Open Journal of Medical Microbiology
Vol.3 No.1(2013), Article ID:28667,10 pages DOI:10.4236/ojmm.2013.31011
Interaction of Helicobacter pylori Cell Membrane with Non-Esterified Cholesterol and Other Steroids
Division of Bacteriology, Department of Infection and Immunity, Jichi Medical University, Tochigi, Japan
Email: shimo@jichi.ac.jp
Received December 10, 2012; revised January 19, 2013; accepted February 2, 2013
Keywords: Helicobacter pylori; Cholesterol-α-glucosyltransferase; Phosphatidylethanolamine
ABSTRACT
Helicobacter pylori performs the unique action of assimilating exogenous non-esterified cholesterol into its cell membrane. This bacterium aggressively incorporates non-esterified cholesterol into the membrane, induces its glucosylation, and uses both non-esterified cholesterol and glucosylated cholesterols as membrane lipid compositions. The reason for this assimilation of non-esterified cholesterol into the cell membrane of H. pylori has eluded investigators for many years. Recent hypotheses posit that the sterol-uptake and sterol-glucosylation contribute to the survival of H. pylori cells in different ways. The incorporation of the non-esterified cholesterol into the cell membrane fortifies the resistance of H. pylori against the antibacterial actions of phosphatidylcholines, antibiotics, and bile salts. In parallel, the glucosylation of the non-esterified cholesterol incorporated into the cell membrane serves H. pylori in two ways. First, it helps the bacterium evade host immune responses, such as phagocytosis by macrophages and activation of antigen-specific T cells. Second, it detoxifies sterols fatal to the bacterium via a novel action of sterol glucosylation recently described in another report from our group. The reluctance of H. pylori to absorb esterified cholesterol remains unexplained. A recent study by our group has demonstrated that the phosphatidylethanolamine (PE) in the outer membrane of H. pylori serves as a steroid-binding lipid the incorporation of non-esterified cholesterol into the membrane. We have also discovered that the myristic acid (C14:0) molecule attached to the PE of this bacterium plays an important role in the selective binding of non-esterified cholesterol but not esterified cholesterol.
1. Introduction
Helicobacter pylori, a Gram-negative curved rod, is a pathogen responsible for chronic gastritis and peptic ulcers in humans [1,2]. In addition, the colonization of the stomach by this pathogen for many years contributes to the development of gastric cancer and marginal zone B-cell lymphoma [3-6]. Approximately half of population in the world is infected with H. pylori, and the majority of infected persons develop atrophic gastritis with or without symptoms. Among H. pylori-infected individuals, about 10% persons develop gastric and duodenal ulcers, 1% to 3% persons develop gastric adenocarcinoma, and 0.1% or less person develops gastric mucosa-associated lymphoid tissue (MALT) lymphoma [3,4, 7-9].
While sterols are generally unnecessary for the growth or viability of bacteria, a few bacterial species assimilate exogenous non-esterified cholesterol into their cell membranes and glucosylate it once assimilated. As of this writing, five unique bacterial genera are known to exhibit this cell membrane assimilation of non-esterified cholesterol: Borrelia spp., Acholeplasma axanthum, Spiroplasma spp., Mycoplasma gallinarum, and Helicobacter spp. [10-15]. Unlike plants and fungi, which produce glucosyl sterols such as glucosyl sitosterol and glucosyl ergosterol universally, bacterial species produce glucosyl sterols only in rare and special cases [16-21]. Plants and fungi biosynthesize the sterols by themselves and attach D-glucose molecules to the sterols via the catalytic action of sterol-β-glucosyltransferase. In contrast, Helicobacter pylori and the other bacteria lack the synthetic pathway for sterols. Thus, bacterial cells can only biosynthesize glucosyl cholesterol by ingesting nonesterified cholesterol from outside the cells.
While H. pylori attaches a D-glucose molecule to a cholesterol molecule absorbed into the cell membrane via α-glucosidic linkage, Borrelia hermsii attaches its sugar molecule to a cholesterol molecule via β-glucosidic linkage [22], as with the glucosyl sterols in plants and fungi. Recent studies by other groups have demonstrated that Borrelia burgdorferi, B. garinii, and B. afzelii attach a D-galactose molecule to a cholesterol molecule via β- galactosidic linkage in place of a D-glucose molecule [22-24]. Put simply, the O-glycoside bond between sugar molecule and sterol molecule in H. pylori differs from the O-glycoside bond between the same two molecules in other organisms.
It has long been unclear why H. pylori needs to assimilate non-esterified cholesterol into its cell membrane. Recent investigations by our group and others have helped us more fully understand the purposes of this cell membrane assimilation of non-esterified cholesterol in the bacterium. This review article describes the biological significance of the cholesterol uptake and glucosylation for the survival of H. pylori, and the interaction of the H. pylori cell membrane with other steroid compounds.
2. Biosynthesis and Membrane Localization of Glucosyl Cholesterols
H. pylori like most other bacteria, is capable of growing in vitro without help from non-esterified cholesterol. Yet, when non-esterified cholesterol is added into a culture medium, H. pylori aggressively absorbs the cholesterol into the cell membrane, glucosylates the cholesterol, and uses both non-esterified cholesterol and glucosylated cholesterol as cell membrane lipid components. Lebrun et al. (2006) identified the enzyme involved in the biosynthesis of glucosyl cholesterol in H. pylori as a cholesterol-α-glucosyltransferase (CGT) encoded by hp0421 gene on chromosomal DNA. CGT uses the uridine diphosphate glucose (UDP-Glc) as a sugar source, catalyzes the dehydration reaction between the 1α-hydroxyl (OH) group in a D-glucose molecule and the 3β-OH group in a cholesterol molecule, and synthesizes cholesteryl-α-D-glucopyranoside (CGL) [25].
H. pylori is known to produce at least three types of glucosyl cholesterols, namely CGL, cholesteryl-6-O-tetradecanoyl-α-D-glucopyranoside (CAG), and cholesteryl- 6-O-phosphatidyl-α-D-glucopyranoside (CPG), but the enzymes catalyzing the acylation and phosphatidylation of the CGL molecule involved in the biosynthesis of CAG and CPG have yet to be identified [26]. A study by our group in 2009 revealed that glucosyl cholesterols are more potent lipid constituents in the outer membrane than in the inner membrane, but that the non-esterified cholesterol absorbed into the H. pylori cell is present in both the inner membrane and the outer membrane (Figure 1) [27].
