APP [109,110]. Retinoic acid and DMSO increased total APP expression; KPI+ isoforms constituting most of this increase [111].
Penetrating needle, local head impact and other traumatic brain injuries stimulate APP immunoreactivity in damaged axons and reactive glial cells [112-115]. Focal cerebral ischaemia in the rat enhanced APP KPI+ gene expression as measured by northern analysis [116]. Stimulation of APP after rat focal ischaemia was confirmed immunocytochemically in adult and neonatal animals [117,118]. Similar increases in APP immunoreactivity have been found after global ischaemia in gerbils [119]. This degree of immunoreactivity has been related to the duration of ischaemia in the gerbil [120]. The deposition of APP has been provisionally localized to astrocytes in the ischaemic gerbil hippocampus [121]. Propentofylline, an adenosine A2 receptor agonist, decreased APP immunoreactivity in microglial cells and protected the gerbil hippocampus from ischaemia [122]. Propentofylline may enhance the neuroprotective functions of APP in ischemia [123].
Axotomy of the sciatic nerve increased APP 695 and APP KPI+ mRNAs in dorsal root ganglia [124]. After reinnervation, APP 695 levels decreased and KPI+ isoforms remained elevated. In the spinal cord, APP 695 returned to control levels after reinnervation. Rat sciatic nerve exposed to APP KPI+ and other protease inhibitors produced axonal sprouts [125]. Heat inactivated KPI+ and KPI– isoforms did not share this property. When rat sciatic nerve was treated with KPI+ Schwann cell mitosis and demyelination were found [126]. These findings give weight to in vitro evidence of the neurite-promoting properties of protease inhibitors.
Water deprivation and hypertonic saline injections stimulated APP immunoreactivity in the supraoptic and paraventricular hypothalamic nuclei [121], suggesting that APP may have broader physiological functions in response to stress.
Brain damage augments the proteoglycan form of APP (APPICAN) suggesting a novel proteolytic processing mechanism following injury [127].
Neonatal rats given anti-NGF antibodies developed neuronal degeneration in the superior cervical ganglion, associated with reductions in APP 695 mRNA and increases in APP 751 and 770 [77]. Multiple intracerebroventricular injections of NGF into rat pups did not induce changes in APP mRNA in the septal region and striatum [128]. Aging may decrease KPI+ mRNA isoforms correlating with lower NGF in the neocortex [129].
APP deficient mice have shown reactive gliosis and decreased locomotor activity indicating a role for APP in motor function and astrocytic responses [130,131]. When the mouse APP promoter was disrupted by insertion of an exon 2 cassette containing a transcription termination sequence, the animals produced one-tenth of the usual amounts of APP [132,133]. This led to an increased frequency of agenesis of the corpus callosum and severe impairment of spatial learning and exploratory behaviour. Mice containing the double knockout (KO) APLP2 and APP die within the first week of life and the 20% of surviving mice have low weight, poor righting reflexes, ataxia, spinning behaviour and head tilt [134]. Single APLP2 KO mice were normal indicating substitution of function with APP. Hippocampal neurons from APP knockout mice have diminished viability and decreased neurite development [135]. Mice lacking APP had enhanced age related cognitive decline, compromised longterm potentiation, and gliosis in the cerebral cortex and hippocampus [136] suggesting that APP is involved in the enhancement of synaptic function.
Infusions of sAPPα 695 and sAPPα 751 intracerebroventricularly in mice, trained to perform learning tasks assessing both short term and long term memory, had memory enhancement effects and reduced the deficits induced by pretreatment with scopolamine [137]. These effects were observed at low doses, blocked by antibodies to APP, and not a result of impaired motor performance or exploratory behaviour. These data suggests that sAPP may strengthen synapses involved in memory. The findings also support the concept of APP modulating the function of cholinergic neurons.
A 17 amino acid peptide fragment immediately C-terminal to the KPI domain is probably responsible for the memory enhancing properties of APP and may lead to an increase in the number of presynaptic terminals [138].
In order to elucidate the role of APP in AD transgenic mice have been developed which over-express normal and mutational human APP. A transgenic mouse expressing human APP 751 deposited Aβ in their brains with increasing age [139]. Production of a transgenic mouse with a human missense mutation for familial AD, with overexpression of mutant APP, led to neuronal loss, senile plaques but no neurofibrillary tangles [140]. Other transgenic models have been produced with similar findings. [10,141-157]. These transgenic mice also provide further data on the function of APP and confirm the findings in other model systems of the role of APP in age-related impairment of learning and memory, neuronal survival, neuritic changes, amyloid deposition, and abnormal phosphorylation of tau.
Phenotypic expression is influenced by the genetic background of the mouse line. APP transgenes in FUB/N or C57 BL/GJ mice are lethal whereas outbred lines develop amyloid plaques suggesting that the genetic background of the host modifies the functional expression of APP [158].
A transgenic mouse line expressing a double mutation in APP and PS1 had extensive amyloidosis with a reduction in the density of cholinergic synapses in the frontal cortex [100]. An interaction between APP and the cholinergic system is further strengthened by studies of the transgenic mouse over-expressing the C-terminal fragment of APP in which the activity of acetylcholinesterase was increased [159].
Transgenic mice may have a functional disturbance in glutamate neurotransmission. However, the glutamate receptors NMDA and AMPA are unchanged [160,161]. Serotonergic agonists were able to suppress aggressive behaviour in these transgenic animals [160].
Transgenic mice possessing a mutation at α-secretase site of APP displayed abnormal behaviour and neurotoxicity confirming that absence of α-secretase activity enhances Aβ neurotoxicity [162].
Dietary cholesterol may modify the expression of APP transgenes such that animals receiving a high cholesterol diet have reduced secreted forms of APP and Aβ [163]. Transgenic mouse models have enhanced microglial activation [164].
Brain transplants exposed to increased levels of APP695 showed plaque formation [165].
Inhibition of phosphatases by intraventricular infusions of okadaic acid into rats alters phosphorylation/ dephosphorylation homeostasis and results in amnesia. These animals have hyperphosphorylation of tau and increased Aβ deposition pointing to phosphorylation as a fundamental biochemical step in the metabolism of APP [166]. Chronic forebrain lesions, which reduce cholinergic activation of the neocortex, increase the expression of APP providing additional evidence for the role of the cholinergic system in APP metabolism [147]. An experimental model of autoimmune encephalomyelitis revealed an increase in APP expression in microglia providing further support for a role of inflammatory mediators in APP expression [167].
Hormonal manipulations may affect the function of APP. Oestrogens may reduce APP mRNA in focal ischaemia [168], and adrenalectomy augments APP immunoreactivity in the hippocampus [169]. Oestrogens and progestagens may both influence the age related decline APP695 mRNA in the hippocampus [170].
Drosophila possesses an APP-like gene encoding a protein with over 90% homology to human APP, termed APPL. Flies delete of APPL have defective fast phototaxis. This phenomenon was reversible with transgenic flies containing an APPL deletion with a normal human APP gene, suggesting functional homology and a broader role for APP in motor behaviour [171,172]. The development of transgenic Drosophila containing normal and mutant human APP possess a blistered-wing phenotype confirming the impression from other models that APP is involved in cell adhesion and signalling [173].
Caenorhabditis elegans also has an APP-like protein with significant homology to APP [174].
2. Functions of Aβ
2.1. In Vitro Studies: Aβ
The amyloidogenic fragment Aβ is found in soluble form in blood, CSF and supernatants from cultured human brain cells [4,7,175-178]. Phosphatases may stimulate the production of Aβ through the β-secretase pathway [179]. Aggregation and insolubility of Aβ is accompanied by toxicity, which can be reproduced by synthetic fragments containing amino acids 29 - 35 [180]. This part of the sequence is critical for initial aggregation and formation of the β-pleated sheet structure.
Aβ has a hydrophobic C-terminal domain, which takes a beta strand structure, and the N-terminal region adopts an alpha-helical or beta-strand conformation depending on the environmental conditions such as acidic pH, peptide concentration and incubation time [181-184]. Two dimensional nuclear magnetic resonance spectroscopy and sequence data has revealed that the helical regions of Aβ have sequence homology to the helical domains of prion proteins [185]. There may also be homology to the paired helical filaments which make up neurofibrillary tangles [186]. The hydrophilic residues in Aβ contribute to the specificity of the interactions between Aβ peptides and may influence the transformation from a β pleated sheet structure to a random coil [187]. Aβ deposits are not found in rodent brain and this might relate to differences in the amino acid sequence between rodents, and other species—unknown factors independent of the primary structure might also influence fibril formation [188- 191]. Amyloid structures in mammalian tissues might encourage covalent polymerization of reactive small molecules into larger molecules like melanin with cytoprotective properties. Amyloid minimizes toxicity by sequestering and limiting diffusion of toxic molecules, as found in Pmel17 amyloidogenesis in melanin [192].
Insertion of proline residues at positions 17 - 23 within the Aβ sequence inhibits aggregation [193] and replacement of a glutamine for a glutamate at position 22 accelerates fibril formation [194].
Aβ interacts with other proteins. It binds with high avidity to the N-terminus of its own precursor APP which may have important functional and pathophysiological implications [195]. It also binds to immunoglobulin IgG at the hinge region of the immunoglobulin heavy chain which preserves the antigen binding site [196]. It also forms heteropolymers with the alpha 2 macroglobulin receptor and extracellular matrix molecules [197].
The vulnerability of cultured neurons to glutamate, Nmethyl-D-aspartate and kainate is increased after the addition of Aβ [198]. Neurotrophic effects have been observed with low concentrations of Aβ in culture [199]. The RERMS domain of Aβ is the active region for cell surface binding and neurite extension [25]. This effect was reproduced using a fragment containing amino acids 25 - 35, a region having 90% sequence homology with substance P. Tachykinin antagonists inhibited this effect [199]. These observations require confirmation, as inconsistent reports exist on a possible interaction between Aβ and NK-1 receptors [25,200-202]. When the NK-1 receptor was expressed in Xenopus oocytes, Aβ required excitatory amino acids to activate this NK-1 receptor as measured by Ca2+ uptake and inositol triphosphate stimulation [199]. These observations may account for some of the negative findings of Aβ-tachykinin interactions as excitatory amino acids may need to coexist in the same experimental conditions. At high concentrations in vitro Aβ caused neuronal death in tissue culture experiments [203]. Aβ has shown to promote neuronal viability in primary cultures [204]. Using an organotypic slice culture system from the hippocampus, it was revealed that Aβ inhibits excitatory synapses in response to neuronal activity using NMDA/AMPA and Ca2+ dependent mechanisms [205]. Aβ was shown to act in an automatous manner upon both neurons and neighbouring cells, and as part of a negative feedback loop to regulate the degree of neuronal activity. Aβ also impairs long-term potentiation—a model of synaptic plasticity useful to understand learning and memory [205].
Aβ (25 - 35) has a vasoconstrictor response at the level of the microvasculature operating through endothelin and protein kinase C [206]. This effect might also involve an interaction with calcitonin gene related polypeptide (CGRP), substance P, and the serpin enzyme complex (SEC) [202]. Aβ may mimic the effects of substance P on nicotine secretion [207]. Aβ disrupts muscarinic receptor coupling to G proteins which mediate carbachol inositol phosphate stimulation and Ca2+ release [208].
Resting Ca2+ levels and Ca2+ responses to excitatory amino acids were enhanced after Aβ treatment in human cerebral cortical cells [209]. Aβ probably destabilizes Ca2+ regulation through an effect on Ca2+ channels [210]. Aβ (25-35) may also influence Ca2+ levels in astrocytes [211]. In astrocytes Aβ reduces intracellular Ca2+ concentrations; in neurons it leads to an increase [212]. Potassium channel openers such as diazoxide and levocromakalim protect neurons against Aβ toxicity [213]. Aβ (25-35) increases Ca2+ concentrations in microglial cells via L-type voltage gated channels similar to prion proteins [214]. Aβ stimulates the synthesis of the third component of complement in microglial cells [215].
Neurotoxic effects may be a result of oxidative injury and free radical formation [216-218]. The damaging effects of Aβ are believed to be both necrotic and apoptotic [219,220]. The cells may need to be post mitotic before being vulnerable to the effects of Aβ [221]. Calretinin containing neurons have been shown to be resistant to Aβ [222]. The mechanism of toxicity may require an interaction with tau protein kinase I, as antisense oligodeoxynucleotides to the mRNA of this enzyme prevented Aβ-induced death of hippocampal neurons [223]. Aβ has been shown to reduce cell membrane fluidity and induce membrane ruffling [224]. Apoptosis has been induced by Aβ in tissue culture of CNS neurons [220,225]. Ceramide, a membrane-permeable activator of NF kappa β, protects neurons against the toxicity of Aβ [226]. The toxicity of Aβ (25 - 35) may be mediated by phospholipase D activation [227]. Oestrogens protect neurons from Aβ induced neurotoxicity [228] and reduce the production of Aβ in neurons [229].
Stimulation of astrocytic and microglial IL-1 and bFGF was found after exposure to Aβ [230,231]. The microglia proliferated and transformed morphologically. Microglial cells were activated synergistically by Aβ (25 - 35) and γ-interferon [232]. Aβ had no obvious toxic effects on astrocytes, which envelop and probably phagocytose Aβ [231]. Macrophages may be directly stimulated by Aβ [233].
Functional Ca2+ channels have been formed by spontaneous association of Aβ in artificial membrane bilayers [234]. Nimodipine-sensitive Ca conductance was enhanced by Aβ in neuroblastoma cells [235].
When the low affinity NGF receptor p75 is expressed in PC12 cells, Aβ toxicity is potentiated [153]. NGF was preventive, suggesting an interaction between the neurotrophins and Aβ. The non-selective neurotrophin receptor p75 NTR, a member of the death receptor family binds Aβ and leads to neuronal death [236].
The toxicity of Aβ requires fibril formation. Transthyretin is the major Aβ binding protein in human and animal CSF, when added to the CSF of patients and controls it sequesters Aβ [237]. The molecular chaperones antichymotrypsin and APOE increase Aβ aggregation, with APOEε4 having the highest catalytic activity [238]. This observation providing a possible mechanism for the association between APOEε4 and late onset AD [239, 240]. The neurotoxicity of Aβ may be modified by cholesterol [241].
The formation of β-pleated sheets is also promoted by aluminium and inhibited by silicates [242,243]. Metal induced oxidation favours amyloidogenesis of Aβ [244]. Zinc binds human Aβ more avidly than that from the rat; this may contribute to differences in Aβ deposition between these two species [301]. Iron ions also promote Aβ aggregation [245]. Congo red inhibits fibril formation as does thioflavin T [246-248]. The Nand C-terminal of Aβ 25-35 contribute to aggregation, but amino acids 33 - 35 seem critical [177,223].
Aβ interacts with other proteins involved in the pathogenesis of AD such as α-synuclein, which is the major constituent of the non Aβ component of AD amyloid plaques (NACP), as Aβ stimulates the oligomerization of α-synuclein via the action of transglutaminase [249-251]. Presenilin 1 mutations may enhance Aβ neurotoxicity via apoptosis [252]. Aβ inhibits phosphofructokinase and decreases glycolysis, which contribute to neuronal death through reduced energy metabolism [253]. Aβ has also been shown to cause abnormal intercellular communication and alter gap junction formation through augmentation of connexins, changes which may enhance neuronal toxicity [254].
Recent evidence has shown that the receptor for advanced glycation end products (RAGE), relevant to the pathogenesis of diabetes mellitus, binds synthetic Aβ (both 1 - 40 and 1 - 42) [255]. This receptor interaction stimulates oxidant stress in endothelial cells and neurons, leading to microglial activation. RAGE is a member of immunoglobulin superfamily of cell surface molecules which includes neural cell adhesion molecule (NCAM). RAGE may be upregulated in AD and the possibility exists that agents may be developed which block Aβ- RAGE interaction and may be of therapeutic benefit. Aβ is also a ligand for an intracellular protein known as ERAB which is found in endoplasmic reticulum and mitochondria [256]. This protein is a hydroxysteroid dehydrogenase which enhances the toxicity of Aβ; if ERAB is blocked Aβ toxicity is prevented, if ERAB expression is enhanced Aβ toxicity is increased.
2.2. Anti Microbial Functions
Recent in vitro studies with Aβ have compared its antimicrobial activities with that of LL-37, a typical human antimicrobial peptide (AMP). Aβ exerted antimicrobial activities against eight common clinically relevant microorganisms with a potency equivalent to LL-37. These workers also showed that AD whole brain homogenates delivered high antimicrobial activity than aged matched non-AD, so that AMP action correlates with tissue Aβ levels. The Aβ mediated activity was ablated by immunodepletion of AD brain homogenates with anti-Aβ antibodies. This work has found that Aβ is an unrecognised AMP and suggests that Aβ might be part of the innate immune system—the body’s generic cells and chemicals to fight infection. This is in contrast to the adaptive immune system with specific lymphocytes and antibodies. The blood brain barrier functions as part of this immune system and blocks adaptive cells. Innate immunity is therefore important [257].
2.3. Synaptic Functions
Stimulated hippocampal brain slices increase Aβ secretion, probably by APP trafficking to the cell membrane with β secretase cleavage. Physiological levels of APP and Aβ secretion reduce synaptic activity. That is, under physiological conditions APP and Aβ act as neuromodulators [258]. In vitro studies have shown that stimulation of NMDA receptors inhibits secretase action, increases APP, reduces alpha secretase and enhances Aβ production [259]. These findings suggest that APP and Aβ work as a coupled peptide system involved in neuromodulation. This is supported by the finding that kainic acid induced seizures are increased in APP deficient mice [260]. That is, without the inhibitory effects of Aβ, excitotoxicity is enhanced in the central nervous system.
Aβ1-42 binds with high affinity to the α7 nicotinic acetylcholine receptor (α7 nAChR) [261,262]. Aβ1-42 is able to reversibly block neuronal α7 nAChR receptors and might be relevant to some of the cognitive impairment observed in AD.
2.4. Soluble Oligomers of Aβ
The neuritic plaques of Alzheimer’s disease, which contain insoluble Aβ, do not correlate with the severity of dementia. It has been hypothesised that soluble Aβ is important and studies have shown a correlation with cognitive impairment [263]. Soluble Aβ is defined as Aβ that remains in solution after high speed centrifugation of brain extracts. Monomers and oligomers of Aβ are secreted by neural activity. The soluble oligomers are active in the synaptic cleft. Monomers and oligomers, especially trimers, seem to be active at peak of soluble concentrations and inhibit hippocampal long-term presentation (LTP), a model of learning [264]. These observations have been extended to the effects of oligomers on long-term potentiation in vivo [265]. These in vitro and in vivo studies of LTP suggest that exposure to Aβ oligomers leads to a NMDAR blockade by either reducing NMDAR activation, impairing NMDAR calcium influx or enhancing NMDAR influence on calcineuron. LTP has an effect on neuronal spine enlargement and Aβ oligomers might impair dendritic spines and the functions of excitatory neurotransmission [266]. These conclusions have been supported by studies on the glutamate AMPA receptor in hippocampus slices, where AMPA activation by Aβ might lead to synaptic depression [267]. Understanding the functions of these oligomers is important as some of the recent interest in passive immunisation treatments for Alzheimer’s disease might have their effect on binding the soluble form of Aβ and might lead to a disturbance of synaptic function. In vitro studies found that physiological concentrations of Aβ dimers and trimers, but not monomers, of human Aβ trigger synaptic loss that can be reversed by therapeutic agents [268]. APP transgenic mice expressing the E693D mutation led to Aβ oligomerization without fribrillization. These mice had age-dependent intraneuronal Aβ without extracellular amyloid deposits. Hippocampal plasticity and memory were decreased and the presynaptic marker synaptophysin was low. Tau phosphorylation and microglial increase was observed with neuronal loss. This experimental study suggests that Aβ oligomers affect synaptic function, the processing of presynaptic proteins and of the microtubular associated protein tau [269]. An oral compound Denantiometric peptide D3 has been shown to improve the cognitive performance of AD transgenic mice probably by binding Aβ oligomers and converting them to nonamyloidogenic, non-fibrillar and non-toxic aggregates without increasing the concentration of mononeuric Aβ [270].
PiB PET scanning highlights Aβ plaques in the brains of patients with Alzheimer’s disease which might be useful in the diagnosis and monitoring effects of treatment. It is possible that monoclonal antibodies that bind Aβ might be operational upon soluble oligomers and not Aβ deposited in neuritic plaques [271]. Bapineuzumab, a humanized anti Aβ monoclonal, reduced Aβ deposition using PiB PET imaging but seemed to have no effect on cognition suggesting that the soluble oligomers might be important in the cognitive effects of Alzheimer’s disease [272].
2.5. In Vivo Studies: Aβ
Bilateral hippocampal injection of Aβ did not influence reinforcement or delayed conditional discrimination in rats [273]. Aβ immunoactivity in rabbit brain has been induced by a high cholesterol diet and confirmed in pigs; Aβ deposition was found in neurons and accompanied by microgliosis [274]. Intrahippocampal injections of Aβ induced errors in a radial maze test in rats as a test of short-term memory [275].
