American Journal of Analytical Chemistry
Vol.07 No.01(2016), Article ID:63118,34 pages
10.4236/ajac.2016.71011
Drug-Excipient Interactions: Case Studies and Overview of Drug Degradation Pathways
Kishore Kumar Hotha*, Swapan Roychowdhury, Veerappan Subramanian
Novel Laboratories Inc., Somerset, NJ, USA

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 16 December 2015; accepted 24 January 2016; published 28 January 2016
ABSTRACT
The objective of the current research article is to provide a comprehensive review of excipients impact on the stability of the drug product and their implications during the product development. Recent developments in the understanding of the degradation pathways further impact methodologies used in the pharmaceutical industry for potential stability assessment. The formation of drug excipient adducts was very common based on the sensitive chemical moieties in the drugs and the excipients. The formation of the impurities was not limited to drug related impurities but there were several possibilities of the drug-excipient adduct formations as well as excipient impurities reaction with Active Pharmaceutical Ingredients. Identification of drug degradation in presence of excipients/excipient impurities requires extensive knowledge and adequate analytical characterization data. Systematic literature review and understanding about the drug formulation process, give you a smooth platform in establishing the finished product in the drug market. This paper discusses mechanistic basis of known drug-excipient interactions with case studies and provides an overview of common underlying themes in solid, semisolid and parenteral dosage forms.
Keywords:
Drug, Excipients, Forced Degradation, Impurities, Adducts, Degradation Pathways, HPLC, LC-MS/MS, Synthesis, Chemistry, Characterization

1. Introduction
Excipients are included in dosage forms to aid manufacture, administration or absorption. Excipients can initiate, propagate or participate in chemical or physical interactions with an active, possibly leading to compromised quality or performance of the medication. Chemical interaction can lead to degradation of the active ingredient, theory by reducing the amount available for the therapeutic effect; reaction products may compromise safety or tolerance. Physical interactions can affect the rate of dissolution, uniformity of dose or ease of administration. Understanding the chemical and physical nature of excipients, the impurities or residues associated with them and their interaction with actives as well as with each other is the important phenomenon in the drug excipient interaction process [1] -[3] . Excipients may have functional groups that interact directly with active pharmaceutical ingredients. Alternatively, they may contain impurities or residues or form degradation products that in turn cause decompositions of the drug substance. Analysis of excipients and their purity and quality shall be ensured prior to product development. Analytical development should ensure any unknown peaks formed during the drug excipient compatibility studies. Thorough monitoring of unknown impurities and its origin during drug development process reduces the delay in the product filings [4] [5] .
Excipients form several reactions based on the formulation process design [6] [7] . The chemical nature of the excipient, drug-to-excipient ratio, moisture, micro environmental pH of the drug excipient mixture, temperature, and light, on dosage form stability could be the possible factors for the drug degradation process and the formation of the reaction products. The incompatibilities with the functional groups shall be kept in mind while evaluating the unknown impurities formed in the drug product (Table 1) [8] -[10] .
Many of the reported drug-excipient reactions involve hydrolysis, oxidation, or specific interaction of drugs with reactive impurities in excipients. Depending on the application of a specific excipient as well as other formulation and processing factors, the presence of these impurities in excipients even in trace amounts could influence the safety and efficacy of the drug products, especially for highly potent Active Pharmaceutical Ingredients (API), which have low dose and high excipient/API ratio in the drug product. A robust formulation is one that is able to accommodate the typical variability in API, excipients, and processes. The choice of excipients in the design of formulation was made based on their function as well as chemical compatibility with the drug substance [10] -[12] . In a typical drug-excipient compatibility experiment, drug stability at accelerated temperature was assessed in the presence of single or multiple excipients (either as powder blend or as compact) with or without humidity/water [13] -[16] . Although these experiments are useful as a first step to eliminate incompatible excipient(s), the chemical compatibility of drugs with different lots of excipients or excipients from different vendors is usually never studied prospectively. The excipient lot-to-lot variability in drug compatibility might arise from the variability in the levels of reactive impurities in excipients (Table 2) [17] .
In this present article, certain kinds of drugs and their interactions with the most commonly used excipients were discussed with case studies. This review paper focuses on these drug-excipient interactions in solid, semisolid and parenteral dosage forms. Excipients interactions and drug degradation due to flavors, lactose, microcrystalline cellulose, povidone, crospovidone, hydroxypropyl cellulose, stearic acid, magnesium stearate and silicon dioxide were explained. Some specific examples of drug-excipient impurity interaction from internal research (famotidine, buprenorphine, benzyl alcohol, fluphenazine decanoate, dexamethasone sodium phosphate, methylphenidate and cetirizine) and important case studies from the literature were reported. Unpublished research