3. A Role of Cholesterol Glucosylation in the Survival of H. pylori
Wunder et al. (2006) were the first to identify a role of cholesterol glucosylation in H. pylori. Specifically, they demonstrated that H. pylori cells glucosylate the nonesterified cholesterol extracted from the lipid raft of
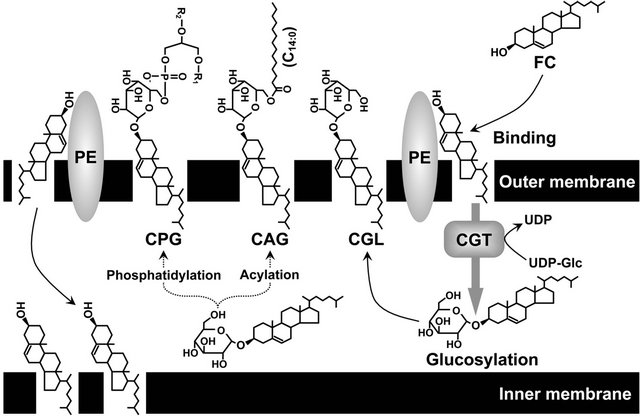
Figure 1. Localization of non-esterified cholesterol and glucosylated cholesterols in H. pylori cell membranes. FC, non-esterified cholesterol; CGL, cholesteryl-α-D-glucopyranoside; CAG, cholesteryl-6-O-tetradecanoyl-α-D-glucopyranoside; CPG, cholesteryl-6-O-phosphatidyl-α-D-glucopyranoside; PE, phosphatidylethanolamine; UDP-Glc, uridine diphosphate glucose; CGT, cholesterol-α-glucosyltransferase
gastric epithelial cells to evade the host immune responses [28]. The glucosylation of the non-esterified cholesterol absorbed into the cell membrane allows the bacterium to colonize the gastric mucosa tissues of hosts for long periods by conferring resistance against the phagocytosis of macrophages and regulating the activation of antigen-specific T cells. Yet an hp0421 genedisrupted H. pylori mutant that regained only the nonesterified cholesterol without glucosylation was strong activator of macrophages and antigen-specific T cells and was promptly banished from the gastric mucosa tissues of hosts. The same pattern was observed in an abnormal H. pylori strain bioengineered to artificially incorporate excessive amounts of non-esterified cholesterol into the cell membrane, a strain that turned out to be incapable of biosynthesizing sufficient amounts of glucosyl cholesterol. We can thus infer that non-esterified cholesterol itself poses a threat to the survival of any H. pylori cells that fail to glucosylate it. If H. pylori behaved like other general bacterial species and had a cell membrane impervious to exogenous non-esterified cholesterol, its cells would have no need to glucosylate the non-esterified cholesterol. In sum, no one has yet unraveled why the cell membrane of H. pylori actively assimilates non-esterified cholesterol.
4. Biological Significance of Cholesterol Uptake by H. pylori
Our study in 2009 revealed that H. pylori cells without non-esterified cholesterol or other cholesterol analogues succumb to the bacteriolytic action of phosphatidylcholines (PCs), while the H. pylori cells with absorbed nonesterified cholesterol or the cholesterol analogue estrone acquire resistance against the bactericidal action of PCs (Figure 2) [27]. H. pylori was incapable of glucosylating estrone absorbed into the cell membrane. Hence, its resistance against the bacteriolytic action of the PCs was acquired independently of the glucosylation of the steroid compounds. PC is the most prevalent glycerophospholipid in mammals and exists in sufficient abundance to kill H. pylori in gastric mucosa and gastric juice of humans [29,30]. In sum, our study demonstrated that H. pylori aggressively incorporates exogenous non-esterified cholesterol into the cell membrane, in order to survive in the presence of PCs.
In separate experiments, McGee et al. (2011) and Trainor et al. (2011) inactivated the CGT of an H. pylori strain by inserting a chloramphenicol-resistant (cat) gene cassette into the hp0421 gene. With this strain, they found that non-glucosylated non-esterified cholesterol absorbed into the cell membrane fortified this bacterium’s resistance to antibacterial agents such as ciprofloxacin, tetracycline, and clarithromycin, and to bile salts and ceragenins [31,32]. The studies by our group and others indicate that non-esterified cholesterol strengthens the cell membrane lipid barrier of H. pylori. Incidentally, McGee et al. have also proposed that the glucosylation of the non-esterified cholesterol absorbed by H. pylori confers further resistance against the bactericidal action of antibacterial peptides such as polymixin B and colistin, at least in part [31].
5. Response of H. pylori Cell Membrane to Other Steroid Compounds
It has been unclear whether the H. pylori cell glucosylates only the non-esterified cholesterol. Steroid hormones such as sex hormones (steroid compounds) and corticoids, typical sterol analogues in mammals, are derived from non-esterified cholesterol. Earlier investigations by other groups have shown that the enzymes
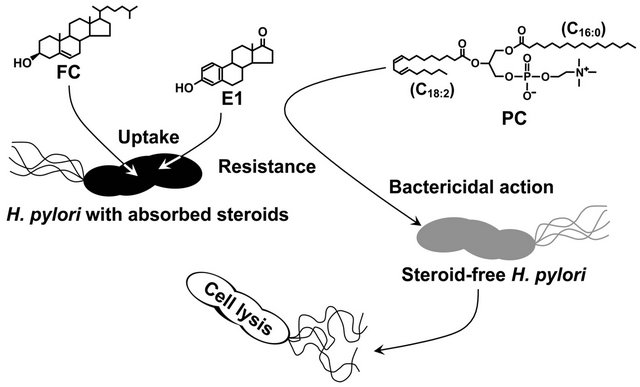
Figure 2. How H. pylori acquires resistance to the bacteriolytic action of phosphatidylcholine by incorporating steroid compounds into the cell membrane. FC, non-esterified cholesterol; E1, estrone; PC, phosphatidylcholine.
involved in the biosynthesis and activation of sex hormones are also expressed in human gastric tissues [33- 36]. Still other studies have revealed the expression of sex hormone receptors in gastric cancer [37-39]. These studies, taken together, indirectly demonstrate that sex hormones exist in the human stomach, an environment that H. pylori colonizes and inhabits for many years. Our group has looked closer at this issue by investigating how the cell membrane of H. pylori interacts with sex hormones. In 2009 we found that H. pylori glucosylates not only non-esterified cholesterol, but also various steroid compounds with a 3β-OH group, such as pregnenolone, dehydroepiandrosterone, and epiandrosterone [40]. Yet again, as mentioned earlier, estrone with a 3-OH group was incorporated into the cell membrane of H. pylori but not glucosylated once absorbed [27]. These results indicate that the 3β-OH group in a steroid molecule is a crucial conformation for glucosylation by the enzymatic action of CGT regardless of the differences of other structures among the steroid compounds.
Our group also found that some steroid compounds impair the viability of H. pylori. In particular, progesterone with an oxo group at the carbon 3-position of the steroid framework destabilized the cell membrane structure of H. pylori and ultimately destroyed the cells [41]. The cell membrane of H. pylori apparently interacted with a number of steroid compounds, some of which were even toxic to this bacterium (Figure 3). Intriguingly, a synthetic progesterone derivative acylated by a caproic acid (C6:0) at the carbon 17-position in a progesterone molecule had conspicuously stronger anti-H. pylori activity than the progesterone, whereas a natural progesterone derivative hydroxylated at the same carbon position of a progesterone molecule lacked this activity. This may open the way to the development of a new class of steroidal medicines that lack the hormonal activity but maintain the anti-H. pylori activity, using the progesterone molecule as a basic structure (Figure 4).