Infusions of Aβ into the carotid arteries of rats have been shown to increase the permeability of the blood brain barrier (BBB) [276,277] and alter lectin binding sites on endothelial cells [276]. Aβ crosses the BBB in primates in an aged dependent manner [278]. These studies suggest that Aβ influences the integrity of the BBB, findings which may be important in the mechanism by which inflammatory mediators influence AD.
Fragments of Aβ (1 - 28, 12 - 28, 12 - 20) given by intracerebroventricular injection had an amnestic effect on footshock-active avoidance training in mice; impaired learning was also seen when Aβ was injected into limbic structures [279]. Aβ peptides containing the VVG domain were active for amnesia in this model; peptides with the sequences QFVG, DFFVG, AIFT blocked this effect, again suggesting the possibility of endogenous antagonists [273].
After injection of Aβ into the cerebral cortex of rats neuronal loss and degenerating neurites were observed; this was prevented by substance P administered systemically [280]. Retroviral expression of a C-terminal fragment containing the Aβ region in PC12 cells, when injected into the brains of newborn mice, caused atrophy of the cerebral cortex [281].
Transformed cells over-expressing Aβ transplanted into the suprachiasmatic nuclei of adult rats altered their circadian rhythms suggesting a physiological role for Aβ in hypothalamic function [282].
2.6. Human Studies
The human APP gene is found on chromosome 21 [283]. The gene and its product have been found in most human tissues. Apart from brain and spinal cord, APP has been found in peripheral nerve, dorsal root ganglia, sympathetic ganglia, anterior nervous system, and the neuromuscular junction [283].
The 5’ end of the promoter region of human APP has been sequenced and shares many properties with the rat APP promoter [284-286]. These are: 1) The promoter has 79.5% C+G regions; 2) No TATA or CAAT box; and 3) The promoter recognizes SP1, AP1, AP2, AP4, GCF and heat-shock elements [287]. The following elements are found in the promoter region of APP in humans and primates: APOE-β1, AP2, immediate early genes, heat shock elements, and AP-1 [288]. The Aβ region is identical in humans and microcebus monkeys [289].
Electrophoresis mobility shift assays showed that the promoter is heavily occupied by nuclear protein factors [285]. The interaction of the promoter with protein extracts from HeLa cells and nuclei obtained from human cortical neurons was different, suggesting the chromatin conformation of the promoter region may vary with cell type and origin; this possibly relates to regional differences in gene expression. Only minor differences in chromatin structure were observed between AD and non-AD neocortical nuclei. This finding suggested that a posttranscriptional process, or regulatory elements elsewhere in the APP gene, caused the abnormal accumulation of the gene product [285]. Frameshift mutations at a transcriptional level or at the post-transcriptional editing of APP RNA may be important in the development of sporadic AD and may be secondary to aging [290]. Somatic mutations do not account for sporadic AD [291].
Over expression of APP due to a gene dosage effect probably explains the Aβ deposition and AD pathology seen in trisomy 21 (Down’s syndrome, DS) [292,293]. The over expression of APP is detectable at 21 weeks gestation [294]. Duplication of the APP gene does not occur in familial and sporadic AD [283]. Increased expression of APP in cell lines is associated with cytotoxicity and increased generation of C-terminal fragments containing the complete Aβ sequence [295].
Other studies of APP in DS have not revealed differences in its splice variants in mononuclear cells from peripheral blood [296,297]. The secretory processing of APP may be heterogeneous in DS generating multiple amyloidogenic C-terminal fragments [298]. Fibroblasts from DS patients secrete more APP and have reduced responsiveness to protein kinase stimulation [299]. There may be an increase in KPI+ isoforms in young DS patients in comparison to old [300]. Lymphocytes contain higher APP content from DS patients than controls [301].
Three point mutations of APP 770 at valine 717, 3 residues carboxyl to Aβ, account for a small proportion of familial AD patients: Val717 to isoleucine [302], Val717 to phenylalanine [249], Val717 to glycine [303]. Human neuroblastoma cells transfected with these mutations produce more amyloidogenic Aβ (1 - 42) and less Aβ (1 - 40) than control cells [304,305]. Insertion of the Val717 to isoleucine mutation into mice produced Alzheimer-like pathology [140]. Other mutations in APP also result in familial AD and these involve the Swedish double mutation at codons 670 and 671 in which LysMet ® Asn-Leu [251]. A Gly-Ala substitution at codon 692 is also associated with presenile dementia and cerebral haemorrhages [306,307]. A glutamine substitution for glutamic acid at position 22 of Aβ and codon 693 of APP is associated with autosomal dominant hereditary cerebral haemorrhage with amyloid angiopathy and paradoxically is not associated with dementia [308,309].
Neurons containing the 670/671 mutations do not produce sAPPα indicating reduced alpha-secretase activity [310]. Neuronal nicotinic receptors are reduced in the brains of patients with the Swedish double mutation again providing further evidence for a relationship between APP and cholinergic neurotransmission [311].
APP mutations probably account for less than 2% of familial AD—other mechanisms must therefore be involved in the deposition of Aβ and the neuropathology of AD. Mutations in presenilin genes located on chromosome 14 (presenilin 1) and chromosome 1 (presenilin 2) are also associated with familial AD and their contribution to the pathophysiology of AD must be considered [312,313]. Over 60 missense and deletion mutations in PS1 have been observed in families worldwide and PS1 mutations amount to over 60% of familial AD [314]. Other genes causing familial AD will probably be identified in the future. Analysis of the APP promoter in fibroblasts from patients with chromosome 14-related familial AD showed hyperactivity but normal gene structure and revealed that fibroblasts from these patients produced two times the amount of APP mRNA, APP, and Aβ in comparison to controls when grown in low (0.5%) serum [315].
Cell lines expressing PS1 mutations have an increased ratio of Aβ (1-43) to Aβ (1-40) and the brains of double transgenic animals possessing both APP and PS1 mutations contain elevated levels Aβ (1-43) to Aβ (1-40) confirming that PS1 is involved in the metabolism of APP as part of the γ-secretase complex [316,317]. PS1 and the γ- secretase complex have other important functions in the processing of other membrane bound proteins such as the products of Notch genes [318]. Cell lines and knock-out mice not containing PS1 do not produce Aβ [319].
The long forms of Aβ (1-42, 1-43) are initially deposited in sporadic and familial AD, but not in APP mutations associated with autosomal dominant cerebral haemorrhages without Alzheimer pathology [320-322]. APP mutations have been shown to activate the G protein Go (composed of beta and gamma subunits) which mediate apoptosis; observations which may be inhibited by a β-adrenergic receptor kinase [323].
The tissue factor pathway inhibitor-1 (TFPI-1), a KPI inhibitor, is localized to microglia and neuritic plaques and may play a role in the protease regulation of human APP [324].
The CSF concentration of sAPPα is low and correlates with impaired neuropsychological scores in AD and ageing; findings which suggest impaired APP metabolism is related to cognitive deficits [325].
There are conflicting reports on changes in APP gene expression in normal aging brains and AD. Rnase protection assay showed that the ratio of APP KPI+ to KPI– increased 1.5 times with age and AD [326,327]. An elevated KPI+/KPI– ratio correlated directly with plaque density [328]. These findings were confirmed using immunocytochemistry and in situ hybridisation [329]. Nuclease protection assays showed APP 695 was strongly expressed in fetal brain. Adults, the aged, and patients with AD had similar splicing patterns [330]. An increase in the APP transcript lacking the Aβ sequence has been reported [281].
Lymphoid cells from patients with familial AD have reduced levels of APP KPI+ which might influence the balance of function of the secretases favouring Aβ deposition [331]. Northern analysis suggests APP mRNA might decrease with age [332].
A combined immunoreactivity and Western analysis detected no difference in APP isoforms between AD and controls [333]. Nonisotopic in situ hybridization also found no trends in APP mRNA isoform expression when correlated with neurofibrillary tangles or senile plaques, even when neurons up to 100 µm from a senile plaque were examined [334,335].
In situ hybridization of AD frontal cortex revealed reductions in both KPI– and KPI+ isoforms, not found in other neurodegenerative diseases [336]. Other studies have suggested differences in neuronal subpopulation expression of APP mRNA [190]. An effect of age and anatomical region is supported by a study of cynomolgus monkeys in which APP 695 and 751 were increased 2 - 7 times in the thalamus in 17-year-old monkeys when compared with 3-year olds [337]. Solution hybridization studies have not demonstrated changes in APP in ageing and AD [338].
An immunocytochemical study showed that APP KPI+ correlates with neuritic plaque density [339]. There was a relative increase in the purified soluble fraction of KPI+ protein in AD using Western blots implying that APP may be involved in the regulation of its own metabolism [149].
These variable findings in APP isoforms in AD might relate to the interval of time from death to brain sampling, agonal state, and techniques employed (e.g. nuclease protection assays versus in situ hybridization). A relatively recent study using radioactive in situ hybridization analysis of APP isoforms and larger numbers of AD patients and neurodegenerative controls has revealed no differences in differential mRNA expression of APP isoforms [340]. An investigation using PT-PCR in AD post-mortem brains showed an increase in the KPI+ isoforms controlling for age at death, brain pH and gender [341]. Caution must be exercised in investigating changes in gene expression at death in a slowly evolving neurological process, since critical changes in gene expression may have occurred some time before.
Aβ deposition is not unique to AD but is also found in aging, head injury, and other neurodegenerative disorders [342]. Twenty-four hours after head injury enhanced Aβ immunoreactivity has been observed throughout the cortical mantle, not just in the area adjacent to the impact [343]. Even after concussion, the mildest form of head injury not associated with macroscopic or microscopic evidence of brain damage, increased APP immunoreactivity has been found in axons within the fornix and may be a marker of diffuse axonal injury [344]. The immediate early gene c-jun may be associated with amyloid deposition in blood vessels and glial cells [345,346]. The APOEε4 isoform has been associated with increased Aβ deposition after head injury [34]. Microglia with IL-1a immuno-reactivity were found after head injury adjacent to neurons expressing APP, suggesting a role in its production [347].
Interleukin 1 expression has been demonstrated in the vicinity of senile plaques, possibly in microglial cells, in AD and DS [348]. Microglial IL-1 was associated with increased neuronal APP in temporal lobectomy specimens from patients with refractory epilepsy [349]. APP KPI+ isoforms were increased in comparison to controls, without any change in APP KPI–. An IL-1 antagonist
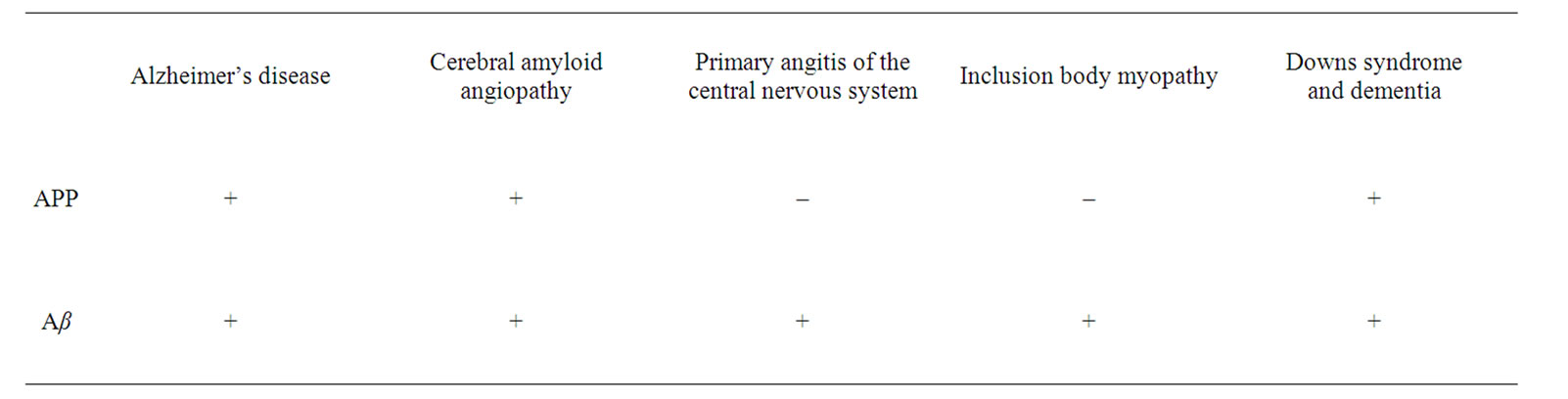
Table 1. The Disease Associations of APP and Aβ.
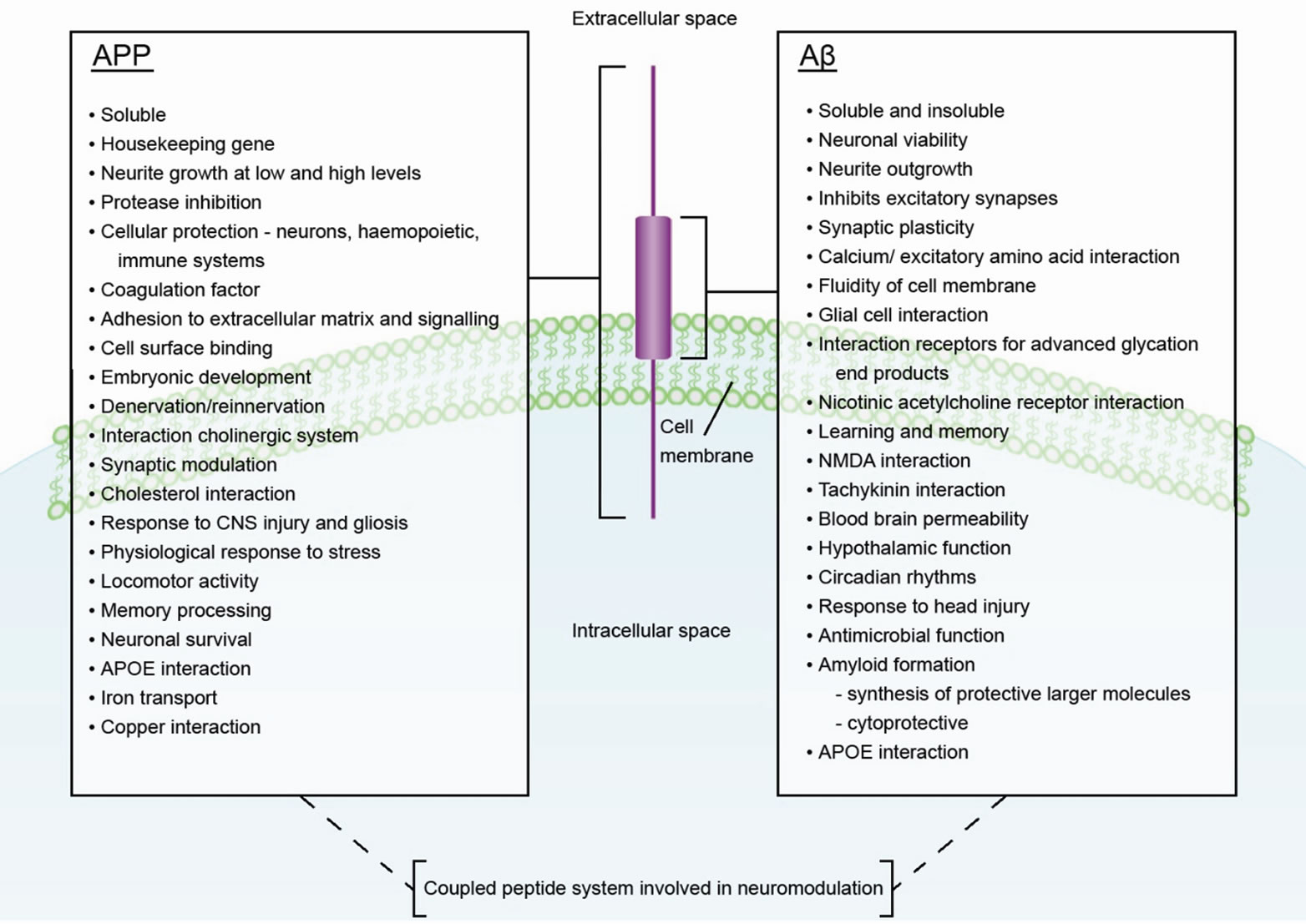
Figure 1. The Functions of APP and Aβ.
rhIL-1ra prevented excitotoxicity induced changes in APP [102]. These observations provide evidence for an interaction between neuronal injury and loss, microglial activation, cytokines—in particular IL-1—and changes in APP isoform expression. The synthesis of APP mRNA in cultured human endothelial cells has been shown to be influenced by IL-1 [350].
Human platelets may have differential expression of APP isoforms in AD and APP might function to inhibit platelet activation post-granulation [351,352].
Aβ deposition is associated with some forms of primary angiitis of the CNS and is found in the presence of cerebral amyloid angiopathy [353]. In the muscle disease known as inclusion body myopathy (IBM), Aβ is deposited near the end plate of the acetylcholine receptor. IBM is an uncommon neuromuscular disease in adults and is the only disorder outside of the central nervous system associated with Aβ deposition [354] (Table 1).
The functions of APP and Aβ are summarised in the figure (Figure 1).
REFERENCES
- Z. Zhong, J. Higaki, K. Wang, R. Catalano, D. Quon and B. Cordell, “Secretion of β-Amyloid Precursor Protein Involves Multiple Cleavage Sites,” Journal of Biological Chemistry, Vol. 269, No. 1, 1994, pp. 627-632.
- S. B. Roberts, J. A. Ripellino, K. M. Ingalls, N. K. Robakis and K. M. Felstein, “Non-Amyloidogenic Cleavage of the B-Amyloid Precursor Protein by an Integral Membrane Metalloendopeptidase,” Journal of Biological Chemistry, Vol. 269, No. 4, 1994, pp. 3111- 3116.
- M. R. Palmert, M. B. Podlisny, D. S. Witker, T. Oltersdorf, L. H. Younkin, D. J. Selkoe and S. G. Younkin, “The B-Amyloid Protein Precursor of Alzheimer Disease Has Soluble Derivatives Found in Human Brain and Cerebrospinal Fluid,” Proceedings of the National Acadamy of Sciences of the United States of America, Vol. 86, 1989, pp. 6338-6342. doi:10.1073/pnas.86.16.6338
- P. Seubert, C. Vigo-Pelfrey, F. Esch, M. Lee, H. Dovey, D. Davis, S. Sinha, M. Schlossmacher, J. Whaley and C. Swindlehurst, “Isolation and Quantification of Soluble Alzheimer’s B-Peptide from Biological Fluids,” Nature, Vol. 359, No. 6393, 1992, pp. 325-327. doi:10.1038/359325a0
- F. S. Esch, P. S. Keim, E. C. Beattie, R. W. Blacher, A. R. Culwell, T. Oltersdorf, D. McClure and P. J. Ward, “Cleavage of Amyloid B-Peptide during Constitutive Processing of Its Precursor,” Science, Vol. 248, 1990, pp. 1122-1124. doi:10.1126/science.2111583
- C. Haass, E. H. Koo, A. Mellon, A. Y. Hung and D. J. Selkoe, “Targeting of Cell-Surface B-Amyloid Precursor Protein to Lysosomes: Alternative Processing into Amyloid-Bearing Fragments,” Nature, Vol. 357, No. 6378, 1992, pp. 500-502. doi:10.1038/357500a0
- C. Haass, M. G. Schlossmacher, A. Y. Hung, C. VigoPelfrey, A. Mellon, B. L. Ostaszewski, I. Lieberburg, E. H. Koo, D. Schenk and D. B. Teplow, “Amyloid BPeptide is Produced by Cultured Cells During Normal Metabolism,” Nature, Vol. 359, 1992, pp. 322-325. doi:10.1038/359322a0
- P. Seubert, T. Oltersdorf, M. G. Lee, R. Barbour, C. Blomquist, D. L. Davis, K. Bryant, L. C. Fritz, D. Galasko and L. J. Thal, “Secretion of β-Amyloid Precursor Protein Cleaved at the Aminoterminus of the β-Amyloid Peptide,” Nature, Vol. 361, 1993, pp. 260-263. doi:10.1038/361260a0
- C. Haass and D. J. Selkoe, “Cellular Processing of β- Amyloid Precursor and the Genesis of Amyloid BPeptide,” Cell, Vol. 75, No. 6, 1993, pp. 1039-1042. doi:10.1016/0092-8674(93)90312-E
- M. L. Oster-Granite, D. L. McPhie, J. Greenan and R. L. Neve, “Age-Dependent Neuronal and Synaptic Degeneration in Mice Transgenic for the C Terminus of the Amyloid Precursor Protein,” Journal of Neuroscience, Vol. 16, No. 21, 1996, pp. 6732-6741.
- M. N. Pangalos, J. Shioi, S. Efthimiopoulos, A. Wu and N. K. Robakis, “Characterization of Appican, the Chondroitin Sulfate Proteoglycan Form of the Alzheimer Amyloid Pre-cursor Protein,” Neurodegeneration, Vol. 5, No. 4, 1996, pp. 445-451. doi:10.1006/neur.1996.0061
- B. Allinquant, K. L. Moya, C. Bouillot and A. Prochiantz, “Amyloid Precursor Protein in Cortical Neurons: Existence of Two Pools Differentially Distributed in Axons and Dendrites and Association with Cytoskeleton,” Journal of Neuroscience, Vol. 14, 1994, pp. 6842-6854.