Table 1. Interaction and incompatabilities of the excipients with functional groups.
Table 2. Impurities present in the excipients.
work of drug excipient interactions from our internal findings is also reported as case study examples for methylphenidate and fluphenazine decanoate. The origin of these reactive impurities and their potential chemical reactions with the susceptible APIs were identified. Finally, stratagems to mitigate the potential incompatibilities are also reviewed.
2. Experimental
2.1. Chemical and Reagents
Some specific examples of drug-excipient impurity interactions performed internally. Benzyl alcohol, glycerin, hydrogen peroxide and reference standards were procured from sigma Aldrich, USA. HPLC grade Acetonitrile, methanol, and ortho phosphoric acid, Ammonium acetate, acetic acid were purchased from Merck, Darmstadt, Germany. Water used was obtained by using Millipore MilliQ Plus water purification system. Novel Laboratories Inc., Somerset, New Jersey, supplied drug product/Drug substance samples.
2.2. Equipment
Water HPLC with 2695 separation module equipped with 2996 PDA detector used for the HPLC method development. The output signal was monitored and processed using Empower-3 software. LC-MS/MS system (Acquity UPLC coupled with TQD mass spectrometer with empower software, Waters Corporation, Milford, USA) was used for the identification of unknown compounds formed during forced degradation and stability testing studies.
3. Aldehydes
Many commonly used pharmaceutical excipients contain trace level aldehydes. Aldehyde impurities in excipients are often due to excipient degradation. With organic excipients, degradation to aldehydes is generally associated with oxidation. A second source of aldehydes involves functional additives present in the excipients, either as aldehydes themselves or as materials that oxidize or hydrolyze to give aldehydes. Aldehydes even in trace amounts adversely affect the stability and efficacy of drugs via direct reaction with the active pharmaceutical ingredient. Formaldehyde, acetaldehyde, benzaldehyde, furfural are most often responsible for stability issues in drug products [18] -[21] .
3.1. Famotidine-Benzaldehyde Adduct Formation Due to Cherry-Flavor Excipient
Flavors used in the formulation process are the most possible origins to form impurities due to the trace level presence of aldehydes and ketones that apparently used for its aroma. Famotidine oral solution forms unknown impurity due to Benzaldehyde, which is present in the excipient of cherry flavor used in the formulation process. Cherry flavor used in the famotidine oral solution contains very small amount of benzaldehyde that formed imine reaction with the terminal nitrogen of the famotidine (Figure 1). Hotha et al. described the identification and characterization of this unknown impurity by UPLC-MS/MS and NMR (Figure 1 and Figure 2). Molecular ions of 187 and 236 confirm the formation of sulfynyl amine attached to the benzaldehyde moiety. Aldehyde free cherry flavor recommended for the preparation of oral solutions for amine containing compounds [18] .

Figure 1. Famotidine interaction with benzaldehyde present in the cherry flavor.

Figure 2. MS/MS chromatogram of the famotidine impurity in negative ion mode.
3.2. Benzyl Alcohol Oxidation in Parenteral Formulations: Benzaldehyde-Dibenzyl Acetal
Benzyl alcohol used as a bacteriostatic preservative at low concentration in intravenous medications, cosmetics and topical drugs. The use of benzyl alcohol as a 5% solution has been approved by the US FDA in the treatment of head lice in children older than 6 months and in adults. Majority of the parenteral solutions use Benzyl alcohol as a preservative. There were several possible degradants associated with the oxidation of benzyl alcohol (Figure 3). Benzaldehyde and benzoic acid are the possible degradants observed in the parenteral solutions, which contains benzyl alcohol (Figure 4) [19] -[21] . These oxidation products of benzyl alcohol react in a solution, which will form benzaldehyde dibenzyl acetal (Figure 5).
3.3. Vanillin Reaction with Amine Containing Compound
Flavor containing Vanillin reacts with the Active Pharmaceutical Ingredient (API), having primary amine to form imines. The finished product is a ready to use liquid and oil based formulation [10] [17] . The aldehyde functionality of vanillin leads to form cis/trans-imines and to inversion of the chiral center (Figure 6).
3.4. BMS-204352 Formaldehyde Adduct Impurity Due to Polysorbate 80 and PEG 300
Munnir N. Nasser et al. reported a degradation product in the BMS-204352 clinical formulation identified as the formaldehyde adduct of BMS-204352 (hydroxymethyl derivative, BMS-215842) (Figure 7). The formaldehyde spiking study supports the hypothesis that formaldehyde in the Polysorbate 80 and PEG 300 excipients used in the formulation result in formation of the formaldehyde adduct degradant [22] [23] . Limits may need to be established for reactive impurities in formulation excipients, to ensure acceptable levels of degradants and adequate product shelf life [24] .
3.5. Phenylephrine Reaction with Formaldehyde and 5-HMF
Michal Dousa et al. [25] studied different pharmaceutical preparations against the common cold containing phenylephrine and saccharose. The formulations containing both phenylephrine and saccharose found to be susceptible

Figure 3. Photochemical oxidation products of benzyl alcohol.
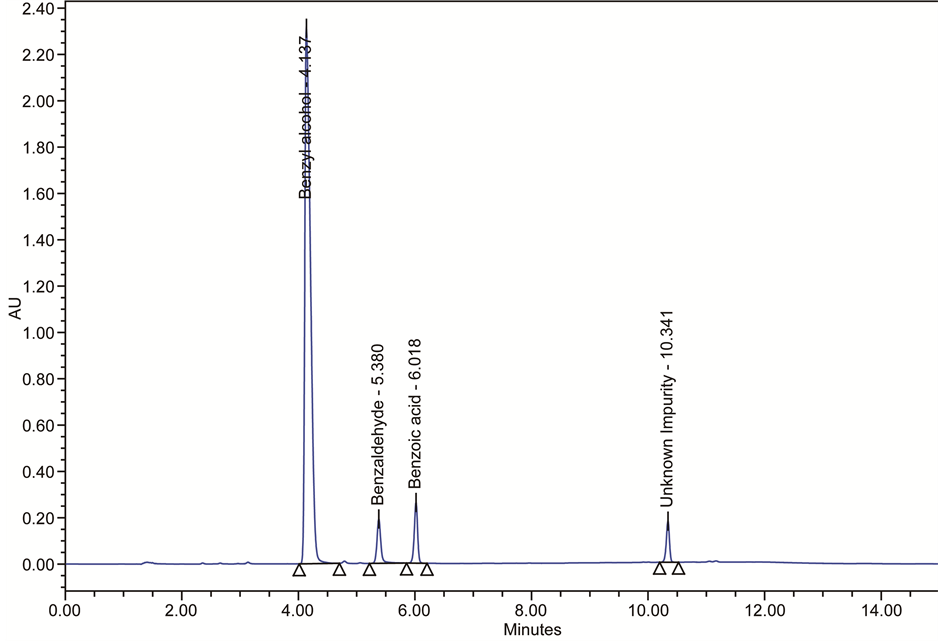
Figure 4. HPLC chromatogram of benzyl alcohol degradation in parenteral solution.