Progesterone non-reversibly bound to the cell membrane of H. pylori and inhibited the interaction of non-
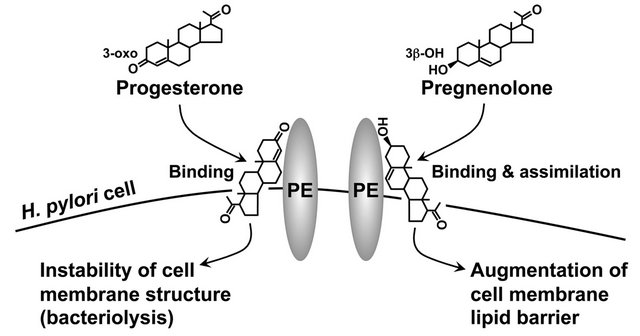
Figure 3. Cell membrane of H. pylori interacting with steroid compounds regardless of toxicity. PE, phosphatidylethanolamine.
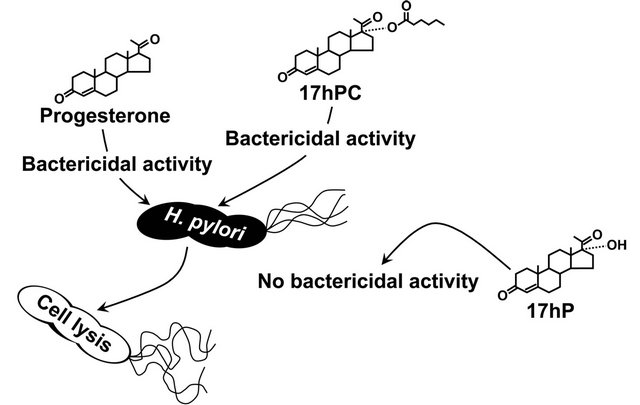
Figure 4. Anti-H. pylori activity of progesterone and its derivatives. 17hPC, 17α-hydroxyprogesterone caproate; 17hP, 17α-hydroxyprogesterone.
esterified cholesterol to the bacterial cell membrane. Conversely, non-esterified cholesterol protected the H. pylori cells from the bacteriolytic action of progesterone to some degree. This suggested that non-esterified cholesterol and progesterone bind to the identical component on the cell membrane of H. pylori and thereby obstruct each other’s effects on the bacterial cell. We were unable, however, to identify which of the cell membrane components of H. pylori took part in the binding of the steroid compounds in that series of experiments.
6. Uptake Mechanism of Non-Esterified Cholesterol by H. pylori Cell Membrane
When H. pylori was incubated in a serum-supplemented culture medium, the cell membrane of this bacterium incorporated only the non-esterified cholesterol, even though esterified cholesterol was more abundant. Year after year we were unable to explain why the cell membrane of H. pylori absorbs non-esterified cholesterol but not esterified cholesterol. In an earlier investigation we found that the components of glucosyl cholesterols hardly changed in H. pylori cells undergoing growth phase changes from the logarithmic phase to the decline phase in a serum-supplemented culture medium. The level of CGL, a basic structure of glucosyl cholesterols, decreased in close correlation with the increases in CAG and CPG [42]. This meant that the active uptake of nonesterified cholesterol by the H. pylori cells was only observable in the logarithmic growth phase. Hence, the decrease in the CGL reflected a reduction of non-esterified cholesterol uptake by the H. pylori cell. Intriguingly, the amount of phosphatidylethanolamine (PE) in the membrane lipid composition decreased remarkably, from about 66% to 29%, in H. pylori cells undergoing growth phase changes. Further, the decline curve of PE along the time axis of the growth phase in H. pylori cells matched the decline curve of the CGL almost exactly. In contrast to the PE, the amounts of phosphatidylglycerol-cardiolipin (PG-CL), other glycerophospholipids of H. pylori, negligibly altered in its bacterial cells undergoing growth phase changes from the logarithmic phase to the decline phase. We therefore assumed that the PE of H. pylori regulated the uptake of non-esterified cholesterol by the cells. PE is the most prevalent glycerophospholipid in H. pylori, as in many other commonplace bacteria, yet the function of PE is still incompletely understood.
In a more recent study in 2012 we found that PE took part in the incorporation of non-esterified cholesterol into the cell membrane of H. pylori [43]. H. pylori PE clearly bound to non-esterified cholesterol more potently than PG-CL of this bacterium, or a reference Escherichia coli PE. Surprisingly, E. coli PE bound to both esterified cholesterol and non-esterified cholesterol at similar levels, whereas H. pylori PE bound to far less esterified cholesterol than to non-esterified cholesterol. In sum, H. pylori PE bound non-esterified cholesterol selectively and left esterified cholesterol alone. H. pylori PE also turned out to be involved in the incorporation of nonesterified steroids, that is, pregnenolone and dehydroepiandrosterone, other steroid compounds with a 3β-OH group, into the cell membrane.
When the heat-killed cells of H. pylori and E. coli were treated with 2,6-di-O-methyl-β-cyclodextrin (dMβCD), a lipid solubilizer, the conspicuous elution of PE from the cells was observed in H. pylori. Meanwhile, the elution of PE from the heat-killed E. coli cells was negligible levels, whereas the elution of PG-CL from the same cells was conspicuous. Given that dMβCD makes its first contact with the bacterial cell in the outermost layer of the outer membrane, and given that the conspicuous elution of PE from the H. pylori cells is induced via the action of dMβCD, we can assume that the outermost layer of the outer membrane of H. pylori contained more PE than PG-CL. Conversely, the outermost layer of the outer membrane of E. coli was seemed to contain more PG-CL than PE. E. coli cells show no signs of interaction with either esterified cholesterol or non-esterified cholesterol. Two of our results may partly explain why no cholesterol is absorbed into the cell membrane of E. coli: first, relatively less PE is expressed on the cell surface of E. coli than on the cell surface of H. pylori; second, H. pylori PE clearly binds to non-esterified cholesterol more efficiently than E. coli PE binds to non-esterified cholesterol or esterified cholesterol.
In one set of experiments we analyzed the fatty acid compositions of H. pylori PE to identify which structure is most closely involved in the selective binding of non-esterified cholesterol. The predominant saturated fatty acid component of H. pylori PE was a myristic acid (C14:0) molecule, whereas that of E. coli PE turned out to be a palmitic acid (C16:0) molecule. Several studies by others have identified a palmitic acid (C16:0) as the predominant saturated fatty acid component of PE in typical Gram-negative bacteria such as E. coli, Klebsiella pneumoniae, Salmonella enterica serovar Typhimurium, and Pseudomonas aeruginosa [44-47]. These findings, taken in sum, proved that the saturated fatty acid composition of H. pylori PE was quite distinct from that of other bacterial species PEs.