- Y. H. Suh, Y. H. Chong, S. H. Kim, W. Choi, K. Min, S. J. Jeong, S. P. Fraser and M. B. Djamgoz, “Molecular Physiology, Biochemistry and Pharmacology of Alzheimer’s Amyloid Precursor Protein (APP),” Annals of the New York Acadamy of Sciences, Vol. 786, 1996, pp. 169- 183. doi:10.1111/j.1749-6632.1996.tb39060.x
- D. Schubert, L. W. Jin, T. Saitoh and G. Cole, “The Regulation of Amyloid B Protein Precursor Secretion and its Modulatory Role in Cell Adhesion,” Neuron, Vol. 3, 1989, pp. 689-694. doi:10.1016/0896-6273(89)90237-7
- C. J. Smith, D. Widon and P. Brachet, “Nerve Growth Factor-Induced Neuronal Differentiation is Accompanied by Differential Splicing of B-Amyloid Precursor mRNAs in the PC12 Cell Line,” Molecular Brain Research, Vol. 10, 1991, pp. 351-354. doi:10.1016/0169-328X(91)90095-F
- E. A. Milward, R. Papadopoulos, S. J. Fuller, R. D. Moir, D. Small, K. Beyreuther and C. L. Masters, “The Amyloid Protein Precursor of Alzheimer’s Disease is a Mediator of the Effects of Nerve Growth Factor on Neurite Outgrowth,” Neuron, Vol. 9, No. 1, 1992, pp. 129-137. doi:10.1016/0896-6273(92)90228-6
- J. J. Luo, W. Wallace, T. Riccioni, D. K. Ingram, G. S. Roth and J. W. Kusiak, “Death of PC12 Cells and Hippocampal Neurons Induced by Adenoviral-Mediated FAC Human Amyloid Precursor Protein Gene Expression,” Journal of Neuroscience Research, Vol. 55, 1999, pp. 629-642. doi:10.1002/(SICI)1097-4547(19990301)55:5<629::AID-JNR10>3.0.CO;2-Y
- H. J. Clarris, V. Nurcombe, D. H. Small, K. Beyreuther and C. L. Masters, “Secretion of Nerve Growth Factor From Septum Stimulates Neurite Outgrowth and Release of the Amyloid Protein Precursor of Alzheimer’s Disease From Hippocampal Explants,” Journal of Neuroscience Research, Vol. 38, No. 3, 1994, pp. 248-258. doi:10.1002/jnr.490380303
- S. Rossner, U. Ueberham, R. Schliebs, J. R. Perez-Polo and V. Bigl, “Neurotrophin Building to the p75 Neurotrophin Receptor is Necessary but not Sufficient to Mediate NGF-Effects on APP Secretion in PC-12 Cells,” Journal of Neural Transmission, Vol. 54, 1998, pp. 279-285.
- K. Yoshikawa, T. Aizawa and Y. Hayashi, “Degeneration In Vitro of Post-Mitotic Neurons Over Expressing the Alzheimer Amyloid Protein Precursor,” Nature, Vol. 359, 1992, pp. 64-67. doi:10.1038/359064a0
- N. Y. Barnes, L. Li, K. Yoshikawa, L. M. Schwartz, R. W. Oppenheim and C. E. Milligan, “Increased Production of Amyloid Precursor Protein Provides a Substrate for Caspase-3 in Dying Motoneurons,” Journal of Neuroscience, Vol. 18, No. 15, 1998, pp. 5869-5880.
- D. Schubert and C. Behl, “The Expression of Beta Amyloid Protein Precursor Protects Nerve Cells From B-Amyloid and Glutamate Toxicity and Alters Their Interaction with the Extracellular Matrix,” Brain Research, Vol. 629, No. 2, 1993, pp. 275-282. doi:10.1016/0006-8993(93)91331-L
- D. H. Small, V. Nurcombe, G. Reed, H. Clarris, R. Moir, K. Beyreuther and C. L. Masters, “A Heparin-Binding Domain in the Amyloid Protein Precursor of Alzheimer’s Disease Is Involved in the Regulation of Neurite Outgrowth,” Journal of Neuroscience, Vol. 14, No. 4, 1994, pp. 2117-2127.
- W. Q. Qiu, A. Ferreira, C. Miller, E. H. Koo and D. J. Selkoe, “Cell-Surface B-Amyloid Precursor Protein Stimulates Neurite Outgrowth of Hippocampal Neurons in an Isoform-Dependent Manner,” Journal of Neuroscience, Vol. 15, 1995, pp. 2157-2167.
- L. W. Jin, H. Ninomiya, J. M. Rocn, D. Schubert, E. Maslian, D. A. Otero and T. Saitoh, “Peptides Containing the RERMS Sequence of Amyloid β/A4 Protein Precursor Bind Cell Surface and Promote Neurite Extension,” Journal of Neuroscience, Vol. 14, 1994, pp. 5461-5470.
- F. G. Klier, G. Cole, W. Stallcup and D. Schubert, “Amyloid B-Protein Precursor Is Associated with Extracellular Matrix,” Brain Research, Vol. 515, 1990, pp. 336-342. doi:10.1016/0006-8993(90)90619-M
- J. Hoffmann, C. U. Pietrzik, M. P. Kummer, C. Twiesselmann, C. Bauer and V. Herzog, “Binding and Selective Detection of the Secretory N-Terminal Domain of the Alzheimer Amyloid Precursor Protein on Cell Surfaces,” Journal of Histochemistry and Cytochemistry, Vol. 47, No. 3, 1999, pp. 373-382. doi:10.1177/002215549904700311
- K. Barber, S. A. Enam, S. Bodovitz, M. Falduto, D. Frail and W. L. Klein, “Particulate Forms of APP in the Extracellular Milieu of Cultured Cells,” Experimental Neurology, Vol. 132, No. 1, 1995, pp. 42-53. doi:10.1016/0014-4886(95)90057-8
- S. Narindrasorasak, D. Lowery, P. Gonzalez-deWhitt, R. A. Poorman, B. Greenberg and R. Kisilevsky, “High Affinity Interactions Between the Alzheimer’s BAmyloid Precursor Proteins and the Basement Membrane form of Heparin Sulfate Proteoglycan,” Journal of Biological Chemistry, Vol. 266, No. 20, 1991, pp. 12878-12883.
- K. C. Breen, “APP-Collagen Interaction is Mediated by a Heparin Bridge Mechanism,” Molecular and Chemical Neuropathology, Vol. 16, No. 1-2, 1992, pp. 109-121. doi:10.1007/BF03159964
- M. C. Kibbey, M. Jucker, B. S. Weeks, R. L. Neve, W. E. Van Nostrand and H. K. Kleinman, “β-Amyloid Precursor Protein Binds to the Neurite-Promoting IKVAV Site of Laminin,” Proceedings of the National Acadamy of Sciences of the United States of America, Vol. 90, No. 2, 1993, pp. 10150-10153. doi:10.1073/pnas.90.21.10150
- L. Buee, W. Ding, J. P. Anderson and S. Narindrasorasak, “Binding of Vascular Heparan Sulfate Proteoglycan to Alzheimer’s Amyloid Precursor Protein Is Mediated in part by the N-terminal Region of A4 Peptide,” Brain Research, Vol. 627, No. 2, 1993, pp. 199-204. doi:10.1016/0006-8993(93)90321-D
- L. Buee, W. Ding, A. Delacourte and H. Fillit, “Binding of Secreted Human Neuroblastoma Proteoglycans to the Alzheimer’s Amyloid A4 Peptide,” Brain Research, Vol. 601, No. 1-2, 1993, pp. 154-163. doi:10.1016/0006-8993(93)91706-X
- H. Ninomiya, J. M. Roch, L. W. Jin and T. Saitoh, “Secreted Form of Amyloid Beta/A4 Protein Precursor (APP) Binds to Two Distinct APP Binding Sites on Rat B103 Neuron-Like Cells through Two Different Domains, But Only One Site Is Involved in Neuritotropic Activity,” Journal of Neurochemistry, Vol. 63, No. 2, 1994, pp. 495-500. doi:10.1046/j.1471-4159.1994.63020495.x
- U. Monning, R. Sandbrink, A. Weidemann, R. B. Banati, C. L. Masters and K. Beyreuther, “Extracellular Matrix Influences the Biogenesis of Amyloid Precursor Protein in Microglial Cells,” Journal of Biological Chemistry, Vol. 270, No. 13, 1995, pp. 7104-7110. doi:10.1074/jbc.270.13.7104
- G. Multhaup, H. Mechler and C. L. Masters, “Characterization of the High Affinity Heparin Binding Site of the Alzheimer’s Disease Beta A4 Amyloid Precursor Protein (APP) and its Enhancement by Zinc (II),” Journal of Molecular Recognition, Vol. 8, No. 4, 1995, pp. 247- 257.
- S. S. Mok, G. Sberna, D. Heffernan, R. Cappai, D. Galatis, H. J. Clarris, W. H. Sawyer, K. Beyreuther, C. L. Masters and D. H. Small, “Expression and Analysis of Heparin-Binding Regions of the Amyloid Precursor Protein of Alzheimer’s Disease,” FEBS Letters, Vol. 415, No. 3, 1997, pp. 303-307. doi:10.1016/S0014-5793(97)01146-0
- H. J. Clarris, R. Cappai, D. Heffernan, K. Beyreuther, C. L. Masters and D. H. Small, “Idenification of HeparinBinding Domains in the Amyloid Precursor Protein of Alzheimer’s Disease by Deletion Mutagenesis and Peptide Mapping,” Journal of Neurochemistry, Vol. 68, No. 3, 1997, pp. 1164-1172. doi:10.1046/j.1471-4159.1997.68031164.x
- T. Yamazaki, E. H. Koo and D. J. Selkoe, “Cell Surface Amyloid Beta-Protein Precursor Colocalizes with Beta 1 Integrins at Substrate Contact Sites in Neural Cells,” Journal of Neuroscience, Vol. 17, No. 3,1997, pp. 1004- 1010.
- S. Fisher, J. D. Gearhart and M. L. Oster-Granite, “Expression of the Amyloid Precursor Protein Gene in Mouse Oocytes and Embryos,” Proceedings of the National Acadamy of Sciences of the United States of America, Vol. 88, No. 5, 1991, pp. 1779-1782. doi:10.1073/pnas.88.5.1779
- A. Y. Hung, E. H. Koo, C. Haass and D. J. Selkoe, “Increased Expression of B-Amyloid Precursor Protein During Neuronal Differentiation Is not Accompanied by Secretory Cleavage,” Proceedings of the National Acadamy of Sciences of the United States of America, Vol. 89, 1992, pp. 9439-9443. doi:10.1073/pnas.89.20.9439
- K. Yoshikawa, T. Aizawa and K. Maruyama, “Neural Differentiation Increases Expression of Alzheimer Amyloid Protein Precursor in Murine Embryonal Carcinoma Cells,” Biochemical and Biophysical Research Communications, Vol. 171, 1990, pp. 204-209. doi:10.1016/0006-291X(90)91377-5
- W. Schubert, R. Prior, A. Weidmann, H. Dirksen, G. Multhaup, C. L. Masters and K. Beyreuther, “Localization of Alzheimer BA4 Amyloid Precursor Protein at Central and Peripheral Sites,” Brain Research, Vol. 563, 1991, pp. 184-194. doi:10.1016/0006-8993(91)91532-6
- K. Fukuchi, K. Kamino, S. S. Deeb, A. C. Smith, T. Dang and G. M. Martin, “Overexpression of Amyloid Precursor Protein Alters Its Normal Processing and Is Associated With Neurotoxicity,” Biochemical and Biophysical Research Communications, Vol. 182, No. 1, 1992, pp. 165- 173. doi:10.1016/S0006-291X(05)80126-3
- T. Morimoto, I. Ohsawa, C. Takamura, M. Ishiguro, Y. Nakamura and S. Kohsake, “Novel Domain-Specific Actions of Amyloid Precursor Protein on Developing Synapses,” Journal of Neuroscience, Vol. 18, No. 22, 1998, pp. 9386-9393.
- M. Usami, W. Yamao-Harigaya and K. Maruyama, “The Triplet of Lysine Residues (Lys724-Lys725-Lys726) of Alzheimer’s Amyloid Precursor Protein Plays an Important Role in Membrane Anchorage and Processing,” Journal of Neurochemistry, Vol. 61, No. 1,1993, pp. 239- 246. doi:10.1111/j.1471-4159.1993.tb03560.x
- E. H. Koo, S. S. Sisodia, D. R. Archer, L. J. Martin, A. Weidemann, K. Beyreuther, P. Fischer, C. L. Masters and D. L. Price, “Precursor of Amyloid Protein in Alzheimer Disease Undergoes Fast Anterograde Axonal Transport,” Proceedings of the National Acadamy of Sciences of the United States of America, Vol. 87, No. 4, 1990, pp. 1561-1565. doi:10.1073/pnas.87.4.1561
- J. G. Calvenor, A. Friedhuber, S. J. Fuller, K. Beyreuther and C. L. Masters, “Expression of the Amyloid Precursor Protein of Alzheimer’s Disease on the Surface of Transfected HeLa Cells,” Experimental Cell Research, Vol. 220, No. 2, 1995, pp. 474-481. doi:10.1006/excr.1995.1339
- A. I. Bush, R. N. Martins, B. Rumble, R. Moir, S. Fuller, E. Milward, J. Currie, D. Ames, A. Weidemann and P. Fischer, “The Amyloid Precursor Protein of Alzheimer’s Disease Is Released by Human Platelets,” Journal of Biological Chemistry, Vol. 265, No. 26, 1990, pp. 15977- 15983.
- R. P. Smith, D. A. Higuchi and G. J. Broze, “Platelet Coagulation Factor XIa-Inhibitor, a Form of Alzheimer Amyloid Precursor Protein,” Science, Vol. 248, No. 4959, 1990, pp. 1126-1128. doi:10.1126/science.2111585
- M. J. Bullido, M. A. Munoz-Fernandez, M. Recuero, M. Fresno and F. Valdivieso, “Alzheimer’s Amyloid Precursor Protein Is Expressed on the Surface of Hematopoietic Cells upon Activation,” Biochimica et Biophysica Acta, Vol. 1313, No. 1, 1996, pp. 54-62. doi:10.1016/0167-4889(96)00015-8
- N. Jahroudi, J. Kitney, J. S. Greenberger and R. Bowser, “Endothelial Cell Dysfunction in Response to Intracellular Overexpression of Amyloid Precursor Protein,” Journal of Neuroscience Research, Vol. 54, No. 6, 1998, pp. 828-839. doi:10.1002/(SICI)1097-4547(19981215)54:6<828::AID-JNR11>3.0.CO;2-M
- J. R. Ciallella, V. V. Rangnekar and J. McGillis, “Heat Shock Alters Alzheimer’s Beta Amyloid Precursor Protein Expression in Human Endothelial Cells,” Journal of Neuroscience Research, Vol. 37, No. 6, 1994, pp. 769- 776. doi:10.1002/jnr.490370611
- T. A. Davies, A. M. Billingslea, H. J. Long, H. Tibbles, J. M. Wells, P. B. Eisenhauer, S. J. Smith, D. H. Cribbs, R. E. Fine and E. R. Simons, “Brain Endothelial Cell Enzymes Cleave Platelet-Retained Amyloid Precursor Protein,” Journal of Laboratory and Clinical Medicine, Vol. 132, No. 4, 1998, pp. 341-350. doi:10.1016/S0022-2143(98)90048-8
- S. Gloor, K. Odink, J. Guenther, H. Nick and D. Monard, “A Glia-Derived Neurite Promoting Factor With Protease Inhibitory Activity Belongs to the Protease Nexins,” Cell, Vol. 47, No. 5, 1986, pp. 687-693. doi:10.1016/0092-8674(86)90511-8
- J. Guenther, H. Nick and D. Monard, “A Glial-Derived Neurite-Promoting Factor with Protease Inhibitory Activity,” EMBO Journal, Vol. 4, No. 8, 1985, pp. 1963-1966.
- H. Tanahashi and T. Tabira, “X11L2, a New Member of the X11 Protein Family, Interacts with Alzheimer’s Beta-Amyloid Precursor Protein,” Biochemical and Biophysical Research Communications, Vol. 255, No. 3, 1999, pp. 663-667. doi:10.1006/bbrc.1999.0265
- D. Monard, E. Niday, A. Limat and F. Solomon, “Inhibition of Protease Activity Can Lead to Neurite Extension in Neuroblastoma Cells,” Progress in Brain Research, Vol. 58, 1983, pp. 359-365. doi:10.1016/S0079-6123(08)60037-0
- T. B. Shea, “Role of Glial-Derived Nexin in Neuronal Differentiation and in Acute Brain Injury and Potential Involvement in Exacerbation of Neurodegeneration in Alzheimer’s Disease,” Brain Research Reviews, Vol. 20, No. 2, 1995, pp. 171-184. doi:10.1016/0165-0173(94)00010-M
- R. Joseph, W. Tsang, E. Han and G. M. Saed, “Neuronal B-Amyloid Precursor Protein Gene Expression: Regulation by Aurintricarboxylic Acid,” Brain Research, Vol. 625, 1993, pp. 244-255. doi:10.1016/0006-8993(93)91065-Z
- C. J. Smith, E. M. Johnson, P. Osborn, R. S. Freman, I. Neveu and P. Brachet, “NGF Deprivation and Neuronal Degeneration Trigger Altered β-Amyloid Precursor Proetin Expression in the Rat Superior Cervical Ganglion in Vivo and in Vitro,” Molecular Brain Research, Vol. 17, No. 3-4, 1993, pp. 328-354. doi:10.1016/0169-328X(93)90018-K
- K. Abe, P. H. St George-Hyslop, R. E. Tanzi and K. Kogure, “Induction of Amyloid Precursor Protein mRNA After Heat Shock in Cultured Human Lymphoblastoid Cells,” Neuroscience Letters, Vol. 125, No. 2, 1991, pp. 169-171. doi:10.1016/0304-3940(91)90019-P
- J. D. Buxbaum, M. Oishi, H. I. Chen, R. PinkasKramarski, E. A. Jaffe, S. E. Gandy and P. Greengard, “Cholinergic Agonists and Interleukin-1 Regulate Processing and Secretion of the Alzheimer B/A4 Amyloid Protein Precursor,” Proceedings of the National Acadamy of Sciences of the United States of America, Vol. 89, 1992, pp. 10075-10078. doi:10.1073/pnas.89.21.10075
- G. Forloni, F. Demicheli, S. Giorgi, C. Bendotti and N. Angeretti, “Expression of Amyloid Precursor Protein mRNAs in Endothelial, Neuronal and Glial Cells: Modulation by Interleukin-1,” Molecular Brain Research, Vol. 16, No. 1-2, 1992, pp. 128-134. doi:10.1016/0169-328X(92)90202-M
- C. W. Gray and A. J. Patel, “Regulation of B-Amyloid Precursor Protein Isoform mRNAs by Transforming Growth Factor-B1 and Interleukin-1B in Astrocytes,” Molecular Brain Research, Vol. 19, 1993, pp. 251-256. doi:10.1016/0169-328X(93)90037-P
- U. Monning, R. Sandbrink, R. B. Banati, C. L. Masters and K. Beyreuther, “Transforming Growth Factor Beta Mediates Increase of Mature Transmembrane Amyloid Precursor Protein in Microglial Cells,” FEBS Letters, Vol. 342, No. 3, 1994, pp. 267-272. doi:10.1016/0014-5793(94)80514-8
- F. Yang, L. Jansen, W. E. Friedrichs, J. M. Buchanan and B. H. Bowman, “IL-1B Decreases Expression of Amyloid Precursor Protein Gene in Human Glioma Cells,” Biochemical and Biophysical Research Communications, Vol. 191, 1993, pp. 1014-1019. doi:10.1006/bbrc.1993.1318
- J. P. Vasilakos, R. T. Carroll, M. R. Emmerling, P. D. Doyle, R. E. Davis, K. S. Kim and B. D. Shivers, “Interleukin-1 Beta Dissociates Beta-Amyloid Precursor Protein and Beta-Amyloid Peptide Secretion,” FEBS Letters, Vol. 354, No. 3, 1994, pp. 289-292. doi:10.1016/0014-5793(94)01142-7
- R. Del Bo, N. Angeretti, E. Lucca, M. G. De Simoni and G. Forloni, “Reciprocal Control of Inflammatory Cytokines, IL-1 and IL-6, and B-Amyloid Production in Cultures,” Neuroscience Letters, Vol. 188, No. 1, 1995, pp. 70-74. doi:10.1016/0304-3940(95)11384-9
- J. T. Rogers, L. M. Leiter, J. McPhee, C. M. Cahill, S. S. Zhan, H. Potter and L. N. Nilsson, “Translation of the Alzheimer Amyloid Precursor Protein mRNA is UpRegulated by Interleukin-1 Through 5’ Untranslated Region Sequences,” Journal of Biological Chemistry, Vol. 274, No. 10, 1999, pp. 6421-6431. doi:10.1074/jbc.274.10.6421
- I. Nishimoto, T. Okamoto, Y. Matsuura, S. Takahashi, T. Okamoto, Y. Murayama and E. Ogata, “Alzheimer Amyloid Protein Precursor Complexes with Brain GTPBinding Protein G,” Nature, Vol. 362, No. 6415, 1993, pp. 75-79. doi:10.1038/362075a0
- H. Schulze, A. Schuler, D. Stuber, H. Dobeli, H. Langen and G. Huber, “Rat Brain Glyceraldehyde-3-Phosphate Dehydrogenase Interacts with the Recombinant Cytoplasmic Domain of Alzheimer’s Beta-Amyloid Precursor Protein,” Journal of Neurochemistry, Vol. 60, No. 5, 1993, pp. 1915-1922. doi:10.1111/j.1471-4159.1993.tb13420.x
- S. W. Barger and M. P. Mattson, “Isoform-Specific Modulation by Apolipoprotein E of the Activities of Secreted Beta-Amyloid Precursor Protein,” Journal of Neurochemistry, Vol. 69, No. 1, 1997, pp. 60-67. doi:10.1046/j.1471-4159.1997.69010060.x
- S. Hass, F. Fresser, S. Kochl, K. Beyreuther, G. Utermann and G. Baier, “Physical Interaction of ApoE with Amyloid Precursor Protein Independent of the Amyloid Abeta Region in Vitro,” Journal of Biological Chemistry, Vol. 273, No. 22, 1998, pp. 13892-13897.