Figure 5. Benzaldehyde and benzyl alcohol interaction to form benzaldehyde dibenzyl acetal.
to loss of phenylephrine activity. There is evidence that the degradation of PHE resulted from condensation reactions of phenylephrine with aldehydes (such as formaldehyde or 5-HMF) (Figure 8 and Figure 9). The results indicate that phenylephrine is molecule, which should handle with special care during pharmaceutical processing to avoid decomposition and interactions. The new identified degradation products of phenylephrine found in pharmaceutical preparations against the common cold containing phenylephrine and saccharose.
3.6. Benzaldehyde Induced Oxidation of Cyclic Heptapeptide
David C. Dubost et al. reported degradation of a cyclic peptide drug in a lyophilized formulation occurs in the

Figure 6. Vanillin reaction with primary amine in the API drug substance.

Figure 7. Formaldehyde present in the PEG 300 and polysorbate 80 interaction with BMS204352.
solid state to give an aldehyde as an oxidation product [26] . The oxidative pathway does not require oxygen but does depend on the presence of impurities in the pharmaceutical excipient used in the formulation. A mechanism involving Schiff base formation, double bond isomerization, and subsequent hydrolysis has been proposed to account for the formation of the degradant. The extent of the formation of the impurity is influenced by the amount of mannitol used as an excipient in the formulation (Figure 10).
4. Transesterification
Transesterification is the process of exchanging the organic group of an ester with the organic group of an alcohol. These reactions are often catalyzed by the addition of an acid or base catalyst [27] [28] . The reaction can

Figure 8. Phenylephrine reaction with formaldehyde present in saccharose.

Figure 9. Phenylephrine reaction with 5-HMF.

Figure 10. Mannitol induced oxidation of heptapeptide in lyophilized injection.
also be accomplished with the help of enzymes (biocatalysts) particularly lipases. Strong acids catalyze the reaction by donating a proton to the carbonyl group, thus making it a more potent electrophile, whereas bases catalyze the reaction by removing a proton from the alcohol, thus making it more nucleophilic. Esters with larger alkoxy groups can be made from methyl or ethyl esters in high purity by heating the mixture of ester, acid/base, and large alcohol and evaporating the small alcohol to drive equilibrium. Several drug excipients reactions were accompanied by transesterification process to give different impurities during the stability conditions [29] [30] .
4.1. Transesterification Reactions between Two Parabens and Sugar Alcohols
Sugar alcohols and parabens are commonly used ingredient in oral suspension formulations. Parabens are commonly used antimicrobial agents in pharmaceuticals, food and cosmetics. However, their possible incompatibility because of transesterification reaction is a concern during formulation development. During the stability, study of an experimental batch of an oralsuspension formulation that contained sorbitol and paraben preservatives shows approximately 1% degradation for the samples stored at 30˚C for one year (Figure 11). 3, 4, 5 carbon sugar alcohols reacts with methyl paraben to form 6 isomer products were identified by HPLC and NMR by Minhui Ma et al. [28] -[30] .
4.2. Ester and Amide Formation between Citric Acid and 5-Aminosalicylic Acid
The reaction between 5-aminosalicylic acid (5-ASA) and the excipient citric acid (Figure 12) during storage of an experimental enema preparation observed three isobaric reaction products of an ester and an amide with non-symmetrically substituted citric acid moieties and a symmetrical amide. Larsen et al identified and characterized these products [31] . Approximately 5% of the 5-Aminosalicylic acid present in the formulation was transformed into these impurities. Because of the relatively rapid and extensive formation of reaction, production between 5-Aminosalicylic acid and citric acid the use of citric acid in liquid 5-Aminosalicylic acid formulations not recommended. Thus based on these findings further development of a formulation 5-Aminosalicylic acid contains citric acid was abandoned and replaced with another formulation.
4.3. Reaction of Citric Acid with 6-Aminocaproic Acid
Drug excipient interactions between 6-aminocaproic acid and the excipients citric acid were found in highly concentrated solutions at pH 4.0 and pH 5.0 [32] . In addition to that, several degradation products of the 6-aminocaproic acid reacting with itself, e.g., dimers trimers and cyclized forms observed (Figure 13). The utilization of citric acid as buffering species in liquid formulations requires careful evaluation on a case-by-case basis.
4.4. Ester Formation of Cetirizine with Sorbitol and Glycerol
Cetirizine with the sorbitol and glycerol to form monoesters (Figure 14 and Figure 15). At a temperature as low as 40˚C more than 1% of the cetirizine content was transformed into a monoester within 1 week using concentrations similar to those used in marketed preparations [33] .

Figure 11. Interaction between methyl paraben and sorbitol in formulations.

Figure 12. 5-Aminosalicylic acid degradation with citric acid.
4.5. Transesterification Reaction of Glycerin and Methylphenidate
Transesterification reaction of methylphenidate with glycerin forms different structural isomeric products at the specified RRT of 0.75 and 0.77 in a HPLC method. The formation of these impurities monitored during forced degradation studies, excipient compatibility studies and in the stability conditions. Quantification of the stability samples were analyzed and observed that about 0.6% of the Methylphenidate content was transformed into methylphenidate-glycerin isomers within 3 Months at 40˚C/75% RH and 18 Months at 25˚C/60% RH conditions. Identificaiton and characterization of these impurities (Figure 16, Figure 17) by LC-MS/MS was performed [34] -[36] . The LC-MS/MS method was developed using Acquity UPLC BEH C18 1.7 µm, 2.1 × 150 mm column as stationary phase. The mobile phase A is a 0.1% of acetic acid in water. The mobile phase B is acetonitrile. The UPLC gradient program was set as: Time (min) % solution B: 00/15, 5.58/15, 26.92/40, 26.98/15, 35.00/15. The column temperature maintained at 40˚C and the injection volume was 15 μL Milli-Q water used as Diluent. The mobile phase pumped at 0.64 mL/min−1. The eluted compounds monitored at 210 nm. The run time was 35 min. Mass spectrometric conditions optimized as cone gas 10 V, Collision flow 70 L/Hr., Ion energy 0.7 Entrance and Exit Potentials 1 V, source temperature 150˚C, Desolovation gas 800 L/hr., Desolvation

Figure 13. 6-Aminocaproic acid reaction with citric acid.