Noting this, we decided to examine the importance of the myristic acid (C14:0) molecule attached to PE in the selective binding of non-esterified cholesterol by dimyristoyl PE. As expected, dimyristoyl PE bound to nonesterified cholesterol selectively and bound to esterified cholesterol to only a negligible degree. In contrast, dipalmitoyl PE bound to both esterified cholesterol and non-esterified cholesterol. Hence, the myristic acid (C14:0) molecule in H. pylori PE played an important role in the binding of non-esterified cholesterol. These results demonstrate how H. pylori PE contributes as a steroid-binding lipid functionally important to the assimilation of non-esterified cholesterol and 3β-OH steroids (Figure 5).
As described above, progesterone non-reversibly binds to the H. pylori cells and inhibits the absorption of nonesterified cholesterol into its cell membrane. On this basis, H. pylori PE is also thought to be involved in the binding of progesterone to H. pylori cells.
7. A Novel Role of Cholesterol Glucosylation in the Survival of H. pylori
Several studies have clarified the respective roles of cholesterol-uptake and cholesterol-glucosylation in H. pylori cells, as mentioned earlier. The uptake of non-esterified cholesterol into the cell membrane is important to H. pylori’s resistance to the antibacterial activity of phosphatidylcholines (PCs), antibiotics, and bile salts. Likewise, the glucosylation of the non-esterified cholesterol absorbed into the cell membrane is important to the regulation of host immune responses against H. pylori.
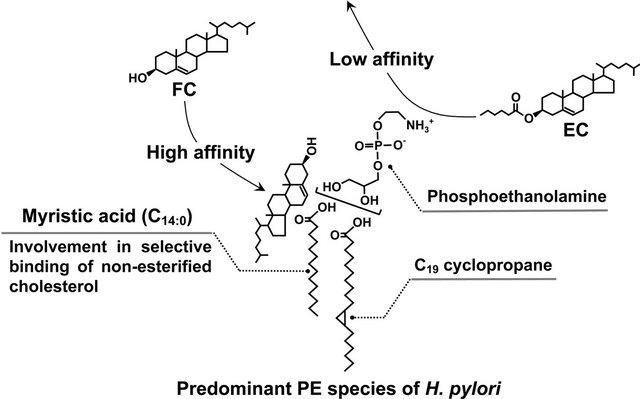
Figure 5. Selective binding of non-esterified cholesterol to H. pylori phosphatidylethanolamine. FC, non-esterified cholesterol; EC, esterified cholesterol; PE, phosphatidylethanolamine.
The most recent study by our group revealed a novel role of cholesterol glucosylation in the survival of H. pylori cells, a role distinct from host immune system evasion [48].
The sterol 7-dehydrocholesterol is the direct precursor of non-esterified cholesterol in the cholesterol biosynthetic pathway in mammalian tissues. The non-esterified cholesterol and 7-dehydrocholesterol differ in the number of hydrogen atoms in the steroid framework. The precursor lacks the two hydrogen atoms at carbon positions 7 and 8 of the non-esterified cholesterol molecule. Yet no studies have shown whether 7-dehydrocholesterol is useful or harmful for the growth and/or survival of the H. pylori cell.
When H. pylori at a low cell density (106.5 CFU/ml) was incubated in the presence of 7-dehydrocholesterol at a 100 µM concentration, we were intrigued to find that the non-esterified cholesterol precursor induced the bacteriolysis of this bacterium. Put simply, 7-dehydrocholesterol proved to be fatally toxic to H. pylori. Incidentally, non-esterified cholesterol at the same concentration had no influence on the viability or growth of this bacterium. In contrast, when H. pylori at a high cell density (109 CFU/ml) was incubated in the presence of 7-dehydrocholesterol (100 µM), we were surprised to find that the H. pylori cells lasted somehow to the bacteriolytic action of 7-dehydrocholesterol, glucosylated its toxic sterol, and admitted the glucosylated 7-dehydrocholesterols as membrane lipid components. Observing this, we assumed that the H. pylori detoxified the 7-dehydrocholesterol via the glucosylation. In our next experiment we examined the glucosylation-induced abolishment of the anti-H. pylori activity of 7-dehydrocholesterol by incubating the H. pylori at a low cell density (106.5 CFU/ ml) in the presence of glucosyl 7-dehydrocholesterol (100 µM). As expected, the glucosylated 7-dehydrocholesterol had no effect on the viability or growth of H. pylori cells. In addition, the hp0421 gene-disrupted H. pylori mutants with inactivated CGT and no ability to glucosylate sterols were clearly more susceptible to the bactericidal action of 7-dehydrocholesterol than the wild type H. pylori cells. These results indicate that the cholesterol-α-glucosyltransferase (CGT) encoded by the hp0421 gene plays an important role in the inactivation of sterols toxic to H. pylori (Figure 6).
Non-esterified cholesterol is biosynthesized from 7-dehydrocholesterol via the catalytic action of 7-dehydrocholesterol reductase encoded by a gene on chromosome 11 in humans [49]. A mutation of the gene for 7-dehydrocholesterol reductase leads to an accumulation of the sterol in plasma and tissues and has a role in the development of Smith-Lemli-Opitz (SLO) syndrome [49,50]. The plasma concentration of 7-dehydrocholesterol is vastly higher in SLO patients (from about 100 µM to 400
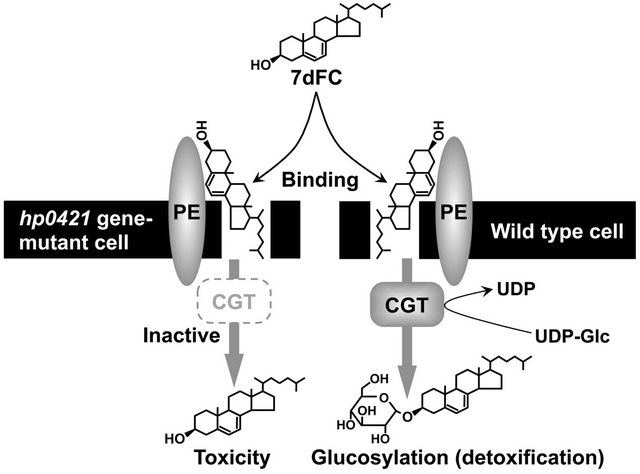
Figure 6. Detoxification of 7-dehydrocholesterol via the enzymatic action of H. pylori cholesterol-α-glucosyltransferase. 7dFC, 7-dehydrocholesterol; UDP-Glc, uridine diphosphate glucose; PE, phosphatidylethanolamine; CGT, cholesterol-α-glucosyltransferase.
µM), than in normal subjects (10 µM) [50-53].