- T. Ohshita, A. Nakatani and Y. Hiroi, “Purification by Column Chromatographies of Beta-Amyloid Precursor Proteins and Their Association with Other 95 kDa Protein in Rat Brain,” Journal of Chromatography B: Biomedical Sciences and Applications, Vol. 716, No. 1-2, 1998, pp. 107-118. doi:10.1016/S0378-4347(98)00320-X
- M. Trommsdorff, J. P. Borg, B. Margolis and J. Herz, “Interaction of Cytosolic Adaptor Proteins with Neuronal Apolipoprotein E Receptors and the Amyloid Precursor Protein,” Journal of Biological Chemistry, Vol. 273, No. 50, 1998, pp. 33556-33560. doi:10.1074/jbc.273.50.33556
- J. P. Borg, S. W. Straight, S. M. Kaech, M. de Taddeo-Borg, D. E. Kroon, D. Karnak, R. S. Turner, S. K. Kim and B. Margolis, “Identification of an Evolutionarily Conserved Heterotrimeric Protein Complex Involved in Protein Targeting,” Journal of Biological Chemistry, Vol. 273, No. 48, 1998, pp. 31633-31636. doi:10.1074/jbc.273.48.31633
- J. P. Borg, Y. Yang, M. De Taddeo-Borg, B. Margolis and R. S. Turner, “The X11Alpha Protein Slows Cellular Amyloid Precursor Protein Processing and Reduces Abeta40 and Abeta42 Secretion,” Journal of Biological Chemistry, Vol. 273, No. 24, 1998, pp. 14761-14766. doi:10.1074/jbc.273.24.14761
- S. L. Sabo, L. M. Lanier, A. F. Ikin, O. Khorkova, S. Sahasrabudhe, P. Greengard and J. D. Buxbaum, “Regulation of Beta-Amyloid Secretion by FE65, an Amyloid Protein Precursor-Binding Protein,” Journal of Biological Chemistry, Vol. 274, No. 12, 1999, pp. 7952-7957. doi:10.1074/jbc.274.12.7952
- Z. Kouchi, H. Sorimachi, K. Suzuki and S. Ishiura, “Proteasome Inhibitors Induce the Association of Alzheimer’s Amyloid Precursor Protein with Hsc73,” Biochemical and Biophysical Research Communications, Vol. 254, No. 3, 1999, pp. 804-810. doi:10.1006/bbrc.1998.9977
- K. Miyazaki, M. Hasegawa, K. Funahashi and M. Umeda, “A Metalloproteinase Inhibitor Domain in Alzheimer Amyloid Protein Precursor,” Nature, Vol. 362, No. 6423, 1993, pp. 839-841. doi:10.1038/362839a0
- A. Odaka, T. Tsukahara, M. Momoi and T. Momoi, “cjun Inhibited the Alternative Splicing of Neuron-Specific Amyloid Precursor Protein, but Stimulated the NonNeuron Type One in P19 EC Cells,” Biochemical and Biophysical Research Communications, Vol. 206, No. 3, 1995, pp. 831-828. doi:10.1006/bbrc.1995.1117
- D. Muller, H. Wiegmann, U. Langer, S. Motlzen-Lenz and R. M. Nitsch, “Lu 25-109, a Combined m1 Agonist and m2 Antagonist, Modulates Regulated Processing of the Amyloid Precursor Protein of Alzheimer’s Disease,” Journal of Neural Transmission, Vol. 105, No. 8-9, 1998, pp. 1029-1043. doi:10.1007/s007020050110
- S. Rossner, U. Ueberham, R. Schliebs, J. R. Perez-Polo and V. Bigl, “The Regulation of Amyloid Precursor Protein Metabolism by Cholinergic Mechanisms and Neurotrophin Receptor Signaling,” Progress in Neurobiology, Vol. 56, No. 5, 1998, pp. 541-569. doi:10.1016/S0301-0082(98)00044-6
- B. Belandia, M. J. Latasa, A. Villa and A. Pascual, “Thyroid Hormone Negatively Regulates the Transcriptional Activity of the Beta-Amyloid Precursor Protein Gene,” Journal of Biological Chemistry, Vol. 273, No. 46, 1998, pp. 30366-30371. doi:10.1074/jbc.273.46.30366
- R. K. Lee, S. Knapp and R. J. Wurtman, “Prostaglandin E2 Stimulates Amyloid Precursor Protein Gene Expression: Inhibition by Immunosuppressants,” Journal of Neuroscience, Vol. 19, No. 3, 1999, pp. 940-947.
- R. M. Nitsch, S. A. Farber, J. H. Growdon and R. J. Wurtman, “Release of Amyloid B-Protein Precursor Derivatives from Hippocampal Slices by Electrical Depolarization,” Proceedings of the National Acadamy of Sciences of the United States of America, Vol. 90, 1993, pp. 5191-5193. doi:10.1073/pnas.90.11.5191
- J. G. Beeson, E. R. Shelton, H. W. Chan and F. H. Gage, “Differential Distribution of Amyloid Protein Precursor Immunoreactivity in the Rat Brain Studied by Using Five Different Antibodies,” Journal of Comparative Neurology, Vol. 342, No. 1, 1994, pp. 78-96. doi:10.1002/cne.903420109
- J. P. Card, R. P. Meade and L. G. Davis, “Immunocytochemical Localization of the Precursor Protein for BAmyloid in the Rat Central Nervous System,” Neuron, Vol. 1, No. 9, 1988, pp. 835-846. doi:10.1016/0896-6273(88)90131-6
- T. Ohgami, T. Kitamoto and J. Tateishi, “The Rat Central Nervous System Expresses Alzheimer’s Disease Amyloid Precursor Protein APP695, but Not APP677 (L-APP form),” Journal of Neurochemistry, Vol. 61, No. 4, 1993, pp. 1553-1556. doi:10.1111/j.1471-4159.1993.tb13655.x
- C. Solá, G. Mengod, A. Probst and K. M. Palacios, “Differential Regional and Cellular Distribution of BAmyloid Precursor Protein Messenger RNAs Containing and Lacking the Kunitz Protease Inhibitor Domain in the Brain of Human, Rat and Mouse,” Neuroscience, Vol. 53, 1993, pp. 267-295. doi:10.1016/0306-4522(93)90304-X
- J. D. Buxbaum, G. Thinakaran, V. Koliatsos, J. O’Callahan, H. H. Slunt, D. L. Price and S. S. Sisodia, “Alzheimer Amyloid Protein Precursor in the Rat Hippocampus: Transport and Pressing through the Perforant Path,” Journal of Neuroscience, Vol. 18, 1998, pp. 9629-9637.
- T. Kawarabayashi, M. Shoji, H. Yamaguchi, M. Tanaka, Y. Harigaya, K. Ishiguro and S. Hirai, “Amyloid β Protein Precursor Accumulates in Swollen Neurites Throughout Rat Brain with Aging,” Neuroscience Letters, Vol. 153, No. 1, 1993, pp. 73-76. doi:10.1016/0304-3940(93)90080-5
- Y. Nakamura, M. Takeda, H. Niigawa, F. Kametani, S. Hariguchi, I. Yoshida, S. Kitajima and T. Nishimura, “Accumulation of Amyloid B-Protein Precursor (APP) in Purkinje Cells and Increase of Amino-Terminal Fragments of APP in Cerebrum and Cerebellum of Aged Rat Brain,” Brain Research, Vol. 643, No. 1-2, 1994, pp. 319- 323. doi:10.1016/0006-8993(94)90040-X
- C. A. Sherman and G. A. Higgins, “Regulated Splicing of the Amyloid Precursor Protein during Postnatal Development of the Rat Basal Forebrain,” Developmental Brain Research, Vol. 66, No. 1, 1992, pp. 63-69. doi:10.1016/0165-3806(92)90141-I
- J. V. Nadler, “Kainic Acid: Neurophysiological and Neurotoxic Actions,” Life Sciences, Vol. 24, No. 4, 1979, pp. 289-300. doi:10.1016/0024-3205(79)90325-4
- R. Siman, J. P. Card, R. B. Nelson and L. G. Davis, “Expression of β-Amyloid Precursor Protein in Reactive Astrocytes Following Neuronal Damage,” Neuron, Vol. 3, No. 3, 1989, pp. 275-285. doi:10.1016/0896-6273(89)90252-3
- T. Kawarabayashi, M. Shoji, Y. Harigaya, H. Tamaguchi and S. Hirai, “Expression of APP in the Early Stage of Brain Damage,” Brain Research, Vol. 563, No. 1-2, 1991, pp. 334-338. doi:10.1016/0006-8993(91)91558-I
- C. Solá, F. J. Garcia-Ladona, G. Mengod, A. Probst, P. Frey and J. M. Palacios, “Increased Levels of the Kunitz Protease Inhibitor-Containing BAPP mRNAs in Rat Brain Following Neurotoxic Damage,” Molecular Brain Research, Vol. 17, 1993, pp. 41-52. doi:10.1016/0169-328X(93)90071-V
- T. P. Wong, T. Debeir, K. Duff and A. C. Cuello, “Reorganization of Cholinergic Terminals in the Cerebral Cortex and Hippocampus in Transgenic Mice Carrying Mutated Presenilin-1 and Amyloid Precursor Protein Transgenes,” Journal of Neuroscience, Vol. 19, No. 7, 1999, pp. 2706-2716.
- P. K. Panegyres, “The Effects of Excitotoxicity on the Expression of the Amyloid Precursor Protein Gene in the Brain and its Modulation by Neuroprotective Agents,” Journal of Neural Transmission, Vol. 105, No. 4-5, 1998, pp. 463-478. doi:10.1007/s007020050070
- P. K. Panegyres and J. Hughes, “The Neuroprotective Effects of the Recombinant Interleukin-1 Receptor Antagonist rhIL-1ra after Excitotoxic Stimulation with Kainic Acid and its Relationship to the Amyloid Precursor Protein Gene,” Journal of the Neurological Sciences, Vol. 154, No. 2, 1998, pp. 123-132. doi:10.1016/S0022-510X(97)00214-1
- B. Brugg, Y. L. Dubreuil, G. Huber, E. E. Wollman, N. Delhaye-Bouchaud and J. Mariani, “Inflammatory Processes Induce B-Amyloid Precursor Protein Changes in Mouse Brain,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 92, No. 7, pp. 3032-3035.
- P. K. Panegyres, “A Study of the Relationship between Brain Seizure Activity, Immediate Early Genes and the Amyloid Precursor Protein Gene,” Ph.D Thesis, University of Cambridge, Cambridge, 1995.
- M. Walton, G. MacGibbon, D. Young, E. Sirimanne, C. Williams, P. Gluckman and M. Dragunow, “Do c-Jun, cFos, and Amyloid Precursor Protein Play a Role in Neuronal Death or Survival?” Journal of Neuroscience Research, Vol. 53, No. 3, 1998, pp. 330-342. doi:10.1002/(SICI)1097-4547(19980801)53:3<330::AID-JNR7>3.0.CO;2-B
- J. Trejo, T. Massamiri, T. Deng, N. N. Dewji, R. M. Bayney and J. H. Brown, “A Direct Role for Protein Kinase C and the Transcription Factor jun/AP-1 in the Regulation of the Alzheimer’s B-amyloid Precursor Protein Gene,” Journal of Biological Chemistry, Vol. 269, No. 34, 1994, pp. 21682-21690.
- K. Iverfeldt, S. I. Walaas and P. Greengard, “Altered Processing of Alzheimer Amyloid Precursor Protein in Response to Neuronal Degeneration,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 90, No. 9, 1993, pp. 4146-4150. doi:10.1073/pnas.90.9.4146
- W. Wallace, S. T. Ahlers, J. Gotlib, V. Bragin, J. Sugar, R. Gluck, P. A. Shea, K. L. Davis and V. Haroutunian, “Amyloid Precursor Protein in the Cerebral Cortex is Rapidly and Persistently Induced by Loss of Subcortical Innervation,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 90, No. 18, 1993, pp. 8712-8716. doi:10.1073/pnas.90.18.8712
- Y. Nakamura, M. Takeda, H. Niigawa, S. Hariguchi and T. Nishimura, “Amyloid B-Protein Deposition in Rat Hippocampus Lesioned by Ibotenic Acid Injection,” Neuroscience Letters, Vol. 136, 1992, pp. 95-98. doi:10.1016/0304-3940(92)90656-R
- K. Shigematsu, P. L. McGeer, D. G. Walker, T. Ishii and E. G. McGeer, “Reactive Microglia/Macrophages Phagocytose Amyloid Precursor Protein Produced by Necrosis Following Neuronal Damage,” Journal of Neuroscience Research, Vol. 31, 1992, pp. 443-453. doi:10.1002/jnr.490310306
- J. B. Pan, L. M. Monteggia and T. Giordano, “Altered Levels and Splicing of the Amyloid Precursor Protein in the Adult Rat Hippocampus after Treatment with DMSO or Retinoic Acid,” Molecular Brain Research, Vol. 18, No. 3, 1993, pp. 259-266. doi:10.1016/0169-328X(93)90198-X
- A. Lewen, G. L. Li, P. Nilsson, Y. Olsson and L. Hillered, “Traumatic Brain Injury in Rat Produces Changes of BAmyloid Precursor Protein Immunoreactivity,” NeuroReport, Vol. 6, 1995, pp. 357-360. doi:10.1097/00001756-199501000-00032
- N. Murakami, T. Yamaki, Y. Iwamoto, T. Sakakibara, N. Kobori, S. Fushiki and S. Ueda, “Experimental Brain Injury Induces Expression of Amyloid Precursor Protein, which May Be Related to Neuronal Loss in the Hippocampus,” Journal of Neurotrauma, Vol. 15, No. 11, 1998, pp. 993-1003. doi:10.1089/neu.1998.15.993
- N. Otsuko, M. Tamonaga and K. Ikeda, “Rapid Appearance of B-Amyloid Precursor Protein Immunoreactivity in Damaged Axons and Reactive Glial Cells in Rat Brain Following Needle Stab Injury,” Brain Research, Vol. 568, No. 1-2, 1991, pp. 35-338.
- C. Van Den Heuvel, S. Lewis, M. Wong, J. Manavis, J. Finnie, P. Blumbergs, N. Jones and P. Reilly, “Diffuse Neuronal Perikaryon Amyloid Precursor Protein Immunoreactivity in a Focal Head Impact Model,” Acta Neurochirurgica Supplementum, Vol. 71, 1998, pp. 209-211.
- K. Abe, R. E. Tanzi and K. Kogure, “Selective Induction of Kunitz Type Protease Inhibitor Domain Containing Amyloid Precursor Protein mRNA after Persistent Focal Ischemia,” Neuroscience Letters, Vol. 125, No. 2, 1991, pp. 172-174. doi:10.1016/0304-3940(91)90020-T
- K. Baiden-Amissah, U. Joashi, R. Blumberg, H. Mehmet, A. D. Edwards and P. M. Cox, “Expression of Amyloid Precursor Protein (Beta-APP) in the Neonatal Brain Following Hypoxic Ischaemic Injury,” Neuropathology and Applied Neurobiology, Vol. 24, No. 5, 1998, pp. 346- 352. doi:10.1046/j.1365-2990.1998.00141.x
- D. T. Stephenson, K. Rash and J. A. Clemens, “Amyloid Precursor Protein Accumulates in Regions of Neurodegeneration Following Focal Cerebral Ischemia in the Rat,” Brain Research, Vol. 593, No. 1,1992, pp. 128-135. doi:10.1016/0006-8993(92)91274-I
- H. Wakita, H. Tomimoto, I. Akiguchi, K. Ohnishi, S. Nakamura and J. Kimura, “Regional Accumulation of Amyloid B/A4 Protein Precursor in the Gerbil Brain Following Transient Cerebral Ischemia,” Neuroscience Letters, Vol. 146, 1992, pp. 135-138.
- H. Tomimoto, H. Wakita, I. Akiguchi, S. Nakamura and J. Kimura, “Temporal Profiles of Accumulation of Amyloid B/A4 Protein Precursor in the Gerbil after Graded Ischemic Stress,” Journal of Cerebral Blood Flow and Metabolism, Vol. 14, No. 4, 1994, pp. 565-573. doi:10.1038/jcbfm.1994.70
- G. Palacios, G. Mengod, A. Tortosa, I. Ferrer and J. M. Palacios, “Increased B-Amyloid Precursor Protein Expression in Astrocytes in the Gerbil Hippocampus Following Ischaemia: Association with Proliferation of Astrocytes,” European Journal of Neuroscience, Vol. 7, 1995, pp. 501-510. doi:10.1111/j.1460-9568.1995.tb00346.x
- M. J. McNamara, C. T. Ruff, W. Wasco, R. E. Tanzi, G. Thinakaran and B. T. Hyman, “Immunohistochemical and In Situ Analysis of Amyloid Precursor-Like Protein-1 and Amyloid Precursor-Like Protein-2 Expression in Alzheimer Disease and Aged Control Brains,” Brain Research, Vol. 804, No. 1, 1998, pp. 45-51. doi:10.1016/S0006-8993(98)00653-2
- K. Plaschke, E. Martin and H. J. Bardenheuer, “Effect of Propentofylline on Hippocampal Brain Energy State and Amyloid Precursor Protein Concentration in a Rat Model of Cerebral Hypoperfusion,” Journal of Neural Transmission, Vol. 105, 1998, pp. 1065-1077. doi:10.1007/s007020050112
- J. N. Scott, I. M. Parhad and A. W. Clark, “B-Amyloid Precursor Protein Gene is Differentially Expressed in Axotomized Sensory and Motor Systems,” Molecular Brain Research, Vol. 10, No. 4, 1991, pp. 315-325. doi:10.1016/0169-328X(91)90090-K
- J. Alvarez, R. D. Moreno, O. Llanos, N. C. Inestrosa, E. Brandan, T. Colby and F. S. Esch, “Axonal Sprouting Induced in the Sciatic Nerve by the Amyloid Precursor Protein (APP) and Other Antiproteases,” Neuroscience Letters, Vol. 144, No. 1-2, 1992, pp. 130-134. doi:10.1016/0304-3940(92)90733-N
- O. Salinero, J. J. Garrido and F. Wandosell, “Amyloid Precursor Protein Proteoglycan Is Increased after Brain Damage,” Biochimica et Biophysica Acta, Vol. 1406, No. 3, 1998, pp. 237-250.
- J. Alvarez, R. D. Moreno and N. C. Inestrosa, “Mitosis of Schwann Cells and Demyelination Are Induced by the Amyloid Precursor Protein and Other Protease Inhibitors in the Rat Sciatic Nerve,” European Journal of Neuroscience, Vol. 7, No. 1, 1995, pp. 152-159. doi:10.1111/j.1460-9568.1995.tb01029.x
- G. Forloni, R. Del Bo, N. Angeretti, S. Smiroldo, N. Gabellini and G. Vantini, “Nerve Growth Factor Does Not Influence the Expression of B Amyloid Precursor Protein mRNA in Rat Brain: In Vivo and in Vitro Studies,” Brain Research, Vol. 620, No. 2, 1993, pp. 292-296. doi:10.1016/0006-8993(93)90168-M
- M. D. Lindner, S. I. Dworetzky, C. Sampson and R. Loy, “Relationship of APP mRNA Transcripts and Levels of NGF and Low-Affinity NGF Receptors to Behavioral Measures of Age-related Cognitive Dysfunction,” Journal of Neurosciemce, Vol. 14, No. 4, 1994, pp. 2282-2289.