Figure 14. Cetirizine transesterification reaction with sorbitol.

Figure 15. Cetirizine transesterification reaction with glycerol.

Figure 16. HPLC chromatogram of transesterification products of methylphenidate.

Figure 17. Transesterification mechanism of methylphenidate with glycerin.
temperature 400˚C. Molecular ion [M + H]+ shows intense ion of 294 confirms the addition of glycerin moiety with Methylphenidate (Figure 18).
4.6. Ester and Amide Formation between Citric Acid and Carvedilol
The solid state reaction between citric acid and carvedilol forms three citric acid esters and three citric acid amides with the two diastereomeric pairs RR and RS (Figure 19). When citric acid monohydrate replaced with

Figure 18. Q3-MS/MS (positive ion mode) spectrum of methylphenidate transesterification products.

Figure 19. Carvedilol degradation with citric acid.
anhydrous citric acid, the amount of esters was reduced approximately eight folds. Presence of small amounts of water significantly speeds up the solid-state reaction. Jesper Larsen et al. [37] reported all the degradants and elucidated the structures by HPLC and NMR.
4.7. Buprenorphine Degradation in Presence of Citric Acid
Buprenorphine shows unknown impurity in the Buprenorphine and Naloxone sublingual tablets due to the presence of citric acid in formulation. This was confirmed by the excipient compatibility studies. Characterization performed by LC-MS/MS (Figure 20 and Figure 21). The unknown impurity found in Buprenorphine Naloxone sublingual tablets is Impurity I according to EP monograph [38] . This impurity is formed due to the Dehydrative cyclization of Buprenorphine to form Furanyl ring (Figure 22). The molecular weight of the impurity is 435 whereas the molecular weight of the Buprenorphine is 467. Loss of methoxy group (32 amu) in the cyclization process for Buprenorphine to form the Impurity-I.
5. Sulfate Adduct in the Parenteral Formulation
Incompatibility between APIs and excipients will cause adverse reactions and thus affect drug quality. An unknown impurity at specified RRT 0.21 observed during the routine analysis of the stability samples of Dexamethasone Sodium Phosphate Injections, 4 mg/mL [39] [40] . Investigation revealed that dexamethasone sodium phosphate could react with sodium bisulfite under heat condition and bring in impurity I in the injection (Figure 23). HPLC, MS, IR and NMR analysis confirmed that impurity I was sulfonated product of the nucleophilic addition reaction between dexamethasone sodium phosphate and sodium bisulfite (Figures 24-26). Impurity I reduced drug levels in injections, affecting its efficacy and safety. To conclude, optimization of formulation and process development as well as investigation of drug-excipient compatibility is essential to improve formulation quality and avoid side effects due to incompatibilities.

Figure 20. HPLC chromatogram of the buprenorphine impurity-I due to citric acid.

Figure 21. UPLC-MS chromatogram of the buprenorphine impurity-I.

Figure 22. Dehydrative cyclization of buprenorphine to form furanyl impurity.
6. Magnesium Stearate
Magnesium stearate is a white substance, solid at room temperature, used in the manufacture of pharmaceutical and supplement tablets and capsules. It is composed of magnesium and stearic acid, and oftentimes, palmitic acid as well. The substance is also useful, because it has lubricating properties, preventing ingredients from sticking to manufacturing equipment during the compression of chemical powders into solid tablets; magnesium stearate is the most commonly used lubricant for tablets. Sometimes chemical moieties of the drug can transform into degradants in presence of Magnesium stearate. There were several literature exists for the degradants formed in presence of magnesium stearate [41] -[45] .

Figure 23. Sulfonate adduct formation of dexamethasone sodium phosphate.

Figure 24. HPLC chromatogram of unknown impurity at 2.2 Min due to the sulphate adduct.
6.1. Benzilic Acid Rearrangement of Tacrolimus Due to Magnesium Stearate
Tacrolimus is macrolide drug that is widely used as a potent immune suppressant. Tenja Rozman Peterka et al. [41] reported the formation of alpha-hydroxy acid from the parent tacrolimus through Benzilic acid type rearrangement reaction in the presence of magnesium stearate (Figure 27). This reaction occurred in the presence of divalent metallic cations at higher pH values.
6.2. Stearoyl Rearrangement of Norfloxacin
Norfloxacin reacts with magnesium stearate forms stearamide adduct (Figure 28) through Stearoyl rearrangement [17] [42] [43] . Magnesium stearate is an excipient used in the tablet formulation. This adduct formation was observed after prolonged storage at 60˚C. Since Magnesium stearate derived from multiple sources, the presence of other fatty acids like palmitic acid, arachidic acid and behenic acids can lead to form more than one adducts.

Figure 25. UV spectrum and dexamethasone sodium phosphate of the unknown impurity at 2.2 Min due to the sulphate adduct.

Figure 26. MS spectrum of the unknown impurity at 2.2 min due to the sulphate adduct.
6.3. Metal Induced Degradation of Fosinopril Sodium Due to Magnesium Stearate
Serajuddin et al. reported [44] that Fosinopril sodium forms three degradation products when tablet formulations lubricated with magnesium stearate (Figure 29). There were two distinct pathways of degradation where magnesium ion mediated through hydrolysis. There should be a limited amount of water in the formulation, which will reduce the drug excipient compatibility because it does not depend on the hygroscopicity of the individual components.

Figure 27. Tacrolimus degradation in presence of magnesium stearate through benzylic acid rearrangement.

Figure 28. Stearoyl rearrangement of norfloxacin.