As described above, we have found that phosphatidylcholines (PCs) induce the cell lysis of H. pylori without retained steroid compounds. Given that the SLO patients have a mutation only in the 7-dehydrocholesterol reductase gene, we can assume that PC is present at a normal concentration even in the gastric mucosa tissues of SLO patients. If H. pylori colonizes the gastric mucosa tissues of SLO patients, the cell membrane of this bacterium may ingest the 7-dehydrocholesterol from the gastric mucosa tissues of the hosts in order to survive around the PCs, even though the sterol is toxic to the bacterium. The benefit in this case, survival around the PCs, may outweigh the damage incurred by the toxicity to the bacterium. Hence, H. pylori is thought to be capable of glucosylating, and thereby detoxifying, the 7-dehydrocholesterol. Moreover, given the common occurrence of 3β-OH sterols such as ergosterol, sitosterol, campesterol, and stigmasterol in diverse forms of life, from fungi to mammals and plants, we can safely assume that some exert toxicity against H. pylori much in the same way as 7-dehydrocholesterol. H. pylori cells thus need to inactivate toxic 3β-OH sterols in order to survive in vivo or in vitro. The glucosylation of 3β-OH steroid compounds seems to be an essential mechanism for accomplishing this in H. pylori.
8. Conclusions
This review article has described the interaction of the H. pylori cell membrane with non-esterified cholesterols (including 7-dehydrocholesterol) and other steroid compounds. As described earlier, the inactivation of cholesterol-α-glucosyltransferase (CGT) via the insertion of the chloramphenicol-resistant (cat) gene cassette into the hp0421 gene prevents hp0421 mutant H. pylori cells from glucosylating non-esterified cholesterol. Even so, the cell membrane of the mutant cells absorbed non-esterified cholesterol from serum-supplemented culture medium and incorporated it as a lipid component [48]. This indicates that the hp0421 gene-mutant cells bind to non-esterified cholesterol via the mediation of phosphatidylethanolamine (PE) in the outermost layer of the outer membrane. In sum, the cell membrane of H. pylori glucosylates non-esterified cholesterol and other 3β-OH steroid compounds via at least two pathways: first, via the direct binding of 3β-OH steroid compounds to the CGT protein followed by glucosylation; second, via the transport of 3β-OH steroid compounds to the CGT protein after the binding of those compounds to the PE for glucosylation. Yet it remains unclear whether H. pylori cells possess a transport molecule that carries the 3β-OH steroid compounds from the PE molecules to the CGT proteins. Another unsolved question is the localization of the CGT protein in the outer membrane and inner membrane of the H. pylori cell. Earlier we established that the glucosylated cholesterols (CGL, CAG, and CPG) in H. pylori cells are more predominantly localized in the outer membrane than in the inner membrane, while non-esterified cholesterol in bacterial cells is localized in both the inner and outer membranes. This prompted us to consider the localization of the CGT protein in the inner membrane of H. pylori cell.
Here, we found that the 3β-OH steroid compounds binding to the outer membrane of bacterial cells via the mediation of PE seemed to somehow shift to the inner membrane, and that later, the 3β-OH steroid compounds glucosylated by the catalytic action of CGT in the inner membrane seemed to be carried outward to the outer membrane (Figure 7). Investigations into the carrier molecules involved in the transport of 3β-OH steroid
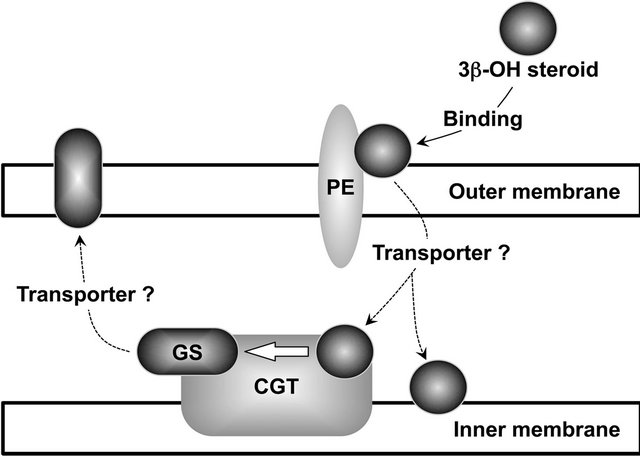
Figure 7. Proposed membrane transport of 3β-OH steroid compounds in H. pylori. PE, phosphatidylethanolamine; CGT, cholesterol-α-glucosyltransferase; GS, glucosyl steroid.
compounds and glucosylated steroid compounds may elucidate the precise mechanisms of the transport systems in H. pylori.
Lipopolysaccharide (LPS) is generally the main membrane lipid component of the outermost layer of the outer membrane of Gram-negative bacteria. LPS is a glycolipid consisting of a long polysaccharide chain and fatty acid molecules. It comes into direct contact with the outsides of the bacterial cells via part of the polysaccharide chain and regulates the permeability of the cell membrane to various lipophilic compounds [54]. Gram-negative bacteria are therefore intrinsically resistant to phosphatidylcholines (PCs) and steroid compounds. Though H. pylori is Gram-negative and naturally possesses LPS, the bacterium succumbs to the bacteriolytic activity of PCs if the cell membrane of the bacterium fails to incorporate the beneficial steroid compounds. The cell membrane of H. pylori also easily ingests a number of steroid compounds, even though some are fatally toxic to the bacterium. As described earlier, the predominant saturated fatty acid molecule attached to the PE of H. pylori is a myristic acid that is composed of 14 carbon atoms, whereas the predominant saturated fatty acid molecule attached to the other Gram-negative bacterial PEs is a palmitic acid that is composed of 16 carbon atoms. Although it is unclear what this means, intriguingly, noticeable acyl groups in H. pylori LPS molecule are fatty acid molecules that are composed of 16 carbon atoms and 18 carbon atoms, while noticeable acyl groups in typical bacterial LPS molecules, such as E. coli and Salmonella enterica serovar Typhimurium, are a fatty acid molecule that is composed of 14 carbon atoms [55-57]. These suggest that the membrane lipid conformation of H. pylori may be quite distinct from those of other Gram-negative bacteria, or that the LPS contents of the outer membrane of H. pylori are conspicuously lower than those of the outer membranes of other Gram-negative bacteria. On account of the unique membrane lipid conformation differing from those of other Gram-negative bacteria, the outer membrane of H. pylori cells without assimilated sterols or steroids is considered to cause the rise of membrane permeability to hydrophobic compounds, and therefore the bacterial cells succumb even to the bacteriolytic action of PCs. Hence, to overcome the membrane collapse induced by the invasion of such glycerophospholipids, H. pylori cells are suggested to allow the contact with various steroid compounds via the mediation of PE in the outer membrane. To clarify whether the steroid compound-free membrane of H. pylori cell is structurally vulnerable to exogenous lipophilic compounds compared with those of other Gram-negative bacteria, it will be necessary to carry out the further analyses for the total lipid compositions of both the outer membrane and inner membrane of the H. pylori cell.