- H. Zheng, M. Jiang and M. E. Trumbauer, “β-amyloid Precursor Protein-Deficient Mice Show Reactive Gliosis and Decreased Locomotor Activity,” Cell, Vol. 81, No. 4, 1995, pp. 525-531. doi:10.1016/0092-8674(95)90073-X
- H. Zheng, M. Jiang, M. E. Trumbauer, R. Hopkins, D. J. Sirinathsinghji, K. A. Stevens, M. W. Conner, H. H. Slunt, S. S. Sisodia, H. Y. Chen and L. H. Van der Ploeg, “Mice Deficient for the Amyloid Precursor Protein Gene,” Annals of the New York Academy of Sciences, Vol. 777, No. 908, 1996, pp. 421-426. doi:10.1111/j.1749-6632.1996.tb34456.x
- U. Muller, N. Cristina, Z. W. Li, D. P. Wolfer, H. P. Lipp, T. Rulicke, S. Brandner, A. Aguzzi and C. Weissmann, “Behavioral and Anatomical Deficits in Mice Homozygous for a Modified B-Amyloid Precursor Protein Gene,” Cell, Vol. 79, No. 5, 1994, pp. 755-765. doi:10.1016/0092-8674(94)90066-3
- U. Muller, N. Cristina, Z. W. Li, D. P. Wolfer, H. P. Lipp, T. Rulicke, S. Brandner, A. Aguzzi and C. Weissman, “Mice Homozygous for a Modified Beta-Amyloid Precursor Protein (Beta APP) Gene Show Impaired Behaviour and High Incidence of Agenesis of the Corpus Callosum,” Annals of the New York Academy of Sciences, Vol. 777, 1996, pp. 65-73. doi:10.1111/j.1749-6632.1996.tb34402.x
- C. S. von Koch, H. Zheng, H. Chen, M. Trumbauer, G. Thinakaran, L. G. van der Ploeg, D. L. Price and S. S. Sisodia, “Generation of APLP2 KO Mice and Early Postnatal Lethality in APLP2/APP Double KO Mice,” Neurobiology of Aging, Vol. 18, No. 6, 1997, pp. 661- 669. doi:10.1016/S0197-4580(97)00151-6
- R. G. Perez, H. Zheng, L. H. T. Van der Ploeg and E. H. Koo, “The B-amyloid Precursor Protein of Alzheimer’s Disease Enhances Neuron Viability and Modulates Neuronal Polarity,” Journal of Neuroscience, Vol. 17, 1997, pp. 9407-9414.
- G. R. Dawson, G. R. Seabrook, H. Zheng, D. W. Smith, S. Graham, G. O’Dowd, B. J. Bowery, S. Boyce, M. E. Trumbauer, H. Y. Chem, L. H. Van der Ploeg and D. J. Sirinathsinghji, “Age-related Cognitive Deficits, Impaired Long-term Potentiation and Reduction in Synaptic Marker Density in Mice Lacking the Beta-amyloid Precursor Protein,” Neuroscience, Vol. 90, No. 1, 1999, pp. 1-13. doi:10.1016/S0306-4522(98)00410-2
- H. Meziane, J. C. Dodart, C. Mathis, S. Little, J. Clemens, S. M. Paul and A. Ungerer, “Memory-Enhancing Effects of Secreted Forms of the B-amyloid Precursor Protein in Normal and Amnestic Mice,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 95, 1998, pp. 12683-12688. doi:10.1073/pnas.95.21.12683
- J. M. Roch, E. Masliah, A. C. Roch-Levecq, M. P. Sundsmo, D. A. Otero, I. Veinbergs and T. Saitoh, “Increase of Synaptic Density and Memory Retention by a Peptide Representing the Trophic Domain of the Amyloid Beta/A4 Protein Precursor,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 91, No. 16, 1994, pp. 7450-7454. doi:10.1073/pnas.91.16.7450
- L. S. Higgins, D. M. Holtzman, J. Rabin, W. C. Mobley and B. Cordell, “Transgenic Mouse Brain Histopathology Resembles Early Alzheimer’s Disease,” Annals of Neurology, Vol. 35, No. 5, 1994, pp. 598-607. doi:10.1002/ana.410350514
- D. Games, D. Adams, R. Alessandrini, R. Barbour, P. Berthelette, C. Blackwell, T. Carr, J. Clemens, T. Donaldson and F. Gillespie, “Alzheimer-type Neuropathology in Transgenic Mice Overexpressing V717F Bamyloid Precursor Protein,” Nature, Vol. 373, No. 6514, 1995, pp. 523-527. doi:10.1038/373523a0
- K. S. Chen, E. Masliah, H. Grajeda, T. Guido, J. Huang, K. Khan, R. Motter, F. Soriano and D. Games, “Neurodegenerative Alzheimer-Like Pathology in PDAPP 717V→F Transgenic Mice,” Progress in Brain Research, Vol. 117, 1998, pp. 327-334.
- C. Czech, C. Masters and K. Beyreuther, “Alzheimer’s Disease and Transgenic Mice,” Journal of Neural Transmission, Vol. 44, 1994, pp. 219-230.
- C. Czech, P. Delaere, A. F. Macq, M. Reibaud, S. Dreisler, N. Touchet, B. Schombert, M. Mazadier, L. Mercken, M. Theisen, L. Pradier, J. N. Octave, K. Beyreuther and G. Tremp, “Proteolytical Processing of Mutated Human Amyloid Precursor Protein in Transgenic Mice,” Molecular Brain Research, Vol. 47, No. 1-2, 1997, pp. 108- 116. doi:10.1016/S0169-328X(97)00039-9
- K. K. Hsiao, D. R. Borchelt, K. Olson, R. Johannsdottir, C. Kitt, W. Yunis, S. Xu, C. Eckman, S. Younkin and D. Price, “Age-Related CNS Disorder and Early Death in Transgenic FVB/N Mice Overexpressing Alzheimer Amyloid Precursor Proteins,” Neuron, Vol. 15, No. 5, 1995, pp. 1203-1218. doi:10.1016/0896-6273(95)90107-8
- K. Hsiao, P. Chapman, S. Nilsen, C. Eckman, Y. Harigaya, S. Younkin, F. Yang and G. Cole, “Correlative Memory Deficits, A Beta Elevation, and Amyloid Plaques in Transgenic Mice,” Science, Vol. 27, 1996, pp. 99-102. doi:10.1126/science.274.5284.99
- M. C. Irizarry, F. Soriano, M. McNamara, K. J. Page, D. Schenk, D. Games and B. T. Hyman, “Abeta Deposition Is Associated with Neruophil Changes, but Not with Overt Neuronal Loss in the Human Amyloid Precursor Protein V717F (PDAPP) Transgenic Mouse,” Journal of Neuroscience, Vol. 17, 1997, pp. 7053-7059.
- G. Leanza, “Chronic Elevation of Amyloid Precursor Protein Expression in the Neocortex and Hippocampus of Rats with Selective Cholinergic Lesions,” Neuroscience Letters, Vol. 257, No. 1, 1998, pp. 53-56. doi:10.1016/S0304-3940(98)00744-7
- J. F. Loring, C. Paszty, A. Rose, T. K. McIntosh, H. Murai, J. E. Pierce, S. R. Schramm, K. Wymore, V. M. Lee, J. Q. Trojanowski and K. R. Peterson, “ Rational Design of an Animal Model for Alzheimer’s Disease: Introduction of Multiple Human Genomic Transgenes to Reproduce AD Pathology in a Rodent,” Neurobiology of Aging, Vol. 17, No. 2, 1996, pp. 173-182. doi:10.1016/0197-4580(95)02076-4
- D. Moechars, K. Lorent, I. Dewachter, V. Baekelandt, B. De Strooper and F. Van Leuven, “Transgenic Mice Expressing an Alpha-Secretion Mutant of the Amyloid Precursor Protein in the Brain Develop a Progressive CNS Disorder,” Behavioural Brain Research, Vol. 95, No. 1, 1998, pp. 55-64. doi:10.1016/S0166-4328(97)00210-6
- L. Mucke, E. Masliah, W. B. Johnson, M. D. Ruppe, M. Alford, E. M. Rockenstein, S. Forss-Petter, M. Pietropaolo, M. Mallory and C. R. Abraham, “Synaptotrophic Effects of Human Amyloid B Protein Precursors in the Cortex of Transgenic Mice,” Brain Research, Vol. 666, 1994, pp. 151-167. doi:10.1016/0006-8993(94)90767-6
- L. Mucke, C. R. Abraham and E. Masliah, “Neurotrophic and Neuroprotective Effects of hAPP in Transgenic Mice,” Annals of the New York Academy of Sciences, Vol. 777, No. 617, 1996, pp. 82-88. doi:10.1111/j.1749-6632.1996.tb34405.x
- I. Nishimura, T. Uetsuki, S. U. Dani, Y. Ohsawa, I. Saito, H. Okamura, Y. Uchiyama and K. Yoshikawa, “Degeneration in Vivo of Rat Hippocampal Neurons by Wild-Type Alzheimer Amyloid Precursor Protein Overexpressed by Adenovirus-Mediated Gene Transfer,” Journal of Neuroscience, Vol. 18, 1998, pp. 2387-2398.
- S. Rabizadeh, C. M. Bitler, L. L. Butcher and D. E. Bredesen, “Expression of the Low-Affinity Nerve Growth Factor Receptor Enhances B-Amyloid Peptide Toxicity,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 91, No. 22, 1994, pp. 10703-10706. doi:10.1073/pnas.91.22.10703
- M. Racchi, J. A. Johnston, F. M. Flood, R. F. Cowburn and S. Govoni, “Amyloid Precursor Protein Metabolism in Fibroblasts from Individuals with One, Two or Three Copies of the Amyloid Precursor Protein (APP) Gene,” Biochemical Journal, Vol. 338, 1999, pp. 777-782. doi:10.1042/0264-6021:3380777
- F. A. Sandhu, M. Salim and S. B. Zain, “Expression of the Human Beta-Amyloid Protein of Alzheimer’s Disease Specifically in the Brains of Transgenic Mice,” Journal of Biological Chemistry, Vol. 266, No. 32, 1991, pp. 21331- 21334.
- C. Sturchler-Pierrat, D. Abramowski, M. Duke, K. H. Wiederhold, C. Mistl, S. Rothacher, B. Ledermann, K. Burki, P. Frey, P. A. Paganetti, C. Waridel, M. E. Calhous, M. Jucker, A. Prosbst, M. Staufenbiel and B. Sommer, “Two Amyloid Precursor Protein Transgenic Mouse Models with Alzheimer Disease-Like Pathology,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 94, No. 24, 1997, pp. 13287-13292. doi:10.1073/pnas.94.24.13287
- F. Yamaguchi, S. J. Richards, K. Beyreuther, M. Salbaum, G. A. Carlson and S. B. Dunnett, “Transgenic Mice for the Amyloid Precursor Protein 695 Isoform Have Impaired Spatial Memory,” NeuroReport, Vol. 2, No. 12, 1991, pp. 781-784. doi:10.1097/00001756-199112000-00013
- G. A. Carlson, D. R. Brochelt, A. Dake, S. Turner, V. Danielson, J. D. Coffin, C. Eckman, J. Meiners, S. P. Nilsen, S. G. Younkin and K. K. Hsiao, “Genetic Modification of the Phenotypes Produced by Amyloid Precursor Protein Overexpression in Transgenic Mice,” Human Molecular Genetics, Vol. 6, No. 11, 1997, pp. 1951-1959. doi:10.1093/hmg/6.11.1951
- C. Schonlein, A. Probst and G. Huber, “Characterization of Proteases with the Specificity to Cleave at the Secretase-Site of Beta-APP,” Neuroscience Letters, Vol. 161, No. 1, 1993, pp. 33-36. doi:10.1016/0304-3940(93)90133-6
- D. Moechars, M. Gilis, C. Kuiperi, I. Laenen and F. Van Leuven, “Aggressive Behaviour in Transgenic Mice Expressing APP is Alleviated by Serotonergic Drugs,” NeuroReport, Vol. 9, No. 16, 1998, pp. 3561-3564. doi:10.1097/00001756-199811160-00004
- F. A. Sandhu, R. H. Porter, R. V. Eller, S. B. Zain, M. Salim and J. T. Greenamyre, “NMDA and AMPA Receptors in Transgenic Mice Expressing Human BetaAmyloid Protein,” Journal of Neurochemistry, Vol. 61, 1993, pp. 2286-2289. doi:10.1111/j.1471-4159.1993.tb07471.x
- D. Moechars, K. Lorent, B. De Strooper, I. Dewachter and F. Van Leuven, “Expression in Brain of Amyloid Precursor Protein Mutated in the Alpha-Secretase Site Causes Disturbed Behaviour, Neuronal Degeneration and Premature Death in Transgenic Mice,” EMBO Journal, Vol. 15, No. 6, 1996, pp. 1265-1274.
- D. S. Howland, S. P. Trusko, M. J. Savage, A. G. Reaume, D. M. Lang, J. D. Hirsch, N. Maeda, R. Siman, B. D. Greenberg, R. W. Scott and D. G. Flood, “Modulation of Secreted Beta-Amyloid Precursor Protein and Amyloid Beta-Peptide in Brain by Cholesterol,” Journal of Biological Chemistry, Vol. 273, No. 26, 1998, pp. 16576- 16582. doi:10.1074/jbc.273.26.16576
- S. A. Frautschy, F. Yang, M. Irrizarry, B. Hyman, T. C. Saido, K. Hsiao and G. M. Cole, “Microglial Response to Amyloid Plaques in APPsw Transgenic Mice,” American Journal of Pathology, Vol. 152, No. 1, 1998, pp. 307-317.
- T. A. Bayer, A. Fossgreen, C. Czech, K. Beyreuther and O. D. Wiestler, “Plaque Formation in Brain Transplants Exposed to Human Beta-Amyloid Precursor Protein 695,” Acta Neuropathologica, Vol. 92, No. 2, 1996, pp. 130-137. doi:10.1007/s004010050500
- T. Arendt, M. Holzer, R. Fruth, M. K. Bruckner and U. Gartner, “Paired Helical Filament-like Phosphorylation of Tau, Deposition of Beta/A4-Amyloid and Memory Impairment in Rat Induced by Chronic Inhibition of Phosphatase 1 and 2A,” Neuroscience, Vol. 69, No. 3, 1995, pp. 691-698. doi:10.1016/0306-4522(95)00347-L
- R. B. Banati, J. Gehrmann, J. Lannes-Vieira, H. Wekerle and G. W. Kreutzberg, “Inflammatory Reaction in Experimental Autoimmune Encephalomyelitis (EAE) is Accompanied by a Microglial Expression of the Beta A4-amyloid Precursor Protein (APP),” Glia, Vol. 14, No. 3, 1995, pp. 209-215. doi:10.1002/glia.440140306
- J. Shi, K. S. Panickar, S. H. Yang, O. Rabbani, A. L. Day and J. W. Simpkins, “Estrogen Attenuates Over-Expression of Beta-Amyloid Precursor Protein Messager RNA in an Animal Model of Focal Ischemia,” Brain Research, Vol. 810, No. 1-2, 1998, pp. 87-92. doi:10.1016/S0006-8993(98)00888-9
- A. Islam, R. N. Kalaria, B. Winblad and A. Adem, “Enhanced Localization of Amyloid Beta Precursor Protein in the Rat Hippocampus Following Long-Term Adrenalectomy,” Brain Research, Vol. 806, 1998, pp. 108- 112. doi:10.1016/S0006-8993(98)00711-2
- H. M. Chao, R. L. Spencer, M. Frankfurt and B. S. McEwen, “The Effects of Aging and Hormonal Manipulation on Amyloid Precursor Protein APP 695 mRNA Expression in the Rat Hippocampus,” Journal of Neuroendocrinology, Vol. 6, 1994, pp. 517-521. doi:10.1111/j.1365-2826.1994.tb00614.x
- T. Tully and K. White, “Human Amyloid Precursor Protein Ameliorates Behavioural Deficit of Flies Deleted for Appl Gene,” Neuron, Vol. 9, 1992, pp. 595-605. doi:10.1016/0896-6273(92)90024-8
- L. Torroja, L. Luo and K. White, “APPL, the Drosophila Member of the APP-family, Exhibits Differential Trafficking and Processing in CNS Neurons,” Journal of Neuroscience, Vol. 16, No. 15, 1996, pp. 4638-4650.
- A. Fossgreen, B. Bruckner, C. Czech, C. L. Masters, K. Beyreuther and R. Paro, “Transgenic Drosophila Expressing Human Amyloid Precursor Protein Show Gammasecretase Activity and a Blistered-Wing Phenotype,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 95, 1998, pp. 13703- 13708. doi:10.1073/pnas.95.23.13703
- I. Daigle and C. Li, “Apl-1, a Caenorhabditis Elegans Gene Encoding a Protein Related to the Human Betaamyloid Protein Precursor,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 90, No. 24, 1993, pp. 12045-12049. doi:10.1073/pnas.90.24.12045
- A. I. Bush, W. H. Pettingal, G. Multhaup, M. D. Paradis, J. P. Vonsattel, J. F. Gusella, K. Beyreuther, C. L. Masters and R. E. Tanzi, “Rapid Induction of Alzheimer AB Amyloid Formation by Zinc,” Science, Vol. 265, 1994, pp. 1464-1467. doi:10.1126/science.8073293
- N. Ida, T. Hartmann, J. Pantel, J. Schroder, R. Zerfass, H. Forstl, R. Sandbrink, C. L. Masters and K. Beyreuther, “Analysis of Heterogeneous A4 Peptides in Human Cerebrospinal Fluid and Blood by a Newly Developed Sensitive Western Blot Assay,” Journal of Biological Chemistry, Vol. 271, No. 37, 1996, pp. 22908-22914. doi:10.1074/jbc.271.37.22908
- M. Simons, B. De Strooper, G. Multhaup, P. J. Tienari, C. G. Dotti and K. Beyreuther, “Amyloidogenic Processing of the Human Amyloid Precursor Protein in Primary Cultures of Rat Hippocampal Neurons,” Journal of Neuroscience, Vol. 16, No. 3, 1996, pp. 899-908.
- C. Vigo-Pelfrey, D. Lee, P. Keim, I. Lieberburg and D. B. Schenk, “Characterization of B-Amyloid Peptide From Human Cerebrospinal Fluid,” Journal of Neurochemistry, Vol. 61, 1993, pp. 1965-1968. doi:10.1111/j.1471-4159.1993.tb09841.x
- G. K. Gouras, H. Xu, J. N. Jovanovic, J. D. Buxbaum, R. Wang, P. Greengard, N. R. Relkin and S. Gandy, “Generation and Regulation of Beta-Amyloid Peptide Variants by Neurons,” Journal of Neurochemistry, Vol. 71, No. 5, 1998, pp. 1920-1925. doi:10.1046/j.1471-4159.1998.71051920.x
- C. J. Pike, A. J. Walencewicz-Wasserman, J. Kosmoski, D. H. Cribbs, C. G. Glabe and C. W. Cotman, “Structureactivity Analyses of B-amyloid Peptides: Contributions of the B25-35 R and Neurotoxicity,” Journal of Neurochemistry, Vol. 64, 1995, pp. 253-265. doi:10.1046/j.1471-4159.1995.64010253.x
- C. J. Barrow, A. Yasuda, P. T. Kenny and M. G. Zagorski, “Solution Conformations and Aggregational Properties of Synthetic Amyloid Beta-Peptides of Alzheimer’s Disease. Analysis of Circular Dichroism Spectra,” Journal of Molecular Biology, Vol. 225, No. 4, 1992, pp. 1075- 1093. doi:10.1016/0022-2836(92)90106-T
- D. Burdick, B. Soreghan, M. Kwon, J. Kosmoski, M. Knauer, A. Henschen, J. Yates, C. Cotman and C. Glabe, “Assembly and Aggregation Properties of Synthetic Alzheimer’s A4/beta Amyloid Peptide Analogs,” Journal of Biological Chemistry, Vol. 267, No. 1, 1992, pp. 546- 554.
- P. E. Fraser, J. T. Nguyen, W. K. Surewicz and D. A. Kirschner, “PH-Dependent Structural Transitions of Alzheimer Amyloid Peptides,” Biophysical Journal, Vol. 60, No. 5, 1991, pp. 1190-1201. doi:10.1016/S0006-3495(91)82154-3
- C. Soto, M. C. Branes, J. Alvarez and N. C. Inestrosa, “Structural Determinants of the Alzheimer’s Amyloid Beta-Peptide,” Journal of Neurochemistry, Vol. 63, No. 4, 1994, pp. 1991-1198.
- H. Stricht, P. Bayer, D. Willbold, S. Dames, C. Hilbich, K. Beyreuther, R. W. Frank and P. Rosch, “Structure of Amyloid A4-(1-40)-peptide of Alzheimer’s Disease,” European Journal of Biochemistry, Vol. 233, No. 1, 1995, pp. 293-298.