Figure 29. Hydrolysis and metal induced degradation of fosinopril sodium.
6.4. Reaction of Duloxetine with HPMCAS
Patric et al. reported Duloxetine to react with polymer degradation products or residual free acids present in the enteric polymers hydroxypropyl methylcellulose acetate succinate (HPMCAS) and hydroxypropyl methylcellulose phthalate (HPMCP) in dosage formulations to form succinamide and phthalamide impurities, respectively (Figure 30) [45] . The rate of formation of the impurities is accelerated by heat and humidity. The structures were deduced using molecular weights obtained from LC comparison of UV spectra, HPLC retention times, and electrospray mass spectra to independently synthesized material. It is proposed that polymer-bound succinic and phthalic substituents can be cleaved from the polymer, resulting in the formation of either the free acids or the anhydrides. It is postulated that the reaction is enabled by migration of either the free acid or anhydride or the parent drug through the formulation. The formation of these impurities was minimized by increasing the thickness of the physical barrier separating the enteric coating from the drug.
7. Maillard Reaction of Lactose
The Maillard reaction is named after Louis Maillard, who reported over 80 years ago that some amines and reducing carbohydrates react to produce brown pigments. It has been extensively studied and reviewed, especially in the food and nutrition literature. The first product of this reaction is a simple glycosylamine, which readily undergoes the Amadori rearrangement to produce 1-amino-1-deoxy-2-ketoses. Reducing carbohydrates such as glucose, maltose, and lactose are substrates for the Maillard reaction since their cyclic tautomers are in equilibrium with their more reactive aldehyde forms; non-reducing carbohydrates such as mannitol, sucrose, and trehalose are not subject to Maillard reactions. Although early scientists believed that only primary aromatic amines were capable of this reaction, subsequent research has shown that nearly all primary and secondary amines, aromatic or aliphatic, are capable of this reaction. Amino acids and proteins are also substrates for the Maillard reaction. The impact of these reactions on the stability of pharmaceuticals has also been known for some time and was recently reviewed [46] -[50] .

Figure 30. Duloxetine reaction with HPMCAS to form amides.
Although the clinical significance of this drug-excipient interaction is unknown, the relevance of these findings to formulation scientists is more straightforward. Namely, the Maillard reaction of secondary amines and lactose should be considered when selecting formulation ingredients and when examining the stability of such products. However, in many cases, the formulation scientist will not know whether a specific nitrogen-containing drug will be compatible with reducing carbohydrates or not, usually due to significant structural variations such as inclusion of the nitrogen within rings or the presence of functionality which would greatly diminish the nucleophilicity of the drug [51] .
7.1. Reaction of Fluoxetine with Lactose
Fluoxetine contains a secondary amine forms the Maillard reaction with lactose to form formyl fluoxetine identified as a major product of this Maillard reaction (Figure 31). Water content, lubricant concentration and temperature founds to influence the degradation. David D. Wirth et al. [46] [52] reported this degradation pattern which and formation of N-formyl fluoxetine through malliard reaction.
7.2. Reaction of Cetirizine with Hydroxy Propyl Cellulose
Cetirizine contains carboxylic acid group and forms lactose esters when reacts with the free hydroxy groups of the lactose present in Hydroxy Propyl Cellulose (HPC), Low Hydroxy Propyl Cellulose (Hyprolose or LHPC-31) and Micro Crystalline Cellulose (MCC) (Figure 32) [53] . LHPC-31 is low-substituted Hydroxypropyl ether of cellulose. It is non-ionic, less reactive to active ingredients, insoluble in water and alcohol but swells in water by holding water molecules around hydroxy propyl groups that are distributed on the cellulose backbone (Figure 33). Based on the Low Molar substitution and in solubility of LHPC-31 in water, the formation of Cetirizine ester with LHPC will be very non-significant levels when compared to HPC. The molar substitution value and number of glucose units attached to the LHPC vs. HPC makes less reactive towards Cetirizine.
7.3. Ceronapril Degradation in Presence of Mannitol and Dibasic Calcium Phosphate Dihydrate (DCP)
Serajuddin et al. [44] reported that Ceronapril is an ACE inhibitor with a primary amine group interaction with mannitol and with dibasic calcium phosphate form the degradation products (Figure 34). Based on the drug excipient compatibility studies lactose and mannitol were excluded from the formulation.

Figure 31. Fluoxetine amadori rearrangement with lactose.
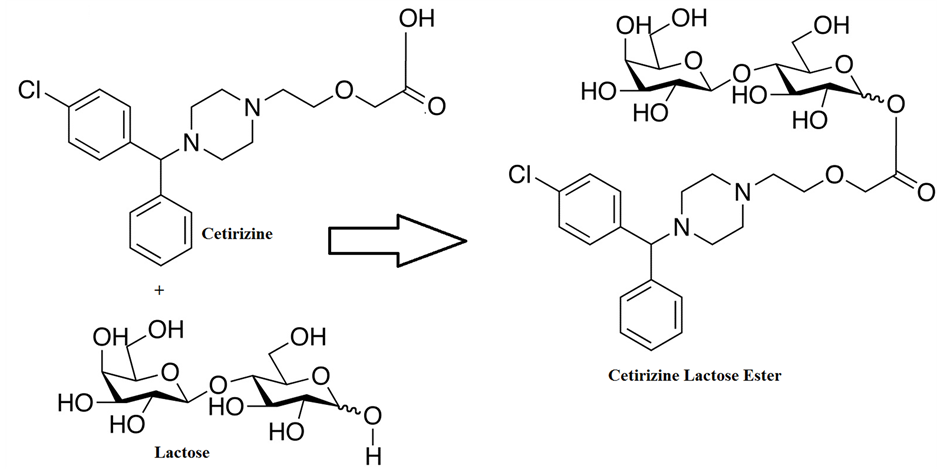
Figure 32. Cetirizine reaction with HPC.
7.4. Olanzapine Stress Studies with Lactose Monohydrate (LM) and Lactose Anhydrous (LA)
Natasa et al. reported the impact of excipients on the stability of olanzapine and confirmed the formation of 2- methyl-5,10-dihydro-4H-thieno[2,3-b][1,5]benzodiazepine-4-one and 2-methyl-4-(4-methyl-4-oxidopiperazin1- yl)-10H-thieno [2,3-b][1,5]benzodiazepine, olanzapine N-oxide (Figure 35). Olanzapine is sensitive to temperature and moisture. In samples protected from moisture, the increase in concentration of impurity was shown to be highly temperature dependent and the degradation followed zero-order kinetics. The physical state of water in drug-excipient mixtures determines its role in drug−excipient interactions. Mannitol, LM and LA anhydrous are crystalline excipients. Upon exposure to humidity, water is adsorbed onto their surfaces and then attached to the polar groups located on them. It could be postulated that degradation of olanzapine in the presence of moisture is excipient mediated. In olanzapine-mannitol mixtures, the molecular mobility is small. In addition, differences found between LM and LA anhydrous could be explained by the fact that water activity in the anhydrous form is higher because it tends to form a hydrate that is thermodynamically more stable. LM contains about 5% (w/w)