REFERENCES
- J. R. Warren and B. Marshall, “Unidentified Curved Bacilli on Gastric Epithelium in Active Chronic Gastritis,” Lancet, Vol. 1, No. 8336, 1983, pp. 1273-1275.
- D. Y. Graham, “Helicobacter pylori: Its Epidemiology and Its Role in Duodenal Ulcer Disease,” Journal of Gastroenterology and Hepatology, Vol. 6, No. 2, 1991, pp. 105-113. doi:10.1111/j.1440-1746.1991.tb01448.x
- K. Fukase, M. Kato, S. Kikuchi, K. Inoue, N. Uemura, S. Okamoto, S. Terano, K. Amagai, S. Hayashi, M. Asaka and Japan Gast Study Group, “Effect of Eradication of Helicobacter pylori on Incidence of Metachronous Gastric Carcinoma after Endoscopic Resection of Early Gastric Cancer: An Open-Label, Randomised Controlled Trial,” Lancet, Vol. 372, No. 9636, 2008, pp. 392-397. doi:10.1016/S0140-6736(08)61159-9
- N. Uemura, S. Okamoto, S. Yamamoto, M. Matsumura, S. Yamaguchi, M. Yamakido, K. Taniyama, N. Sasaki and R. J. Schlemper, “Helicobacter pylori Infection and the Development of Gastric Cancer,” The New England Journal of Medicine, Vol. 345, No. 11, 2001, pp. 829-832. doi:10.1056/NEJMoa001999
- D. Forman and the Eurogast Study Group, “An International Association between Helicobacter pylori Infection and Gastric Cancer,” Lancet, Vol. 341, No. 8857, 1993, pp. 1359-1363. doi:10.1016/0140-6736(93)90938-D
- A. C. Wotherspoon, C. Ortiz-Hidalgo, M. R. Falzon and P. G. Isaacson, “Helicobacter pylori-Associated Gastritis and Primary B-Cell Gastric Lymphoma,” Lancet, Vol. 338, No. 8776, 1991, pp. 1175-1176. doi:10.1016/0140-6736(91)92035-Z
- R. M. Peek Jr. and M. J. Blaser, “Helicobacter pylori and Gastrointestinal Tract Adenocarcinomas,” Nature Reviews Cancer, Vol. 2, No. 1, 2002, pp. 28-37. doi:10.1038/nrc703
- R. M. Peek Jr. and J. E. Crabtree, “Helicobacter Infection and Gastric Neoplasia,” Journal of Pathology, Vol. 208, No. 2, 2006, pp. 233-248. doi:10.1002/path.1868
- M. Stolte, E. Bayerdorffer, A. Morgner, B. Alpen, T. Wundish, C. Thiede and A. Neubauer, “Helicobacter and Gastric MALT Lymphoma,” Gut, Vol. 50, No. 3, 2002, pp. III19-III24. doi:10.1136/gut.50.suppl_3.iii19
- M. Haque, Y. Hirai, K. Yokota and K. Oguma, “Steryl Glucosides: A Characteristic Feature of the Helicobacter spp.?” Journal of Bacteriology, Vol. 177, No. 18, 1995, pp. 5334-5337.
- M. Haque, Y. Hirai, K. Yokota, N. Mori, I. Jahan, H. Ito, H. Hotta, I. Yano, Y. Kanemasa and K. Oguma, “Lipid Profile of Helicobacter spp.: Presence of Cholesteryl Glucoside as a Characteristic Feature,” Journal of Bacteriology, Vol. 178, No. 7, 1996, pp. 2065-2070.
- B. P. Livermore, R. F. Bey and R. C. Johnson, “Lipid Metabolism of Borrelia hermsii,” Infection and Immunity, Vol. 20, No. 1, 1978, pp. 215-220.
- W. R. Mayberry and P. F. Smith, “Structures and Properties of Acyl Diglucosylcholesterol and Galactofuranosyl Diacylglycerol from Acholeplasma axanthum,” Biochimica et Biophysica Acta, Vol. 752, No. 3, 1983, pp. 434- 443. doi:10.1016/0005-2760(83)90273-4
- K. R. Patel, P. F. Smith and W. R. Mayberry, “Comparison of Lipids from Spiroplasma citri and Corn Stunt spiroplasma,” Journal of Bacteriology, Vol. 136, No. 2, 1978, pp. 829- 831.
- P. F. Smith, “Biosynthesis of Cholesteryl Glucoside by Mycoplasma gallinarum,” Journal of Bacteriology, Vol. 108, No. 3, 1971, pp. 986-991.
- Y. K. Kim, Y. Wang, Z. M. Liu and P. E. Kolattukudy, “Identification of a Hard Surface Contact-Induced Gene in Colletotrichum gloeosporioides Conidia as a Sterol Glucosyltransferase, a Novel Fungi Virulence Factor,” Plant Journal, Vol. 30, No. 2, 2002, pp. 177-187. doi:10.1046/j.1365-313X.2002.01284.x
- M. Oku, D. Warnecke, T. Noda, F. Müller, E. Heinz, H. Mukaiyama, N. Kato and Y. Sakai, “Peroxisome Degradation Requires Catalytically Active Sterol Glucosyltransferase with a GRAM Domain,” EMBO Journal, Vol. 22, No. 13, 2003, pp. 3231-3241. doi:10.1093/emboj/cdg331
- L. Peng, Y. Kawagoe, P. Hogan and D. Delmer, “Sitosterol-β-Glucoside as Primer for Cellulose Synthesis in Plants,” Science, Vol. 295, No. 5552, 2002, pp. 147-150. doi:10.1126/science.1064281
- D. C. Warnecke and E. Heinz, “Purification of a Membrane-Bound UDP-Glucose: Sterol-D-Glucosyltransferase Based on Its Solubility in Diethyl Ether,” Plant Physiology, Vol. 105, No. 4, 1994, pp. 1067-1073.