- F. P. Zemlan, G. D. Vogelsang, L. McLaughlin and G. E. Dean, “Alzheimer’s Paired Helical Filaments: Amyloid Precursor Protein Epitope Mapping,” Brain Research Bulletin, Vol. 33, No. 4, 1994, pp. 387-392. doi:10.1016/0361-9230(94)90281-X
- C. Hilbich, B. Kisters-Woike, J. Reed, C. L. Masters and K. Beyreuther, “Human and Rodent Sequence Analogs of Alzheimer’s Amyloid Beta A4 Share Similar Properties and Can be Solubilized in Buffers of pH 7.4,” European Journal of Biochemistry, Vol. 201, No. 1, 1991, pp. 61- 69. doi:10.1111/j.1432-1033.1991.tb16256.x
- B. De Strooper, M. Simons, G. Multhaup, F. Van Leuven, K. Beyreuther and C. G. Dotti, “Production of Intracellular Amyloid-Containing Fragments in Hippocampal Neurons Expressing Human Amyloid Precursor Protein and Protection Against Amyloidogenesis by Subtle Amino Acid Substitutions in the Rodent Sequence,” EMBO Journal, Vol. 14, No. 20, 1995, pp. 4932-4938.
- P. E. Fraser, J. T. Nguyen, H. Inouye, W. K. Surewicz, D. J. Selkoe, M. B. Podlisny and D. A. Kirschner, “Fibril Formation by Primate, Rodent, and Dutch-Hemorrhagic Analogues of Alzheimer Amyloid Beta-Protein,” Biochemistry, Vol. 31, No. 44, 1992, pp. 10716-10723. doi:10.1021/bi00159a011
- G. A. Higgins, D. A. Lewis, S. Bahmanyar, D. Goldgaber, D. C. Gajdusek, W. G. Young, J. H. Morrison and M. C. Wilson, “Differential Regulation of Amyloid-b-protein mRNA Expression within Hippocampal Neuronal Subpopulations in Alzheimer Disease,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 85, 1988, pp. 1297-1301. doi:10.1073/pnas.85.4.1297
- L. Otvos Jr, G. I. Szendrei, V. M. Lee and H. H. Mantsch, “Human and Rodent Alzheimer Beta-Amyloid Peptides Acquire Distinct Conformations in Membrane-Mimicking Solvents,” European Journal of Biochemistry, Vol. 211, No. 1-2, 1993, pp. 249-257. doi:10.1111/j.1432-1033.1993.tb19893.x
- M. D. Fowler, A. V. Koulov, C. Alory-Jost, M. S. Marks, W. E. Balch and J. W. Kelly, “Functional Amyloid Formation with Mammalian Tissue,” PLoS Biology, Vol. 4, No. 1, 2006, p. e6. doi:10.1371/journal.pbio.0040006
- S. J. Wood, R. Wetzel, J. D. Matin and M. R. Hurle, “Prolines and Amyloidogenicity in Fragments of the Alzheimer’s Peptide Beta/A4,” Biochemistry, Vol. 34, No. 3, 1995, pp. 724-730. doi:10.1021/bi00003a003
- T. Wisniewski, J. Ghiso and B. Frangione, “Peptides Homologous to the Amyloid Protein of Alzheimer’s Disease Containing a Glutamine for Glutamic Acid Substitution Have Accelerated Amyloid Fibril Formation,” Biochemical and Biophysical Research Communications, Vol. 179, No. 3, 1991, pp. 1247-1254. doi:10.1016/0006-291X(91)91706-I
- W. J. Strittmatter, D. Y. Huang, R. Bhasin, A. D. Roses and D. Goldgaber, “Avid Binding of Beta A Amyloid Peptide to Its Own Precursor,” Experimental Neurology, Vol. 122, No. 2, 1993, pp. 327-334. doi:10.1006/exnr.1993.1132
- D. Huang, M. Martin, D. Hu, A. D. Roses, D. Goldgaber and W. J. Strittmatter, “Binding of IgG to Amyloid Beta A4 Peptide via the Heavy-Chain Hinge Region with Preservation of Antigen Binding,” Journal of Neuroimmunology, Vol. 48, No. 2, 1993, pp. 199-203. doi:10.1016/0165-5728(93)90192-2
- L. K. Rasmussen, E. S. Sorensen, T. E. Petersen, J. Gliemann and P. H. Jensen, “Identification of Glutamine and Lysine Residues in Alzheimer Amyloid Beta A4 Peptide Responsible for Transglutaminase-Catalysed Homopolymerization and Cross-Linking to Alpha 2M Receptor,” FEBS Letters, Vol. 338, No. 2, 1994, pp. 161-166. doi:10.1016/0014-5793(94)80356-0
- J. Y. Koh, L. L. Yang and C. W. Cotman, “β-amyloid Protein Increases the Vulnerability of Cultured Cortical Neurons to Excitotoxic Damage,” Brain Research, Vol. 533, No. 2, 1990, pp. 315-320. doi:10.1016/0006-8993(90)91355-K
- B. A. Yankner, L. K. Duffy and D. A. Kirschner, “Neurotrophic and Neurotoxic Effects of Amyloid β Protein: Reversal by Tachykinin Neuropeptides,” Science, Vol. 250, No. 4978, 1990, pp. 279-282. doi:10.1126/science.2218531
- P. Rovero, R. Patacchini, A. R. Renzetti, M. Brown, J. Mizrahi, C. A. Maggi and A. Giachetti, “Interaction of Amyloid B Protein (25 - 35) with Tachykinin Receptors,” Neuropeptides, Vol. 22, 1992, pp. 99-101. doi:10.1016/0143-4179(92)90062-2
- Y. Shimohigashi, H. Matsumoto, Y. Takano, R. Saito, T. Iwara, H. O. Kamiya and M. Ohno, “Receptor-mediated Specific Biological Activity of a β-Amyloid Protein Fragment for NK-1 Substance P Receptors,” Biochemical and Biophysical Research Communications, Vol. 193, No. 2, 1993, pp. 624-630. doi:10.1006/bbrc.1993.1670
- V. K. Singh, J. F. Cheng and S. J. C. Leu, “Effect of Substance P and Protein Kinase Inhibitors on β-Amyloid Peptide-Induced Proliferation of Cultured Brain Cells,” Brain Research, Vol. 660, No. 2, 1994, pp. 353-356. doi:10.1016/0006-8993(94)91313-7
- H. Kimura and D. Schubert, “Amyloid β-protein Activates Tachykinin Receptors and Inositol Triphosphate Accumulation by Synergy with Glutamate,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 90, 1993, pp. 7508-7512. doi:10.1073/pnas.90.16.7508
- L. D. Plant, J. P. Boyle, I. F. Smith, C. Peers and H. A. Pearson, “The Production of Amyloid Beta Peptide is a Critical Requirement for the Viability of Central Neurons,” Journal of Neuroscience, Vol. 23, No. 13, 2003, pp. 5531- 5535.
- F. Kamenetz, T. Tomita, H. Hsieh, G. Seabrook, D. Borchelt, T. Iwatsubo, S. Sisodia and R. Malinow, “APP Processing and Synaptic Function,” Neuron, Vol. 37, No. 6, 2003, pp. 925-937. doi:10.1016/S0896-6273(03)00124-7
- Z. Khalil, H. Chen and R. D. Helme, “Mechanisms Underlying the Vascular Activity of Beta-Amyloid Protein Fragment (beta A(4)25-35) at the Level of Skin Microvasculature,” Brain Research, Vol. 736, No. 1-2, 1996, pp. 206-216. doi:10.1016/0006-8993(96)00685-3
- N. S. Cheung, D. H. Small and B. G. Livett, “An Amyloid Peptide, Beta A4 25-35, Mimics the Function of Substance P on Modulation of Nicotine-Evoked Secretion and Desensitization in Cultured Bovine Adrenal Chromaffin Cells,” Journal of Neurochemistry, Vol. 60, No. 3, 1993, pp. 1163-1166. doi:10.1111/j.1471-4159.1993.tb03270.x
- J. F. Kelly, K. Furukawa, S. W. Barger, M. R. Rengen, R. J. Mark, E. M. Blanc, G. S. Roth and M. P. Mattson, “Amyloid Beta-peptide Disrupts Carbachol-Induced Muscarinic Cholinergic Signal Transduction in Cortical Neurons,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 93, No. 13, 1996, pp. 6753- 6758. doi:10.1073/pnas.93.13.6753
- M. P. Mattson, B. Cheng, D. Davis, K. Bryant, I. Lieberburg and R. E. Rydel, “B-Amyloid Peptides Destabilize Calcium Homeostasis and Render Human Cortical Neurons Vulnerable to Excitotoxicity,” Journal of Neuroscience, Vol. 12, No. 2, 1992, pp. 376-389.
- M. P. Mattson, B. Cheng, A. R. Culwell, F. S. Esch, I. Lieberburg and R. E. Rydel, “Evidence for Excitoprotective and Intraneuronal Calcium-Regulating Roles for Secreted Forms of the B-amyloid Precursor Protein,” Neuron, Vol. 10, 1993, pp. 243-254. doi:10.1016/0896-6273(93)90315-I
- B. Stix and G. Reiser, “Beta-Amyloid Peptide 25-35 Regulates Basal and Hormone-Stimulated Ca2+ Levels in Cultured Rat Astrocytes,” Neuroscience Letters, Vol. 243, No. 1-3, 1998, pp. 121-124. doi:10.1016/S0304-3940(98)00106-2
- V. Meske, U. Hamker, F. Albert and T. G. Ohm, “The Effects of Beta/A4-Amyloid and Its Fragments on Calcium Homeostasis, Glial Fibrillary Acidic Protein and S100 beta Staining, Morphology and Survival of Cultured Hippocampal Astrocytes,” Neuroscience, Vol. 85, No. 4, 1998, pp. 1151-1160. doi:10.1016/S0306-4522(98)00008-6
- Y. Goodman and M. P. Mattson, “Ceramide Protects Hippocampal Neurons against Excitotoxic and Oxidative Insults, and Amyloid Beta-Peptide Toxicity,” Journal of Neurochemistry, Vol. 66, No. 2, 1996, pp. 869-872. doi:10.1046/j.1471-4159.1996.66020869.x
- V. Silei, C. Fabrizi, G. Venturini, M. Salmona, O. Bugiani, F. Tagliavini and G. M. Lauro, “Activation of Microglial Cells by PrP and Beta-Amyloid Fragments Raises Intracellular Calcium through L-Type Voltage Sensitive Calcium Channels,” Brain Research, Vol. 818, 1999, pp. 168-170. doi:10.1016/S0006-8993(98)01272-4
- S. Haga, K. Ikeda, M. Sato and T. Ishii, “Synthetic Alzheimer Amyloid Beta/A4 Peptides Enhance Production of Complement C3 Component by Cultured Microglial Cells,” Brain Research, Vol. 601, 1993, pp. 88-94. doi:10.1016/0006-8993(93)91698-R
- C. Behl, J. B. Davis, R. Lesley and D. Schubert, “Hydrogen Peroxide Mediates Amyloid B Protein Toxicity,” Cell, Vol. 77, No. 6, 1994, pp. 817-827. doi:10.1016/0092-8674(94)90131-7
- M. E. Harris, K. Hensley, D. A. Butterfield, R. A. Leedle and J. M. Carney, “Direct Evidence of Oxidative Injury Produced by the Alzheimer’s β-Amyloid Peptide (1-40) in Cultured Hippocampal Neurons,” Experimental Neurology, Vol. 131, No. 2, 1995, pp. 193-202. doi:10.1016/0014-4886(95)90041-1
- D. Schubert, C. Behl, A. Brack, R. Dargusch, Y. Sagara and H. Kimura, “Amyloid Peptides are Toxic via a Common Oxidative Mechanism,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 92, No. 6, 1995, pp. 1989-1993. doi:10.1073/pnas.92.6.1989
- C. Behl, J. B. Davis, F. G. Klier and D. Schubert, “Amyloid B Peptide Induces Necrosis Rather Than Apoptosis,” Brain Research, Vol. 645, 1994, pp. 253- 264. doi:10.1016/0006-8993(94)91659-4
- Y. P. Li, A. F. Bushnell, C. M. Lee, L. S. Perlmutter and S. K. Wong, “Beta-Amyloid Induces Apoptosis in Human-Derived Neurotypic SH-SY5Y Cells,” Brain Research, Vol. 738, No. 2, 1996, pp. 196-204. doi:10.1016/S0006-8993(96)00733-0
- W. Pike and C. W. Cotman, “Calretinin-Immunoreactive Neurons are Resistant to β-Amyloid Toxicity in Vitro,” Brain Research, Vol. 671, No. 2, 1995, pp. 293-298. doi:10.1016/0006-8993(94)01354-K
- M. P. Lambert, G. Stevens, S. Sabo, K. Barber, G. Wang, W. Wade, G. Krafft, S. Snyder, T. F. Holzman and W. L. Klein, “Beta/A4-Evoked Degeneration of Differentiated SH-SY5Y Human Neuroblastoma Cells,” Journal Neuroscience Research, Vol. 39, No. 4, 1994, pp. 377-385. doi:10.1002/jnr.490390404
- A. Takashima, K. Noguchi, Satok, T. Hoshino and K. Imahori, “Tau Protein Kinase I Is Essential for Amyloid β-Protein-Induced Neurotoxicity,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 90, No. 16, 1993, pp. 7789-7793. doi:10.1073/pnas.90.16.7789
- G. E. Maestre, B. A. Tate, R. E. Majocha and C. A. Marotta, “Membrane Surface Ruffling in Cells That Over-express Alzheimer Amyloid β/A4 C-Terminal Peptide,” Brain Research, Vol. 62, 1993, pp. 145-149. doi:10.1016/0006-8993(93)90311-A
- D. T. Loo, A. Copani, C. J. Pike, E. R. Whittemore, A. J. Walencewicz and C. W. Cotman, “Apoptosis Is Induced by B-amyloid in Cultured Central Nervous System Neurons,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 90, No. 17, 1993, pp. 7951-7955. doi:10.1073/pnas.90.17.7951
- Y. Goodman and M. P. Mattson, “K+ Channel Openers Protect Hippocampal Neurons Against Oxidative Injury and Amyloid Beta-Peptide Toxicity,” Brain Research, Vol. 706, No. 2, 1996, pp. 328-332. doi:10.1016/0006-8993(95)01367-9
- I. N. Singh, G. Sorrentino and J. N. Kanfer, “Activation of LA-N-2 Cell Phospholipase D by Amyloid Beta Protein (25 - 35),” Neurochemical Research, Vol. 23, No. 10, 1998, pp. 1225-1232. doi:10.1023/A:1020731813973
- Y. Goodman, A. J. Bruce, B. Cheng and M. P. Mattson, “Estrogens Attenuate and Corticosterone Exacerbates Excitotoxicity, Oxidative Injury, and Amyloid Beta-Peptide Toxicity in Hippocampal Neurons,” Journal of Neurochemistry, Vol. 66, 1996, pp. 1836-1844. doi:10.1046/j.1471-4159.1996.66051836.x
- H. Xu, G. K. Gouras, J. P. Greenfield, B. Vincent, J. Naslund, L. Mazzarelli, G. Fried, J. N. Jovanovic, M. Seeger, N. R. Relkin, F. Liao, F. Checler, J. D. Buxbaum, B. T. Chait, G. Thinakaran, S. S. Sisodia, R. Wang, P. Greengard and S. Gandy, “Estrogen Reduces Neuronal Generation of Alzheimer Beta-Amyloid Peptides,” Nature Medicine, Vol. 4, No. 4, 1998, pp. 447-451. doi:10.1038/nm0498-447
- D. M. Araujo and C. W. Cotman, “Beta-Amyloid Stimulates Glial Cells in Vitro to Produce Growth Factors that Accumulate in Senile Plaques in Alzheimer’s Disease,” Brain Research, Vol. 569, 1992, pp. 141-145. doi:10.1016/0006-8993(92)90380-R
- C. J. Pike, B. J. Cummings, R. Monzani and C. W. Cotman, “β-Amyloid Induced Changes in Cultured Astrocytes Parallel Reactive Astrocytosis Associated With Senile Plaques in Alzheimer’s Disease,” Neuroscience, Vol. 63, No. 2, 1994, pp. 517-531. doi:10.1016/0306-4522(94)90547-9
- L. Meda, M. A. Cassatella, G. I. Szendrei, L. Otvos Jr, P. Baron, M. Villalba, D. Ferrari and F. Rossi, “Activation of Microglial Cells by B-Amyloid Protein and Interferon- γ,” Nature, Vol. 374, 1995, pp. 647-650. doi:10.1038/374647a0
- A. Klegeris, D. G. Walker and P. L. McGeer, “Activation of Macrophages by Alzheimer B Amyloid Peptide,” Biochemical and Biophysical Research Communications, Vol. 199, 1994, pp. 984-991. doi:10.1006/bbrc.1994.1326
- N. Arispe, E. Rojas and H. B. Pollard, “Alzheimer Disease Amyloid B Protein Forms Calcium Channels in Bilayer Membranes: Blockade by Tromethamine and Aluminium,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 90, No. 2, 1993, pp. 567- 571. doi:10.1073/pnas.90.2.567
- R. M. Davidson, L. Shajenko and T. S. Donta, “Amyloid Beta-peptide (ABP) Potentiates a Nimodipine-Sensitive L-Type Barium Conductance in NIE-115 Neuroblastoma Cells,” Brain Research, Vol. 643, 1994, pp. 324-327. doi:10.1016/0006-8993(94)90041-8
- P. Kuner, R. Schubenel and C. Hertel, “Beta-Amyloid Binds to p57NTR and Activates NF KappaB in Human Neuroblastoma Cells,” Journal of Neuroscience Research, Vol. 54, 1998, pp. 798-804. doi:10.1002/(SICI)1097-4547(19981215)54:6<798::AID-JNR7>3.0.CO;2-T
- T. Dyrks, E. Dyrks, U. Monning, B. Urmoneit, J. Turner and K. Beyreuther, “Generation of Beta A4 from the Amyloid Precursor and Fragments Thereof,” FEBS Letters, Vol. 335, 1993, pp. 89-93. doi:10.1016/0014-5793(93)80446-2
- J. Ma, A. Yee, H. B. Brewer, S. Das and H. Potter, “Amyloid-Associated Proteins α, -Antichymotrypsin and Apolipoprotein ε Promote Assembly of Alzheimer BProtein into Filaments,” Nature, Vol. 372, 1994, pp. 92- 94. doi:10.1038/372092a0
- E. H. Corder, A. M. Saunders, W. J. Strittmatter, D. E. Schmechel, P. C. Gaskell, G. W. Small, A. D. Roses, J. L. Haines and M. A. Pericak-Vance, “Gene Dose of Apolipoprotein E Type 4 Allele and the Risk of Alzheimer’s Disease in Late Onset Families,” Science, Vol. 261, No. 5123, 1993, pp. 921-923. doi:10.1126/science.8346443
- W. J. Strittmatter, A. M. Saunders, D. Schmechel, M. Pericak-Vance, J. Enghild, G. S. Salvesen and A. D. Roses, “Apolipoprotein E4 High Avidity Binding to β-amyloid and Increased Frequency of Type 4 Allele in Late-Onset Familial Alzheimer’s Disease,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 90, 1993, pp. 1977-1981.
- H. Hartmann, A. Eckert and W. E. Muller, “Apolipoprotein E and Cholesterol Affect Neuronal Calcium Signalling: The Possible Relationship to Beta-Amyloid Neurotoxicity,” Biochemical and Biophysical Research Communications, Vol. 200, No. 3, 1994, pp. 1185- 1192. doi:10.1006/bbrc.1994.1576
- Y. H. Chong and Y. H. Suh, “Aggregation of Amyloid Precursor Proteins by Aluminium in Vitro,” Brain Research, Vol. 670, 1995, pp. 137-141. doi:10.1016/0006-8993(94)01304-Z
- G. D. Fasman, A. Perczel and C. D. Moore, “Solubilization of β-Amyloid-(1-42)-Peptide: Reversing the B-shed Conformation Induced by Aluminium with Silicates,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 92, 1995, pp. 369- 371. doi:10.1073/pnas.92.2.369
- T. Dyrks, E. Dyrks, T. Hartmann, C. Masters and K. Beyreuther, “Amyloidogenicity of Beta A4 and Beta A4- bearing Amyloid Protein Precursor Fragments By Metalcatalyzed Oxidation,” Journal of Biological Chemistry, Vol. 267, No. 25, 1992, pp. 18210-18217.
- P. W. Mantyh, J. R. Ghilardi, S. Rogers, E. DeMaster, C. J. Allen, E. R. Stimson and J. E. Maggio, “Aluminium, Iron and Zinc Ions Promote Aggregation of Physiological Concentrations of Beta-Amyloid Peptide,” Journal of Neurochemistry, Vol. 61, No. 3, 1993, pp. 1171-1174. doi:10.1111/j.1471-4159.1993.tb03639.x
- H. LeVine Ш, “Thioflavine T Interaction with Synthetic Alzheimer’s Disease Beta-Amyloid Peptides: Detection of Amyloid Aggregation in Solution,” Protein Science, Vol. 2, 1993, pp. 404-410.