Figure 33. Molar substitution of hydroxyl groups in LHPC-31.

Figure 34. Ceronapril degradation due to lactose and DCP.

Figure 35. Olanzapine degradation in presence of lactose.
of water which is incorporated in its crystal structure and therefore, not available for chemical reactions. On the other hand, the LA anhydrous contains only 1% (w/w). It could strongly adsorb water owing to the fact that formation of Lactose Monohydrate is a favored process. Therefore, anhydrous LA uses absorbed water to form the monohydrate thereby decreasing the amount of active water that can contribute towards the degradation of olanzapine. In the course of this study, it was discovered that the choice of tablet filler excipient plays an important role in the formulation of olanzapine film-coated tablets. Degradation of olanzapine film coated tablets was governed by temperature and moisture [54] [55] .
7.5. Condensation Products between Lactose and Hydrochlorothiazide
Paul A. Harmon et al. reported the trace levels of condensation products between lactose and the amine containing Hydrochlorothiazide (HCTZ) formed when a mixture of the two solids containing 30% weight water is heated at 60˚C for 2 weeks [56] [57] . The two most abundant condensation products were characterized (Figure 36) by LCMS. Under the relatively mild conditions of formation, the amine-lactose reaction products are limited to those involving the elimination of only a single molecule or water rather than the multiple water eliminations associated with later stages of Malliard reaction.
7.6. Pregabalin Degradation Products in Presence of Lactose
Michael J. Lovadahl et al. reported that Pregabalin in the presence of lactose undergoes a Maillard reaction over time to form conjugates with lactose in formulated product. Seven of these conjugates, which contain the lactam form of the pregabalin moiety, were generated in milligram to gram quantities by heating pregabalin in the presence of lactose in solution. There were four degradation products (Figure 37) observed with glucose and through B lactose with Amadori rearrangement [58] .
7.7. Acyclovir Degradation Products in Presence of Lactose
Faran et al. reported that low levels of the Acyclovir-lactose condensation product was confirmed by liquid chromatography-tandem mass spectrometry, indicating the formation of a covalent link between Acyclovir and

Figure 36. Hydrochlorothiazide interaction with lactose.
lactose with the loss of a single water molecule (Figure 38). Maillard reaction of acyclovir and lactose in a solid-state formulation is less possible than in the aqueous phase. Thus, there may be some other important factors such as restricted mobility affecting the Maillard reaction in real solid dosage forms. It is also advisable to avoid wet conditions in the formulation process and/or storage of acyclovir solid-state dosage forms containing lactose [59] .
7.8. Degradation of Desloratadine in Presence of Starch and Maltose
Xin Yu et al. reported that Desloratidine reacts with the open chain of maltose to form the maltose adduct through Amadori rearrangement (Figure 39, Figure 40). In one of the formulation, Desloratadine reacted with the isomer of acetylformoin, which is present in one of the starch excipient as an impurity. Xin Yu et al. performed significant efforts in identifying in characterizing this impurity by LCMS and NMR [60] .
7.9. Amlodipine Besylate and Lactose Adduct Formation
Omari et al. found that Amlodipine besylate is unstable in presence of lactose, magnesium stearate, and water in the solid-state formulations. The major degradant observed is amlodipine besylate glycosyl (Figure 41). This is also a conformity with the Maillard reaction between primary amine and lactose [61] .
8. Peroxides
Peroxides are the reactive materials present in a number of excipients. These organic peroxides act as oxidizers and interact with the drugs directly to form the degradants. They thermally generate radicals, which can initiate

Figure 37. Pregabalin-lactose degradation products.
the chain process. They were present in the excipients due to the manufacturing process or due to oxidative instability of the excipients. Majority of the polymeric excipients contains peroxides. Because in the process of manufacturing polymeric materials, it is very difficult to remove peroxides. Polyethylene glycols, oxides, stearates and ethylene oxide based materials contain peroxides. The molecules with sensitive organic labile groups should be taken care in selecting the excipients containing peroxides [62] -[70] .
8.1. Peroxide Impurities in Povidones-Formation of N-Oxide in Raloxifene Hydrochloride
Kerry et al. reported that N oxide derivative of Raloxefine is formed due to the residual peroxides present in the excipient of povidone and crospovidone in the finished product. An oxidative degradation product in a tablet formulation of Raloxefine hydrochloride was identified as a N-oxide derivative (Figure 42). On the basis of applying the ICH guideline for acceptable levels of this degradation product, the data generated in this study can be used to set internal specifications on the level of peroxides for purchased lots of povidone and crospovidone. However, it is also known that in the presence of atmospheric oxygen, the peroxide content of povidone and crospovidone can increase The results in this study suggest that the peroxides in this tablet formulation are not continuing to form appreciably upon aging (from autoxidation), but rather are present in the excipients as process impurities that are consumed by reaction with R-HCl and also possibly by decomposition [62] [63] .
8.2. Peroxide Degradation-Fluphenazine Deaconate
Fluphenazine Deaconate forms several N-oxides due to the Oxidation of the phenothiazine group. This could be

Figure 38. Acyclovir degradation with lactose in wet conditions.