- D. C. Warnecke, M. Baltrusch, F. Buck, F. P. Wolter and E. Heinz, “UDP-Glucose:Sterol Glucosyltransferase: Cloning and Functional Expression in Escherichia coli,” Plant Molecular Biology, Vol. 35, No. 5, 1997, pp. 597-603. doi:10.1023/A:1005806119807
- D. Warnecke, R. Erdmann, A. Fahl, B. Hube, F. Müller, T. Zank, U. Zähringer and E. Heinz, “Cloning and Functional Expression of UGT Gene Encoding Sterol Glucosyltransferase from Saccharomyces cerevisiae, Candida albicans, Pichia pastoris, and Dictyostelium discoideum,” Journal of Biological Chemistry, Vol. 274, No. 19, 1999, pp. 13048-13059. doi:10.1074/jbc.274.19.13048
- G. Stübs, V. Fingerle, B. Wilske, U. B. Gobel, U. Zähringer, R. R. Schumann and N. W. Schröder, “Acylated Cholesteryl Galactosides Are Specific Antigens of Borrelia Causing Lyme Disease and Frequency Induce Antibodies in Late Stage of Disease,” Journal of Biological Chemistry, Vol. 284, No. 20, 2009, pp. 13326-13334. doi:10.1074/jbc.M809575200
- G. Ben-Menachem, J. Kubler-Kielb, B. Coxon, A. Yergey and R. Schneerson, “A Newly Discovered Cholesteryl Galactoside from Borrelia burgdorferi,” Proceedings of the National Academy of Sciences of USA, Vol. 100, No. 13, 2003, pp. 7193-7918. doi:10.1073/pnas.1232451100
- N. W. Schröder, U. Schombel, H. Heine, U. B. Gobel, U. Zähringer and R. R. Schumann, “Acylated Cholesteryl Galactoside as a Novel Immunogenic Motif in Borrelia burgdorferi sensu stricto,” Journal of Biological Chemistry, Vol. 278, No. 36, 2003, pp. 33645-33653. doi:10.1074/jbc.M305799200
- A. H. Lebrun, C. Wunder, J. Hildebrand, Y. Churin, U. Zähringer, B. Lindner, T. F. Mayer, E. Heinz and D. Warnecke, “Cloning of a Cholesterol-α-Glucosyltransferase from Helicobacter pylori,” Journal of Biological Chemistry, Vol. 281, No. 38, 2006, pp. 27765-27772. doi:10.1074/jbc.M603345200
- Y. Hirai, M. Haque, T. Yoshida, K. Yokota, T. Yasuda and K. Oguma, “Unique Cholesteryl Glucosides in Helicobacter pylori: Composition and Structural Analysis,” Journal of Bacteriology, Vo. 177, No. 18, 1995, pp. 5327- 5333.
- H. Shimomura, K. Hosoda, S. Hayashi, K. Yokota, K. Oguma and Y. Hirai, “Steroids Mediate Resistance to the Bactericidal Effect of Phosphatidylcholines against Helicobacter pylori,” FEMS Microbiology Letters, Vol. 301, No. 1, 2009, pp. 84-94. doi:10.1111/j.1574-6968.2009.01807.x
- C. Wunder, Y. Churin, F. Winau, D. Warnecke, M. Vieth, B. Lindner, U. Zähringer, H. J. Mollenkopf, E. Heinz and T. F. Meyer, “Cholesterol Glucosylation Promotes Immune Evasion by Helicobacter pylori,” Nature Medicine, Vol. 12, No. 9, 2006, pp. 1030-1038. doi:10.1038/nm1480
- K. Berstad, A. J. Berstad, R. Sjödahl, R. Weberg and A. Berstad, “Eosinophil Cationic Protein and Phospholipase A2 Activity in Human Gastric Juice with Emphasis on Helicobacter pylori Status and Effects of Antacids,” Scandinavian Journal of Gastroenterology, Vol. 27, No. 12, 1992, pp. 1011-1017. doi:10.3109/00365529209028131
- T. Orihara, H. Wakabayashi, A. Nakaya, K. Fukuta, S. Makimoto, K. Naganuma, A. Entani and A. Watanave, “Effect of Helicobacter pylori Eradication on Gastric Mucosal Phospholipid Content and Its Fatty Acid Composition,” Journal of Gastroenterology and Hepatology, Vol. 16, No. 3, 2001, pp. 269-275. doi:10.1046/j.1440-1746.2001.02440.x
- D. J. McGee, A. E. George, E. A. Trainor, K. E. Horton, E. Hildebrandt and T. L. Testerman, “Cholesterol Enhances Helicobacter pylori Resistance to Antibiotics and LL-37,” Antimicrobial Agents and Chemotherapy, Vol. 55, No. 6, 2011, pp. 2897-2904. doi:10.1128/AAC.00016-11
- E. A. Trainor, K. E. Horton, P. E. Savage, T. L. Testerman and D. J. McGee, “Role of the HefC Efflux Pump in Helicobacter pylori Cholesterol-Dependent Resistance to Ceragenins and Bile Salts,” Infection and Immunity, Vol. 79, No. 1, 2011, pp. 88-97. doi:10.1128/IAI.00974-09
- N. B. Javitt, Y. C. Lee, C. Shimizu, H. Fuda and C. A. Strott, “Cholesterol and Hydroxycholesterol Sulfotransferase: Identification, Distinction from Dehydroepiandrosterone Sulfotransferase, and Differential Tissue Expression,” Endocrinology, Vol. 142, No. 7, 2001, pp. 2978- 2984. doi:10.1210/en.142.7.2978
- Y. Miki, T. Nakata, T. Suzuki, A. D. Darnel, T. Moriya, C. Kaneko, K. Hidaka, Y. Shiotsu, H. Kusaka and H. Sasano, “Systematic Distribution of Steroid Sulfatase and Estrogen Sulfotransferase in Human Adult and Fetal Tissues,” Journal of Clinical Endocrinology & Metabolism, Vol. 87, No. 12, 2002, pp. 5760-5768. doi:10.1210/jc.2002-020670
- J. Takeyama, T. Suzuki, G. Hirasawa, Y. Muramatsu, H. Nagura, K. Iinuma, J. Nakamura, K. I. Kimura, M. Yoshihama, N. Harada, S. Andersson and H. Sasano, “17β- Hydroxysteroid Dehydrogenase Type 1 and 2 Expression in the Human Fetus,” Journal of Clinical Endocrinology & Metabolism, Vol. 85, No. 1, 2000, pp. 410-416. doi:10.1210/jc.85.1.410
- D. Turgeon, J. S. Carrier, E. Levesque, D. W. Hum and A. Belanger, “Relative Enzymatic Activity, Protein Stability, and Tissue Distribution of Human Steroid-Metabolizing UGT2B Subfamily Members,” Endocrinology, Vol. 142, No. 2, 2001, pp. 778-787. doi:10.1210/en.142.2.778
- A. Kominea, P. A. Konstantinopoulos, N. Kapranos, G. Vondoros, M. Gkermpesi, P. Andricopoulos, S. Artelaris, S. Savva, I. Varakis, G. Sotiropoulou-Bonikou and A. G. Papavassiliou, “Androgen Receptor (AR) Expression Is an Independent Unfavorable Prognostic Factor in Gastric Cancer,” Journal of Cancer Research and Clinical Oncology, Vol. 130, No. 5, 2004, pp. 253-258. doi:10.1007/s00432-003-0531-x
- S. Matsuyama, Y. Ohkura, H. Eguchi, Y. Kobayashi, K. Akagi, K. Uchida, K. Nakachi, J. A. Gustafsson and S. Hayashi, “Estrogen Receptor β Is Expressed in Human Stomach Adenocarcinoma,” Journal of Cancer Research and Clinical Oncology, Vol. 128, No. 6, 2002, pp. 219- 324. doi:10.1007/s00432-002-0336-3
- N. Takano, N. Iizuka, S. Hazama, S. Yoshino, A. Tangoku and M. Oka, “Expression of Estrogen Receptor-α and -β mRNAs in Human Gastric Cancer,” Cancer Letter, Vol. 176, No. 2, 2002, pp. 129-135. doi:10.1016/S0304-3835(01)00739-X
- K. Hosoda, H. Shimomura, S. Hayashi, K. Yokota, K. Oguma and Y. Hirai, “Anabolic Utilization of Steroid Hormones in Helicobacter pylori,” FEMS Microbiology Letters, Vol. 297, No. 2, 2009, pp. 173-179. doi:10.1111/j.1574-6968.2009.01685.x
- K. Hosoda, H. Shimomura, S. Hayashi, K. Yokota and Y. Hirai, “Steroid Hormones as Bactericidal Agents to Helicobacter pylori,” FEMS Microbiology Letters, Vol. 318, No. 1, 2011, pp. 68-75. doi:10.1111/j.1574-6968.2011.02239.x
- H. Shimomura, S. Hayashi, K. Yokota, K. Oguma and Y. Hirai, “Alteration in the Composition of Cholesteryl Glucosides and Other Lipids in Helicobacter pylori Undergoing Morphological Change from Spiral to Coccoid Form,” FEMS Microbiology Letters, Vol. 237, No. 2, 2004, pp. 407-413.