- H. LeVine Ⅲ, “Stopped-Flow Kinetics Reveal Multiple Phases of Thioflavin T Binding to Alzheimer Beta (1 - 40) Amyloid Fibrils,” Archives of Biochemistry and Biophysics, Vol. 342, No. 2, 1997, pp. 306-316. doi:10.1006/abbi.1997.0137
- A. Lorenzo and B. A. Yankner, “β-Amyloid Neurotoxicity Requires Fibril Formation and Is Inhibited by Congo Red,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 91, No. 2, 1994, pp. 12243-12247. doi:10.1073/pnas.91.25.12243
- P. H. Jensen, E. S. Sorensen, T. E. Petersen, J. Gliemann and L. K. Rasmussen, “Residues in the Synuclein Consensus Motif of the Alpha-Synuclein Fragment, NAC, Participate in Translgutaminase-catalysed Cross-Linking to Alzheimer-Disease Amyloid Beta A4 Peptide,” Biochemical Journal, Vol. 310, 1995, pp. 91-94.
- S. R. Paik, J. H. Lee, D. H. Kim, C. S. Chang and Y. S. Kim, “Self-Oligomerization of NACP, the Precursor Protein of the Non-Amyloid Beta/A4 Protein (A Beta) Component of Alzheimer’s Disease Amyloid, Observed in the Presence of a C-Terminal A Beta Fragment (Residues 25 - 35),” FEBS Letters, Vol. 42, 1998, pp. 73-76. doi:10.1016/S0014-5793(97)01537-8
- M. Mullan, F. Crawford, K. Axelman, H. Houlden, L. Lilius, B. Winblad and L. Lannfelt, “A Pathogenic Mutation for Probable Alzheimer’s Disease in the APP Gene at the N-Terminus of β-Amyloid,” Nature Genetics, Vol. 1, 1992, pp. 345-347. doi:10.1038/ng0892-345
- Q. Guo, K. Furukawa, B. L. Sopher, D. G. Pham, J. Xie, N. Robinson, G. M. Martin and M. P. Mattson, “Alzheimer’s PS-1 Mutation Perturbs Calcium Homeo-Stasis and Sensitizes PC12 Cells to Death Induced by Amyloid Beta-Peptide,” NeuroReport, Vol. 8, 1996, pp. 379-383. doi:10.1097/00001756-199612200-00074
- M. Bigl and K. Eschrich, “Interaction of Rat Brain Phosphofructokinase with Alzheimer’s Beta A4 Amyloid,” Neurochemistry International, Vol. 26, No. 1, 1995, pp. 69-75. doi:10.1016/0197-0186(94)00100-9
- J. I. Nagy, M. Z. Hossain, E. L. Hertzberg and C. A. Marotta, “Induction of Connexin43 and Gap Junctional Communication in PC12 Cells Overexpressing the Carboxy Terminal Region of Amyloid Precursor Protein,” Journal of Neuroscience Research, Vol. 44, No. 2, 1996, pp. 124-132. doi:10.1002/(SICI)1097-4547(19960415)44:2<124::AID-JNR4>3.0.CO;2-F
- S. D. Yan, X. Chen, J. Fu, M. Chen, H. Zhu, A. Roher, T. Slattery, L. Zhao, M. Nagashima, J. Morser, A. Migheli, P. Nawroth, D. Stern and A. M. Schmidt, “Rage and Amyloid-β Peptide Neurotoxicity in Alzheimer’s Disease,” Nature, Vol. 382, No. 6593, 1996, pp. 685-691. doi:10.1038/382685a0
- S. D. Yan, J. Fu, C. Soto, X. Chen, H. Zhu, F. AlMohanna, K. Collison, A. Zhu, E. Stern, T. Saido, M. Tohyama, S. Ogawa, A. Roher and D. Stern, “An Intracellular Protein that Binds Amyloid-β Peptide and Mediates Neurotoxicity in Alzheimer’s Disease,” Nature, Vol. 389, No. 6652, 1997, pp. 689-695. doi:10.1038/39522
- S. J. Soscia, J. E. Kirby, K. J. Washicosky, S. M. Tucker, M. Ingelsson, B. Hayman, M. A. Burton, L. E. Goldstein, S. Duong, R. E. Tanzi and R. D. Moir, “The Alzheimer’s Disease-Associated Amyloid β-Protein Is an Antimicrobial Peptide,” PloS One, Vol. 5, No. 3, 2010, p. e9505. doi:10.1371/journal.pone.0009505
- F. Kamenetz, T. Tomita, H. Hsieh, G. Seabrook, D. Borchelt, T. Iwatsubo, S. Sisodia and R. Malinow, “APP Processing and Synaptic Function,” Neuron, Vol. 37, No. 6, 2003, pp. 925-937. doi:10.1016/S0896-6273(03)00124-7
- S. Lesne, C. Ali, C. Garbiel, N. Croci, E. T. MacKenzie, C. G. Glabe, et al., “NMDA Receptor Activation Inhibits Alpha-Secretase and Promotes Neuronal Amyloid-beta Production,” Journal of Neuroscience, Vol. 25, No. 41, 2005, pp. 9367-9377. doi:10.1523/JNEUROSCI.0849-05.2005
- J. P. Steinbach, U. Muller, M. Leist, Z. W. Li, P. Nicotera and A. Aguzzi, “Hypersensitivity to Seizures in BetaAmyloid Precursor Protein Deficient Mice,” Cell Death and Differentiation, Vol. 5, No. 10, 1998, pp. 858-866. doi:10.1038/sj.cdd.4400391
- H. Y. Wang, D. H. S. Lee, M. R. D’Andrea, P. A. Peterson, R. P. Shank and A. B. Reitz, “β-amyloid1-42 Binds to α7 Nicotinic Acetylcholine Receptor with High Affinity. Implications for Alzheimer’s Disease Pathology,” Journal of Biological Chemistry, Vol. 275, 2000, pp. 5626-5632. doi:10.1074/jbc.275.8.5626
- H. Y. Wang, D. H. S. Lee, C. B. Davis and P. Shank, “Aβ1-42 Binds Selectively and with Picomolar Affinity to a7 Nicotinic Acetylcholine Receptors,” Journal of Neurochemistry, Vol. 75, 2000, pp. 1155-1161. doi:10.1046/j.1471-4159.2000.0751155.x
- J. M. McDonald, G. M. Savva, C. Brayne, A. T. Welzell, G. Forster, G. M. Shankar, D. J. Selkoe, P. G. Ince and D. M. Walsh, “The Presence of Sodium Dodecyll Sulphatestable A Dimmers Is Strongly Associated with Alzheimer-Type Dementia,” Brain, Vol. 133, 2010, pp. 1328- 1341. doi:10.1093/brain/awq065
- M. Townsend, G. M. Shankar, T. Mehta, D. M. Walsh and D. J. Selkoe, “Effects of Secreted Oligomers of Amyloid β-Protein on Hippocompal Synaptic Plasticity: A Potent Role for Trimers,” Journal of Physiology, Vol. 572, No. 2, 2005, pp. 4767-492.
- D. M. Walsh, I. Klyubin, J. V. Fadeeva, W. K. Cullen, R. Anwyl, M. S. Wolfe, M. J. Rowan and D. J. Selkoe, “Naturally Secreted Oligomers of Amyloid β Protein Potently Inhibit Hippocampal Long-Term Potentiation in Vivo,” Nature, Vol. 416, No. 6880, 2002, pp. 535-539. doi:10.1038/416535a
- D. J. Selkoe, “Soluble Oligomers of the Amyloid BetaProtein Impair Synaptic Plasticity and Behaviour,” Behavioural Brain Research, Vol. 28, 2008, pp. 106-113. doi:10.1016/j.bbr.2008.02.016
- H. Hsieh, J. Boehm, C. Sato, T. Iwatsubo, T. Tomita, S. Sisodia, et al., “AMPAR Removal Underlies Abeta-Induced Synaptic Depression and Dendritic Spine Loss,” Neuron, Vol. 52, No. 5, 2006, pp. 831-843. doi:10.1016/j.neuron.2006.10.035
- G. M. Shanker, B. L. Bloodgood, M. Townsend, et al., “Natural Oligomers of the Alzheimer Amyloid-beta Protein Induce Reversible Synapse Loss of Modulating an NMDA-Type Glutamate Receptor-Dependent Signaling Pathway,” Journal of Neuroscience, Vol. 27, 2007, pp. 2866-2875. doi:10.1523/JNEUROSCI.4970-06.2007
- T. Tomiyama, S. Matsuytama, H. Iso, T. Umeda, H. Takuma, K. Ohnishi, K. Ishibashi, R. Teraoka, N. Sakama, T. Yamashita, K. Nishitsuji, K. Ito, H. Shimada, M. P. Lambert, W. L. Klein and H. Mori, “A Mouse Model of Amyloid β Oligomers: Their Contribution to Synaptic Alteration, Abnormal Tau Phosphorylation, Glial Activation and Neuronal Loss in Vivo,” Journal of Neuroscience, Vol. 30, No. 14, 2010, pp. 4845-4856. doi:10.1523/JNEUROSCI.5825-09.2010
- S. A. Funker, T. van Groen, I. Kadish, D. Bartnik, L. Nagel-Steger, O. Brener, T. Sehl, B. Batra-Saffering, C. Moriscot, G. Schoehn, A. H. C. Horn, A. MallerSchiffmann, C. Korth, H. Sticht and D. Willbold, “Oral Treatment with the D-Enantiomeric Peptide D3 Improves the Pathology and Behaviour of Alzheimer’s Disease Transgenic Mice,” ACS Chemical Neuroscience, Vol. 1, 2010, pp. 639-648.
- S. Salloway, R. Sperling, S. Gilman, et al. “A Phase 2 Multiple Ascending Dose Trial of Bapineuzumab in Mild to Moderate Alzheimer Disease,” Neurology, Vol. 73, No. 24, 2009, pp. 2061-2070. doi:10.1212/WNL.0b013e3181c67808
- J. J. O. Rinne, D. J. Brooks, M. N. Rossor, N. C. Fox, R. Bullock, W. E. Klunk, C. A. Mathis, K. Blennow, J. Barakos, A. A. Okello, S. R. M. de Llano, E. Liu, M. Koller, K. M. Gregg, D. Schenk, R. Black and M. Grundman, “11C-PiB PET Assessment of Change in Fibrillar Amyloid-b Load in Patients with Alzheimer’s Disease Treated with Bapineuzumab: A Phase 2, Doubleblind, Placebo-Controlled, Ascending-Dose Study,” Lancet Neurology, Vol. 9, 2010, pp. 363-372.
- J. Cleary, J. M. Hittner, M. Semotuk, P. Mantyh and E. O’Hare, “Beta-Amyloid (1 - 40) Effects on Behaviour and Memory,” Brain Research, Vol. 682, 1995, pp. 69-74. doi:10.1016/0006-8993(95)00323-I
- D. L. Sparks, “Intraneuronal Beta-Amyloid Immunoreactivity in the CNS,” Neurobiology of Aging, Vol. 17, No. 2, 1996, pp. 291-299. doi:10.1016/0197-4580(95)02067-5
- W. A. Sweeney, J. Luedtke, M. P. McDonald and J. B. Overmier, “Intrahippocampal Injections of Exogenous Beta-amyloid Induce Postdelay Errors in an Eight-Arm Radial Maze,” Neurobiology of Learning and Memory, Vol. 68, No. 1, 1997, pp. 97-101. doi:10.1006/nlme.1997.3770
- G. Jansco, F. Domoki, P. Santha, J. Varga, J. Fischer, K. Orosz, B. Penke, A. Becskei, M. Dux and L. Toth, “Bamyloid (1 - 42) Peptide Impairs Blood-Brain Barrier Function After Intracarotid Infusion in Rats,” Neuroscience Letters, Vol. 253, 1998, pp. 139-141. doi:10.1016/S0304-3940(98)00622-3
- G. C. Su, G. W. Arendash, R. N. Kalaria, K. B. Bjugstad and M. Mullan, “Intravascular Infusions of Soluble Bamyloid Compromise Blood-Brain Barrier, Activate CNS Glial Cells and Induce Peripheral Hemorrhage,” Brain Research, Vol. 818, 1999, pp. 105-117. doi:10.1016/S0006-8993(98)01143-3
- J. Mackic, M. Weiss, W. Mhao, E. Kirkman, J. Ghiso, M. Calero, J. Bading, B. Frangione and B. V. Zlokovic, “Cerebrovascular Accumulation and Increased Bloodbrain Barrier Permeability to Circulating Alzheimer’s Amyloid Beta Peptide in Aged Squirrel Monkey and Cerebral Amyloid Angiopathy,” Journal of Neurochemistry, Vol. 70, 1998, pp. 210-215. doi:10.1046/j.1471-4159.1998.70010210.x
- J. F. Flood, E. Roberts, M. A. Sherman, B. E. Kaplan and J. E. Morley, “Topography of a Binding Site for Small Amnestic Peptides Deduced from Structure-Activity Studies: Relation to Amnestic Effect of Amyloid B Protein,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 91, 1994, pp. 380-384. doi:10.1073/pnas.91.1.380
- H. Komano, M. Seeger, S. Gandy, G. T. Wang, G. A. Krafft and R. S. Fuller, “Involvement of Cell Surface Glycosyl-Phosphatidylinositol-Linked Aspartyl Proteases in Alpha-Secretase-Type Cleavage and Ectodomain Solubilization of Human Alzheimer Beta-Amyloid Precursor Protein in Yeast,” Journal of Biological Chemistry, Vol. 273, No. 48, 1998, pp. 31648-31651. doi:10.1074/jbc.273.48.31648
- R. L. Neve, A. Kammesheidt and C. F. Hohmann, “Brain Transplants of Cells Expressing the Carboxyl-Terminal Fragment of the Alzheimer Amyloid Protein Precursor Cause Neuropathology in Vivo,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 89, 1992, pp. 3448-3452.
- B. Tate, K. S. Aboody-Guterman, A. M. Morris, E. C. Walcott, R. E. Majocha and C. A. Marotta, “Disruption of Circadian Regulation by Brain Grafts that Overexpress Alzheimer Beta/A4 Amyloid,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 89, No. 15, 1992, pp. 7090-7094.
- R. E. Tanzi, J. F. Gusella, P. C. Watkins, G. A. Bruns, P. St George-Hyslop, M. L. Van Keuren, D. Patterson, S. Pagan, D. M. Kurnit and R. L. Neve, “Amyloid Beta Protein Gene: cDNA, mRNA Distribution, and Genetic Linkage Near the Alzheimer Locus,” Science, Vol. 235, No. 4791, 1987, pp. 880-884. doi:10.1126/science.2949367
- D. K. Lahiri and N. K. Robakis, “The Promoter Activity of the Gene Encoding Alzheimer B-Amyloid Precursor Protein (APP) is Regulated by Two Blocks of Upstream Sequences,” Molecular Brain Research, Vol. 9, No. 3, 1991, pp. 253-257. doi:10.1016/0169-328X(91)90009-M
- W. J. Lukiw, E. I. Rogaev, L. Wong, G. Vaula, D. R. C. McLachlan and P. St George Hyslop, “Protein-DNA Interactions in the Promoter Region of the Amyloid Precursor Protein (APP) Gene in Human Neocortex,” Molecular Brain Research, Vol. 22, No. 1-4, 1994, pp. 121-131. doi:10.1016/0169-328X(94)90039-6
- J. M. Salbaum, A. Weidemann, H. G. Lemaire, C. L. Masters and K. Beyreuther, “The Promoter of Alzheimer’s Disease Amyloid A4 Precursor Gene,” EMBO Journal, Vol. 7, No. 9, 1988, pp. 2807-2813.
- K. Beyreuther, P. Pollwein, G. Multhaup, U. Monning, G. Konig, T. Dyrks, W. Schubert and C. L. Masters, “Regulation and Expression of the Alzheimer’s β/A4 Amyloid Protein Precursor in Health, Disease, and Down’s Syndrome,” Annals of the New York Academy of Science, Vol. 695, 1993, pp. 91-102. doi:10.1111/j.1749-6632.1993.tb23035.x
- R. Adroer, C. Lopez-Acedo and R. Oliva, “Conserved Elements in the 5’ Regulatory Region of the Amyloid Precursor Protein Gene in Primates,” Neuroscience Letters, Vol. 226, No. 3, 1997, pp. 203-206. doi:10.1016/S0304-3940(97)00283-8
- S. Silhol, A. Calenda, V. Jallageas, N. Mestre-Frances, M. Bellis and N. Bons, “Beta-Amyloid Protein Precursor in Microcebus Murinus: Genotyping and Brain Localization,” Neurobiology of Disease, Vol. 3, No. 3, 1996, pp. 169-182. doi:10.1006/nbdi.1996.0017
- F. W. van Leeuwen, D. P. de Kleijn, H. H. van den Hurk, A. Neubauer, M. A. F. Sonnemans, J. A. Sluijs, S. Koycu, R. D. J. Ramdjielal, A. Salehi, G. J. M. Martens, F. G. Grosveld, J. P. H. Burbach and E. M. Hol, “Frameshift Mutants of Beta Amyloid Precursor Protein and UbiquitinB in Alzheimer’s and Down Patients,” Science, Vol. 279, No. 5348, 1998, pp. 242-247. doi:10.1126/science.279.5348.242
- H. Reznik-Wolf, J. Machado, V. Haroutunian, L. DeMarco, G. F. Walter, B. Goldman, M. Davidson, J. A. Johnston, L. Lannfelt, S. U. Dani and E. Friedman, “Somatic Mutation Analysis of the APP and Presenilin 1 and 2 Genes in Alzheimer’s Disease Brains,” Journal of Neurogenetics, Vol. 12, No. 1, 1998, pp. 55-65. doi:10.3109/01677069809108555
- Y. Arai, A. Suzuki, M. Mizuguchi and S. Takashima, “Developmental and Aging Changes in the Expression of Amyloid Precursor Protein in Down Syndrome Brains,” Brain and Development, Vol. 19, No. 4, 1997, pp. 290- 294. doi:10.1016/S0387-7604(97)00559-7
- B. Rumble, R. Retallack, C. Hilbich, G. Simms, G. Multhaup, R. Martins, A. Hockey, P. Montgomery, K. Beyreuther and C. L. Masters, “Amyloid A4 Protein and its Precursor in Down’s Syndrome and Alzheimer’s Disease,” New England Journal of Medicine, Vol. 320, No. 22, 1989, pp. 1446-1452. doi:10.1056/NEJM198906013202203
- J. K. Teller, C. Russo, L. M. DeBusk, G. Angelini, D. Zaccheo, F. Dagna-Bricarelli, P. Scartezzini, S. Bertolini, D. M. Mann, M. Tabaton and P. Gambetti, “Presence of Soluble Amyloid Beta-Peptide Precedes Amyloid Plaque Formation in Down’s Syndrome,” Nature Medicine, Vol. 2, No. 1, 1996, pp. 93-95. doi:10.1038/nm0196-93
- Q. X. Li, G. Evin, D. H. Small, G. Multhaup, K. Beyreuther and C. L. Masters, “Proteolytic Processing of Alzheimer’s Disease Beta A4 Amyloid Precursor Protein in Human Platelets,” Journal ofBiological Chemistry, Vol. 270, No. 23, 1995, pp. 14140-14147. doi:10.1074/jbc.270.23.14140
- P. Buckland, S. Tidmarsh, G. Spurlock, F. Kaiser, M. Yates, G. O’Mahony and P. McGuffin, “Amyloid Precursor Protein mRNA Levels in the Mononuclear Blood Cells of Alzheimer’s and Down’s Patients,” Molecular Brain Research, Vol. 18, No. 4, 1993, pp. 316-320. doi:10.1016/0169-328X(93)90095-7
- S. Ledoux, C. Bergeron, J. Nalbantoglu, S. Gauthier and N. R. Cashman, “Polymerase Chain Reaction Quantification of Lymphoid Amyloid Precursor Protein mRNAs in Alzheimer’s Disease and Down’s Syndrome,” Neuroscience Letters, Vol. 193, No. 2, 1995, pp. 137-139. doi:10.1016/0304-3940(95)11681-L
- F. Kametani, K. Tanaka, T. Tokuda and S. Ikeda, “Secretory Cleavage Site of Alzheimer Amyloid Precursor Protein is Heterogeneous in Down’s Syndrome Brain,” FEBS Letters, Vol. 351, No. 2, 1994, pp. 165- 167.
- S. Govoni, S. Bergamaschi, L. Gasparini, C. Quaglia, M. Racchi, E. Cattaneo, G. Binetti, A. Bianchetti, F. Giovetti, F. Battaini and M. Trabuechi, “Fibroblasts of Patients Affected by Down’s Syndrome Oversecrete Amyloid Precursor Protein and Are Hyporesponsive to Protein Kinase C Stimulation,” Neurology, Vol. 47, No. 4, 1996, pp. 1069-1075.
- K. Urakami, J. Kataoka, A. Okada, K. Isoe, Y. Wakutani, Y. Ji, Y. Adachi, K. Ohno and K. Takahashi, “Analysis of Amyloid Precursor Protein mRNAs in Skin Fibroblasts in Down’s Syndrome,” Dementia, Vol. 7, No. 2, 1996, pp. 82-85.