Figure 39. Desloratadine reaction with maltose.

Figure 40. Desloratadine reaction with acetylformoin isomer.

Figure 41. Amlodipine lactose adduct formation.
due to the peroxide present in the excipients or oxidation of the active drug after accelerated conditions. UPLC method developed and peroxide degradation of the impurities were identified by LC-MS/MS (Figures 43-45). The N- and S-oxides produced may be formed by ageing process, which is often the result of photo oxidation. Oxidation of the phenothiazine group of drugs is a well-documented feature of these compounds. The N- and S-oxides produced may be formed by ageing process, which is often the result of photo oxidation [64] . The

Figure 42. N-oxide formation of raloxefine hydrochloride.

Figure 43. Fluphenazine deaconate N- and S-oxidation sites.

Figure 44. UPLC chromatogram of fluphenazine deaconate N- and S-oxidation by peroxide degradation.



Figure 45. MS1 Chromatogram of fluphenazene deaconate N- and S-oxidation by peroxide degradation.
fluphenazine molecule contains four potential oxidation sites, thus fifteen o-tidesare theoretically possible (Table 3). Peroxide content and oxidation due to exposure shall be avoided while manufacturing this product. UPLC-MS/MS with positive ion mode with ammonium acetate buffer with Acquity UPLC column was employed for the identification of these impurities.
9. Controlling Excipient Related Impurities
Excipients were associated with peroxides, which will cause the oxidative degradation of drugs in their formulations. Control of initial peroxide concentration was recommended for drug product stabilization. The compendial limits on peroxide level may or may not be sufficient to assure satisfactory product stability. For example, European Pharmacopoeia does not allow more than 400 ppm of peroxides in crospovidone. Although the crospovidone monograph in USP/NF still has no official limit, some excipient vendors provide “peroxide free”
Table 3. Possible N and S oxides of fluphenazine and fluphenazine deaconate due to oxidation.
crospovidone, which is produced to meet the PhEur or tighter limit. This is because peroxide levels often increase in excipients during storage, which was observed for povidone and PEG. The most practical approach is to conduct a stability study using excipient lots representing a range of concentrations or spiking one lot of excipient with different amount of peroxide. The control strategy for peroxides starts with developing a sensitive analytical method not only to monitor their initial levels in the excipients, but also in the dosage forms. Using a sensitive method, excipients or excipient lots with low HPO scan be selected. The analytical method can be further leveraged to select a manufacturing process that minimizes HPO levels to improve product stability [65] [66] .
Formaldehydes/benzaldehydes present in the excipients react with amine containing compounds to form degradants. Compounds containing amine group should be taken care for the Schiff base formations. Purity and specifications of the excipients should be taken care. Drug excipient compatibility studies give the insight to give the probable degradants. Sensitive and robust analytical methods should be developed to identify the formed impurities in the drug products. Packaging modifications that can improve drug product stability include the use of bottles, which minimize permeation of oxygen from the atmosphere, or canisters, which can absorb oxygen. Reducing moisture in the packages can also play a critical role. This can be achieved by appropriate selection of packaging components including types of bottles, headspace, and number of desiccants inside the bottles. Instead of making these decisions on trial and error basis, they should be made using sound scientific rationale [67] -[71] .
10. Conclusion
Structure and reactivity of the molecules and excipients shall be considered in predicting the drug excipient interactions. Metal ion, peroxide content and manufacturing details should be checked and ensured before finalizing the excipients. This article gives an idea about how the drugs will react with excipients during the storage conditions and its prevention in establishing the good formulation. Analytical development should ensure any unknown peaks formed during the drug excipient compatibility studies. Thorough monitoring of unknown impurities and its origin during drug development process reduces the delay in the product filings.
Acknowledgements
The authors wish to thank the management of Novel Laboratories INC for providing the infrastructure for the supporting of this research work. Cooperation from colleagues Quality Control and Analytical Research & Development of Novel Laboratories is appreciated.
Cite this paper
Kishore KumarHotha,SwapanRoychowdhury,VeerappanSubramanian, (2016) Drug-Excipient Interactions: Case Studies and Overview of Drug Degradation Pathways. American Journal of Analytical Chemistry,07,107-140. doi: 10.4236/ajac.2016.71011
References
- 1. Carstensen, J.T., Osadca, M. and Rubin, S.H. (1969) Degradation Mechanisms for Water-Soluble Drugs in Solid Dosage Forms. Journal of Pharmaceutical Sciences, 58, 549-553.
http://dx.doi.org/10.1002/jps.2600580504 - 2. Crowley, P. and Martini, LG. (2001) Drug-Excipient Interactions. Pharmaceutical Technology, 13, 26-34.
- 3. Katdare, A. and Chaubal, M. (2006) Excipient Development for Pharmaceutical, Biotechnology, and Drug Delivery Systems. Informa Healthcare USA, Inc., 100.