- H. Shimomura, K. Hosoda, S. Hayashi, K. Yokota and Y. Hirai, “Phosphatidylethanolamine of Helicobacter pylori Functions as a Steroid-Binding Lipid in the Assimilation of Free Cholesterol and 3β-Hydroxl Steroids into the Bacterial Cell Membrane,” Journal of Bacteriology, Vol. 194, No. 10, 2012, pp. 2658-2667. doi:10.1128/JB.00105-12
- A. Amine, K. S. Saloua, M. Mouadh, E. I. M. Alya and L. Ahmed, “The Absence of the ‘GATC-Binding Protein SegA’ Affect DNA Replication in Salmonella enterica Serovar Typhimurium,” In: J. Kusic-Tisma, Ed., DNA Replication and Related Cellular Process, InTech, Rijeka, 2011. http//www.intechopen.com/books/dna-replication-and-related-cellar-process/the-absence-of-the-gatc-binding-protein-seqa-affects-dna-replication-in-salmonella-enterica-serovar-typhimurium
- J. K. Dunnick and W. M. O’Leary, “Correlation of Bacterial Lipid Composition with Antibiotic Resistance,” J. Bacteriol., Vol. 101, No. 3, 1970, pp. 892-900.
- D. B. Kearns, J. Robinson and L. J. Shimkets, “Pseudomonas aeruginosa exhibits directed twitching motility up phosphatidylethanolamine gradients,” Journal of Bacteriology, Vol. 183, No. 2, 2001, pp. 763-767. doi:10.1128/JB.183.2.763-767.2001
- A. Shokri and G. Larsson, “Characterisation of the Escherichia coli Membrane Structure and Function during Fed Batch Cultivation,” Microbial Cell Factories, Vol. 3, 2004, pp. 1-9. doi:10.1186/1475-2859-3-1
- H. Shimomura, K. Hosoda, D. J. McGee, S. Hayashi, K. Yokota and Y. Hirai, “Detoxification of 7-Dehydrocholesterol Fatal to Helicobacter pylori Is a Novel Role of Cholesterol Glucosylation,” Journal of Bacteriology, Vol. 195, No. 2, 2013, pp. 359-367. doi:10.1128/JB.01495-12
- N. B. Javitt, “Oxysterols: A New Class of Steroids with Autocrine and Paracrine Functions,” Trends in Endocrinology & Metabolism, Vol. 15, No. 8, 2004, pp. 393-397.
- G. S. Tint, M. Irons, E. R. Elias, A. K. Batta, R. Frieden, T. S. Chen and G. Salen, “Defective Cholesterol Biosynthesis Associated with the Smith-Lemli-Opitz Syndrome,” The New England Journal of Medicine, Vol. 330, No. 2, 1994, pp. 107-113. doi:10.1056/NEJM199401133300205
- I. Björkhem, L. Starck, U. Andersson, D. Lütjohann, S. von Bahr, I. Pikuleva, A. Babiker and U. Diczfalusy, “Oxysterols in the Circulation of Patients with the SmithLemli-Opitz Syndrome: Abnormal Levels of 24Sand 27-Hydroxycholesterol,” Journal of Lipid Research, Vol. 42, No. 3, 2001, pp. 366-371.
- P. E. Jira, J. G. N. De Jong, F. S. M. Janssen-Zijlstra, U. Wendel and R. A. Wevers, “Pitfalls in Measuring Plasma Cholesterol in the Smith-Lemli-Opitz Syndrome,” Clinical Chemistry, Vol. 43, No. 1, 1997, pp. 129-133.
- L. S. Merkens, W. E. Connor, L. M. Linck, D. S. Lin, D. P. Flavell and R. D. Steiner, “Effects of Dietary Cholesterol on Plasma Lipoproteins in Smith-Lemli-Opitz Syndrome,” Pediatric Research, Vol. 56, No. 5, 2004, pp. 726-732. doi:10.1203/01.PDR.0000141522.14177.4F
- E. T. Rietschel, T. Kirikae, F. U. Schade, U. Mamat, G. Schmidt, H. Loppnow, A. J. Ulmer, U. Zähringer, U. Seydel, F. D. Padova, M. Schreier and H. Brade, “Bacterial Endotoxin: Molecular Relationship of Structure to Activity and Function,” FESEB Journal, Vol. 8, No. 2, 1994, pp. 217-225.
- L. Guo, K. B. Lim, J. S. Gunn, B. Bainbridge, R. P. Darveau, M. Hackett and S. I. Miller, “Regulation of Lipid A Modification by Salmonella typhimurium Virulence Genes phoP-phoQ,” Science, Vol. 276, No. 5310, 1997, pp. 250- 253. doi:10.1126/science.276.5310.250
- C. L. Hall and R. S. Munford, “Enzymatic Deacylation of the Lipid A Moiety of Salmonella typhimurium Lipopolysaccharides by Human Neutrophils,” Proceedings of the National Academy of Sciences of USA, Vol. 80, No. 21, 1983, pp. 6671-6675. doi:10.1073/pnas.80.21.6671
- C. M. Stead, A. Beasley, R. J. Cotter and M. S. Trent, “Deciphering the Unusual Acylation Pattern of Helicobacter pylori Lipid A,” Journal of Bacteriology, Vol. 190, No. 21, 2008, pp. 7012-7021. doi:10.1128/JB.00667-08