- C. Pallister, S. S. Jung, I. Shaw, J. Nalbantoglu, S. Gauthier and N. R. Cashman, “Lymphocyte Content of Amyloid Precursor Protein is Increased in Down’s Syndrome and Aging,” Neurobiology of Aging, Vol. 18, No. 1, 1997, pp. 97- 103. doi:10.1016/S0197-4580(96)00207-2
- A. Goate, M. C. Chartier-Harlin, M. Mullan, J. Brown, F. Crawford, L. Fidani, L. Giuffra, A. Haynes, N. Irving and L. James, “Segregation of a Missense Mutation in the Amyloid Precursor Protein Gene with Familial Alzheimer’s Disease,” Nature, Vol. 349, No. 6311, 1991, pp. 704-706. doi:10.1038/349704a0
- M. C. Chartier-Harlin, J. Hardy and M. Mullan, “Early Onset Alzheimer’s Disease Caused by Mutations at Codon 717 of the β-amyloid Precursor Protein Gene,” Nature, Vol. 353, 1991, pp. 844-846. doi:10.1038/353844a0
- X. D. Cai, T. E. Golde and S. G. Younkin, “Excess Amyloid Beta-Protein is Released from a Mutant Amyloid Beta Protein Precursor Linked to Familial Alzheimer’s Disease,” Science, Vol. 259, 1993, pp. 514-516. doi:10.1126/science.8424174
- M. Citron, T. Olterdorf, C. Haass, L. McConlogue, A. Y. Hung, P. Seubert, C. Vigo-Pelfrey, I. Lieberburg and D. J. Selkoe, “Mutation of the Beta-Amyloid Precursor Protein in Familial Alzheimer’s Disease Causes Increased Beta Amyloid Production,” Nature, Vol. 360, 1992, pp. 672- 674. doi:10.1038/360672a0
- L. Hendricks, C. M. van Duijn, P. Cras, M. Cruts, W. Van Hul, F. van Harskamp, A. Warren, M. G. McInnis, S. E. Antonarakis and J. J. Martin, “Presenile Dementia and Cerebral Haemorrhage Linked to a Mutation at Codon 692 of the β-amyloid Precursor Protein Gene,” Nature Genetics, Vol. 1, 1992, pp. 218-221.
- C. Van Broeckhaven, J. Haan, E. Bakker, J. A. Hardy, W. Van Hul, A. Wehnert, M. Vegter-Van der Vlis and R. A. Roos, “Amyloid β Protein Precursor Gene and Hereditary Cerebral Haemorrhage with Amyloidosis (Dutch),” Science, Vol. 248, No. 4959, 1990, pp. 1120-1122. doi:10.1126/science.1971458
- E. Levy, M. D. Cumon, I. J. Fernandez-Madrid, M. D. Power, I. Lieberburg, S. G. van Duinen, G. T. Bots, W. Luyendijk and B. Frangione, “Mutation of the Alzheimer’s Disease Amyloid Gene in Hereditary Cerebral Haemorrhage, Dutch Type,” Science, Vol. 248, 1990, pp. 1124-1126. doi:10.1126/science.2111584
- P. K. Panegyres, J. Kwok, P. Schofield and P. C. Blumbergs, “A Western Australian Kindred with Dutch Cerebral Amyloid Angiopathy,” Journal of Neurological Sciences, Vol. 239, No. 1, 2005, pp. 75-80. doi:10.1016/j.jns.2005.08.002
- M. Simons, P. J. Tienari, C. G. Dotti and K. Beyreuther, “Two-Dimensional Gel Mapping of the Processing of the Human Amyloid Precursor Protein in Rat Hippocampal Neurons,” FEBS Letters, Vol. 368, No. 2, 1995, pp. 363- 366. doi:10.1016/0014-5793(95)00654-R
- A. Marutle, U. Warpman, N. Bogdanovic, L. Lannfelt and A. Nordberg, “Neuronal Nicotinic Receptor Deficits in Alzheimer Patients with the Swedish Amyloid Precursor Protein 670/671 Mutation,” Journal of Neurochemistry, Vol. 72, No. 3, 1999, pp. 1161-1169. doi:10.1046/j.1471-4159.2000.0721161.x
- E. Levy-Lahad, E. M. Wijsman, E. Nemens, I. Anderson, K. A. B. Goddard, J. L. Weber, T. D. Bird and G. D. Schellenberg, “A Familial Alzheimer’s Disease Locus on Chromosome 1,” Science, Vol. 269, No. 5226, 1995, pp. 970-973. doi:10.1126/science.7638621
- R. Sherrington, E. I. Rogaev, Y. Liang, E. A. Rogaeva, G. Levesque, M. Ikeda, H. Chi, C. Lin, G. Li and K. Holman, “Cloning of a Gene Bearing Missense Mutations in Early Onset Familial Alzheimer’s Disease,” Nature, Vol. 375, 1995, pp. 754-760. doi:10.1038/375754a0
- D. Blacker and R. E. Tanzi, “The Genetics of Alzheimer Disease,” Archives of Neurology, Vol. 55, 1998, pp. 294- 296. doi:10.1001/archneur.55.3.294
- H. W. Querfurth, E. M. Wijsman, P. H. St George-Hislop and D. J. Selkoe, “βAPP mRNA Transcription is Increased in Cultured Fibroblasts from the Familial Alzheimer’s Disease-1 Family,” Molecular Brain Research, Vol. 28, 1995, pp. 319-337. doi:10.1016/0169-328X(94)00224-3
- D. R. Borchelt, G. Thinakaran, C. B. Eckman, M. K. Lee, F. Davenport, T. Ratovitsky, C. M. Prada, G. Kim, S. Seekins, D. Yager, H. H. Slunt, R. Wang, M. Seeger, A. I. Levey, S. E. Gandy, N. G. Copeland, N. A. Jenkins, D. L. Price, S. G. Younkin and S. S. Sisodia, “Familial Alzheimer’s Disease-linked Presenilin 1 Variants Elevate Abeta1-42/1-40 Ratio in Vitro and in Vivo,” Neuron, Vol. 17, No. 5, 1996, pp. 1005-1013. doi:10.1016/S0896-6273(00)80230-5
- N. D. Mehta, L. M. Refolo, C. Eckman, S. Sanders, D. Yager, J. Perez-Tur, S. Younkin, K. Duff, J. Hardy and M. Hutton, “Increased Abeta42(43) from Cell Lines Expressing Presenilin 1 Mutations,” Annals of Neurology, Vol. 43, No. 2, 1998, pp. 256-258. doi:10.1002/ana.410430217
- M. Hutton and J. Hardy, “The Presenilins and Alzheimer’s Disease,” Human Molecular Genetics, Vol. 6, No. 10, 1997, pp. 1639-1646. doi:10.1093/hmg/6.10.1639
- B. De Strooper, P. Saftig, K. Craessaerts, H. Vanderstichele, G. Guhde, W. Annaert, K. Von Figura and F. Van Leuven, “Deficiency of Presenilin-1 Inhibits the Normal Cleavage of Amyloid Precursor Protein,” Nature, Vol. 391, No. 6665, 1998, pp. 387-390. doi:10.1038/34910
- C. De Jonghe, C. Zehr, D. Yager, C. M. Prada, S. Younkin, L. Hendriks, C. Van Broeckhoven and C. B. Eckman, “Flemish and Dutch Mutations in Amyloid Beta Precursor Protein Have Different Effects on Amyloid Beta Secretion,” Neurobiology of Disease, Vol. 5, No. 4, 1998, pp. 281-286. doi:10.1006/nbdi.1998.0202
- D. M. Mann, T. Iwatsubo, N. J. Cairns, P. L. Lantos, D. Nochlin, S. M. Sumi, T. D. Bird, P. Poorkaj, J. Hardy, M. Hutton, G. Prihar, R. Crook, M. N. Rosso and M. Haltia, “Amyloid Beta Protein (Abeta) Deposition in Chromosome 14-linked Alzheimer’s Disease: Predominance of Abeta42(43),” Annals of Neurology, Vol. 40, 1996, pp. 149-156. doi:10.1002/ana.410400205
- D. M. Mann, T. Iwatsubo, Y. Ihara, N. J. Cairns, P. L. Lantos, N. Bogdanovic, L. Lannfelt, B. Winblad, M. L. Maat-Schieman and M. N. Rossor, “Predominant Deposition of Amyloid-Beta 42(43) in Plaques in Cases of Alzheimer’s Disease and Hereditary Cerebral Hemorrhage Associated with Mutations in the Amyloid Precursor Protein Gene,” American Journal of Pathology, Vol. 148, 1996, pp. 1257-1266.
- U. Giambarella, T. Yamatsuji, T. Okamoto, T. Matsui, T. Ikezu, Y. Murayama, M. A. Levine, A. Katz, N. Gautam and I. Nishimoto, “G Protein Betagamma Complexmediated Apoptosis by Familial Alzheimer’s Disease Mutant of APP,” EMBO Journal, Vol. 16, No. 16, 1997, pp. 4897-4907. doi:10.1093/emboj/16.16.4897
- R. D. Hollister, W. Kisiel and B. T. Hyman, “Immunohistochemical Localization of Tissue Factor Pathway Inhibitor-1 (TFPI-1), a Kunitz Proteinase Inhibitor, in Alzheimer’s Disease,” Brain Research, Vol. 728, 1996, pp. 13-19. doi:10.1016/0006-8993(96)00384-8
- O. Almkvist, H. Basun, S. L. Wagner, B. A. Rowe, L. O. Wahlund and L. Lannfelt, “Cerebrospinal Fluid Levels of Alpha-Secretase-Cleaved Soluble Amyloid Precursor Protein Mirror Cognition in a Swedish Family with Alzheimer Disease and a Gene Mutation,” Archives of Neurology, Vol. 54, 1997, pp. 641-644.
- H. Tanaka, M. Araki and T. Masuzawa, “Reaction of Astrocytes in the Gerbil Hippocampus Following Transient Ischemia: Immunocytochemical Observations with Antibodies against Glial Fibrillary Acidic Protein, Glutamine Synthetase, and S-100 Protein,” Experimental Neurology, Vol. 116, No. 3, 1992, pp. 264-274. doi:10.1016/0014-4886(92)90006-C
- S. Tanaka, S. Nakamura, J. Kimura and K. Ueda, “Age-Related Change in the Proportion of Amyloid Precursor Protein mRNAs in the Gray Matter of Cerebral Cortex,” Neuroscience Letters, Vol. 163, No. 1, 1993, pp. 19-21. doi:10.1016/0304-3940(93)90219-B
- S. A. Johnson, T. McNeill, B. Cordell and C. E. Finch, “Relation of Neuronal APP-751/APP-695 mRNA Ratio and Neuritic Plaque Density in Alzheimer’s Disease,” Science, Vol. 248, No. 4957, 1990, pp. 854-857. doi:10.1126/science.2111579
- S. A. Johnson, G. M. Pasinetti, P. C. May, P. A. Ponte, B. Cordell and C. E. Finch, “Selective Reduction of mRNA for the B-Amyloid Precursor Protein that Lacks a Kunitztype Protease Inhibitor Motif in Cortex of Alzheimer’s Brains,” Experimental Neurology, Vol. 102, No. 2, 1988, pp. 264-268. doi:10.1016/0014-4886(88)90104-5
- G. Konig, J. M. Salbaum, D. Wiestler, W. Lang, H. P. Schmitt, C. L. Master and K. Beyreuther, “Alternative Splicing of the β/A4 Amyloid Gene of Alzheimer’s Disease in Cortex of Control and Alzheimer’s Disease Patients,” Molecular Brain Research, Vol. 9, 1991, pp. 259-262. doi:10.1016/0169-328X(91)90010-U
- A. Matsumoto and R. Matsumoto, “Familial Alzheimer’s Disease Cells Abnormally Accumulate Beta-AmyloidHarbouring Peptides Preferentially in Cytosol but Not in Extracellular Fluid,” European Journal of Biochemistry, Vol. 225, No. 3, 1994, pp. 1055-1062. doi:10.1111/j.1432-1033.1994.1055b.x
- C. A. Robinson, A. W. Clark, I. M. Parhad, T. S. Fung and S. S. Bou, “Gene Expression in Alzheimer Neocortex as a Function of Age and Pathologic Severity,” Neurobiology of Aging, Vol. 15, No. 6, 1994, pp. 681-690. doi:10.1016/0197-4580(94)90049-3
- M. T. Webster, P. T. Francis, A. W. Procter, G. C. Stratman, R. Doshi, D. M. A. Mann and D. M. Bowan, “Postmortem Brains Reveal Similar but Not Identical Amyloid Precursor Protein-Like Immunoreactivity in Alzheimer Compared with Other Dementias,” Brain Research, Vol. 644, No. 2, 1994, pp. 347-351. doi:10.1016/0006-8993(94)91702-7
- B. T. Hyman, J. J. Wenniger and R. E. Tanzi, “Nonisotopic in Situ Hybridization of Amyloid Beta Protein Precursor in Alzheimer’s Disease: Expression in Neurofibrillary Tangle Bearing Neurons and in the Microenvironment Surrounding Senile Plaques,” Molecular Brain Research, Vol. 18, No. 3, 1993, pp. 253-258. doi:10.1016/0169-328X(93)90197-W
- R. E. Tanzi, J. J. Wenniger and B. T. Hyman, “Cellular Specificity and Regional Distribution of Amyloid β Protein Precursor Alternative Transcripts Are Unaltered in Alzheimer Hippocampal Formation,” Molecular Brain Research, Vol. 18, No. 3, 1993, pp. 246-252. doi:10.1016/0169-328X(93)90196-V
- P. J. Harrison, A. J. L. Barton, A. W. Procter, D. M. Bowen and R. C. A. Pearson, “The Effects of Alzheimer's Disease, Other Dementias, and Premortem Course on B-amyloid Precursor Protein Messenger RNA in Frontal Cortex,” Journal of Neurochemistry, Vol. 62, 1994, pp. 635-644. doi:10.1046/j.1471-4159.1994.62020635.x
- D. J. S. Sirinathsinghji, M. Rigby, R. P. Heavens, D. Smith, J. M. Fernandez, R. Schuligol and R. G. Hill, “Increased Messenger RNA Expression of the 695 and 751 Amino Acid Isoforms of the β-Amyloid Protein Precursor in the Thalamus of 17-Year-Old Cynomolgus (Macaca Fascicularis) Monkeys,” Neuroscience, Vol. 65, No. 1, 1995, pp. 51-57. doi:10.1016/0306-4522(94)00485-N
- F. M. Flood, R. F. Cowburn and J. A. Johnston, “Presenilin-1, Amyloid Precursor Protein and Amyloid Precursor-like Protein 2 mRNA Levels in Human Superior Frontal Cortex During Aging,” Neuroscience Letters, Vol. 235, No. 1-2, 1997, pp. 17-20. doi:10.1016/S0304-3940(97)00697-6
- S. S. Zhan, R. Sandbrink, K. Beyreuther and H. P. Schmitt, “APP with Kunitz Type Protease Inhibitor Domain (KPI) Correlates with Neuritic Plaque Density but Not with Cortical Synaptophysin Immunoreactivity in Alzheimer’s Disease and Non-Demented Aged Subjects: A Multifactorial Analysis,” Clinical Neuropathology, Vol. 14, No. 3, 1995, pp. 142-149.
- P. K. Panegyres, K. Zafiris-Toufexis and B. A. Kakulas, “Amyloid Precursor Protein Gene Isoforms in Alzheimer’s Disease and Other Neurodegenerative Disorders,” Journal of Neurological Sciences, Vol. 173, No. 2, 2000, pp. 81-92. doi:10.1016/S0022-510X(99)00311-1
- [342] P. Preece, D. J. Virley, M. Costandi, R. Coombes, S. J. Moss, A. W. Mudge, E. Jazin and N. J. Cairns, “Amyloid Precursor Protein mRNA Levels in Alzheimer’s Disease Brain,” Molecular Brain Research, Vol. 122, No. 1, 2004, pp. 1-9. doi:10.1016/j.molbrainres.2003.08.022
- E. Cochran, B. Bacci, Y. Chen, A. Patton, P. Gambetti and L. Antiho-Gambetti, “Amyloid Precursor Protein and Ubiquitin Immunoreactivity in Dystrophic Axons is Not Unique to Alzheimer’s Disease,” American Journal of Pathology, Vol. 139, No. 3, 1991, pp. 485-489.
- G. W. Roberts, S. M. Gentleman, A. Lynch and D. I. Graham, “BA4 Amyloid Protein Deposition in Brain after Head Trauma,” Lancet, Vol. 338, 1991, pp. 1422-1423. doi:10.1016/0140-6736(91)92724-G
- P. C. Blumbergs, G. Scott, J. Manavis, H. Wainwright, D. A. Simpson and A. J. McLean, “Staining of Amyloid Precursor Protein to Study Damage in Mild Head Injury,” Lancet, Vol. 344, 1994, pp. 1055-1056. doi:10.1016/S0140-6736(94)91712-4
- A. M. Gleckman, M. D. Bell, R. J. Evans and T. W. Smith, “Diffuse Axonal Injury in Infants with Nonaccidental Craniocerebral Trauma: Enhanced Detection by Beta-amyloid Precursor Protein Immunohistochemical Staining,” Archives of Pathology and Laboratory Medicine, Vol. 123, 1999, pp. 146-151.
- B. Koszyca, P. C. Blumbergs, J. Manavis, H. Wainwright, R. James, J. Gilbert, N. Jones and P. L. Reilly, “Widespread Axonal Injury in Gunshot Wounds to the Head Using Amyloid Precursor Protein as a Marker,” Journal of Neurotrauma, Vol. 15, No. 9 1998, pp. 675-683. doi:10.1089/neu.1998.15.675
- W. S. T. Griffin, J. G. Sheng, S. M. Gentleman, D. I. Graham, R. E. Mrak and G. W. Roberts, “Microglial Interleukin-1a Expression in Human Head Injury: Correlations with Neuronal and Neuritic B-Amyloid Precursor Protein Expression,” Neuroscience Letters, Vol. 176, 1994, pp. 133-136. doi:10.1016/0304-3940(94)90066-3
- W. S. T. Griffin, L. C. Stanley, C. Ling, L. White, V. MacLeod, L. J. Perrot, C. L. White Ш and C. Araoz, “Brain Interleukin 1 and S100 Immunoreactivity Are Elevated in Down Syndrome and Alzheimer Disease,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 86, 1989, pp. 7611-7615. doi:10.1073/pnas.86.19.7611
- J. G. Sheng, F. A. Boop, R. E. Mrak and W. S. T. Griffin, “Increased Neuronal B-Amyloid Precursor Protein Expression in Human Temporal Lobe Epilepsy: Association with Interleukin-1a Immunoreactivity,” Journal of Neurochemistry, Vol. 62, 1994, pp. 1872-1879.
- D. Goldgaber, H. W. Harris, T. Hla, T. Maciag, R. J. Donnelly, J. S. Jacobsen, M. P. Vitek and D. C. Gajdusek, “Interleukin 1 Regulates Synthesis of Amyloid B-protein Precursor mRNA in Human Endothelial Cells,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 86, No. 19, 1989, pp. 7606-7610. doi:10.1073/pnas.86.19.7606
- M. Di Luca, L. Pastorino, A. Bianchetti, J. Perez, L. A. Vignolo, G. L. Lenzi, M. Trabucchi, F. Cattabeni and A. Padovani, “Differential Level of Platelet Amyloid Beta Precursor Protein Isoforms: An Early Marker for Alzheimer Disease,” Archives of Neurology, Vol. 55, No. 9, 1998, pp. 1195-1200. doi:10.1001/archneur.55.9.1195
- A. Henry, Q. X. Li, D. Galatis, L. Hesse, G. Multhaup, K. Beyreuther, C. L. Masters and R. Cappai, “Inhibition of Platelet Activation by the Alzheimer’s Disease Amyloid Precursor Protein,” British Journal of Haematology, Vol. 103, No. 2, 1998, pp. 402-415. doi:10.1046/j.1365-2141.1998.01005.x
- M. C. Sugarman, T. R. Yamasaki, S. Oddo, J. C. Echegoyen, M. P. Murphy, T. E. Golde, M. Jannatipour, M. A. Leissring and F. M. Laferla, “Inclusion Body Myositis-Like Phenotype Induced by Transgenic Overexpression of B APP in Skeletal Muscle,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 99, 2002, pp. 6334-6339. doi:10.1073/pnas.082545599
- N. J. Scolding, F. Joseph, P. A. Kirby, I. Mazanti, F. Gray, J. Mikol, D. Ellison, D. A. Hilton, T. L. Williams, J. M. MacKenzie, J. H. Xuereb and S. Love, “Aβ-Related Angiitis: Primary Angiitis of the Central Nervous System Associated with Cerebral Amyloid Angiopathy,” Brain, Vol. 128, No. 3, 2005, pp. 500-515. doi:10.1093/brain/awh379
NOTES
*Three parts to be published in three consecutive issues