http://dx.doi.org/10.1201/9781420004137 - 4. Akers, M.J. (2002) Excipient-Drug Interactions in Parenteral Formulations. Journal of Pharmaceutical Sciences, 91, 2283-300.
http://dx.doi.org/10.1002/jps.10154 - 5. Pan, C., Liu, F. and Motto, M. (2011) Identification of Pharmaceutical Impurities in Formulated Dosage Forms. Journal of Pharmaceutical Sciences, 100, 1228-1259.
http://dx.doi.org/10.1002/jps.22376 - 6. Bharate, S. and Bharate, A. (2010) Incompatibilities of Pharmaceutical Excipients with Active Pharmaceutical Ingredients: A Comprehensive Review. Journal of Excipients and Food Chemicals, 1, 1-22.
- 7. Antipas, A.S. and Landis, M.S. (2005) Solid-State Excipient Compatibility Testing. Drugs and the Pharmaceutical Sciences, 419-458.
http://dx.doi.org/10.1201/9780849359194.ch13 - 8. Wu, Y., Levons, J., Narang, A.S., Raghavan, K. and Rao, V.M. (2011) Reactive Impurities in Excipients: Profiling, Identification and Mitigation of Drug-Excipient Incompatibility. AAPS PharmSciTech, 12, 1248-1263.
http://dx.doi.org/10.1208/s12249-011-9677-z - 9. Narang, A.S., Desai, D. and Badawy, S. (2012) Impact of Excipient Interactions on Solid Dosage Form Stability. Pharmaceutical Research, 29, 2660-2683.
http://dx.doi.org/10.1007/s11095-012-0782-9 - 10. Li, M. (2012) Organic Chemistry of Drug Degradation. Royal Society Chemistry Publishing, Cambridge.
- 11. Serajuddin, A.T., Thakur, A.B., Ghoshal, R.N., Fakes, M.G., Ranadive, S.A., Morris, K.R. and Varia, S.A. (1999) Selection of Solid Dosage Form Composition through Drug-Excipient Compatibility Testing. Journal of Pharmaceutical Sciences, 88, 696-704.
http://dx.doi.org/10.1021/js980434g - 12. Narang, A.S., Desai, D. and Badawy, S. (2012) Impact of Excipient Interactions on Solid Dosage Form Stability. Pharmaceutical Research, 29, 2660-2683.
http://dx.doi.org/10.1007/s11095-012-0782-9 - 13. Hotha, K.K., Reddy, S.P.K., Raju, V.K. and Ravindranath, L.K. (2013) Forced Degradation Studies: Practical Approach: Overview of Regulatory Guidance and Literature for the Drug Products and Drug Substances. International Research Journal of Pharmacy, 4, 78-85.
http://dx.doi.org/10.7897/2230-8407.04517 - 14. ICH Q2 (R1) (2005) Validation of Analytical Procedures: Text and Methodology.
- 15. ICH Q1A (R2) (2000) Stability Testing of New Drug Substances and Products.
- 16. ICH Guideline Q3A (R) (2002) Impurities in New Drug Substances.
- 17. Baertschi, S.W., Alsante, K.M. and Reed, R.A. (2011) Pharmaceutical Stress Testing: Predicting Drug Degradation. 2nd Edition, Informa Health Care, 210.
- 18. Hotha, K.K., Patel, T., Roychowdhury, S. and Subramanian, V. (2015) Identification, Synthesis, and Characterization of Unknown Impurity in the Famotidine Powder for Oral Suspension Due to Excipient Interaction by UPLC-MS/MS and NMR. Journal of Liquid Chromatography & Related Technologies, 38, 977-985.
http://dx.doi.org/10.1080/10826076.2014.999201 - 19. Abend, A.M., Chung, L., Bibart, R.T., Brooks, M. and McCollum, D.G. (2004) Concerning the Stability of Benzyl Alcohol: Formation of Benzaldehyde Dibenzyl Acetal under Aerobic Conditions. Journal of Pharmaceutical and Biomedical Analysis, 34, 957-962.
http://dx.doi.org/10.1016/j.jpba.2003.11.007 - 20. Sudareva, N.N. and Chubarova, E.V. (2006) Time-Dependent Conversion of Benzyl Alcohol to Benzaldehyde and Benzoic Acid in Aqueous Solutions. Journal of Pharmaceutical and Biomedical Analysis, 41, 1380-1385.
http://dx.doi.org/10.1016/j.jpba.2006.02.028 - 21. Urakami, K., Kobayashi, C., Miyazaki, Y., Nishijima, K., Yoshimura, Y. and Hashimoto, K. (2000) Degradation Products Generated by Sonication of Benzyl Alcohol, a Sample Preparation Solvent for the Determination of Residual Solvents in Pharmaceutical Bulks, on Capillary Gas Chromatography. Chemical and Pharmaceutical Bulletin, 48, 1299-1303.
http://dx.doi.org/10.1248/cpb.48.1299 - 22. Nassar, M.N., Nesarikar, V.N., Lozano, R., Parker, W.L., Huang, Y., Palaniswamy, V., Xu, W. and Khaselev, N. (2004) Influence of Formaldehyde Impurity in Polysorbate 80 and PEG-300 on the Stability of a Parenteral Formulation of BMS-204352: Identification and Control of the Degradation Product. Pharmaceutical Development and Technology, 9, 189-195.
http://dx.doi.org/10.1081/PDT-120030249 - 23. Bindra, D.S., Williams, T.D. and Stella, V.J. (1994) Degradation of O6-Benzylquanine in Aqueous Polyethylene Glycol (PEG 400) Solutions: Concerns with Formaldehyde in PEG 400. Pharmaceutical Research, 11, 1060-1064.
http://dx.doi.org/10.1023/A:1018903908385 - 24. Bergh, M., Magnusson, K., Nilsson, J.L.G. and Karlberg, A.-T. (1998) Formation of Formaldehyde and Peroxides by Air Oxidation of High Purity Polyoxyethylene Surfactants. Contact Dermatitis, 39, 14-20.
http://dx.doi.org/10.1111/j.1600-0536.1998.tb05805.x - 25. Dousa, M., Gibala, P., Havlícek, J., Placek, L., Tkadlecová, M. and Brichác, J. (2011) Drug-Excipient Compatibility Testing-Identification and Characterization of Degradation Products of Phenylephrine in Several Pharmaceutical Formulations against the Common Cold. Journal of Pharmaceutical and Biomedical Analysis, 55, 949-956.
http://dx.doi.org/10.1016/j.jpba.2011.03.027 - 26. Dubost, D.C., Kaufman, M.J., Zimmerman, J.A., Bogusky, M.J., Coddington, A.B. and Pitzenberger, S.M. (1996) Characterization of a Solid State Reaction Product from a Lyophilized Formulation of a Cyclic Heptapeptide. A Novel Example of an Excipient-Induced Oxidation. Pharmaceutical Research, 13, 1811-1814.
http://dx.doi.org/10.1023/A:1016024923002 - 27. Meireles, B.A. and Pereira, V.L.P. (2013) Synthesis of Bio-Additives: Transesterification of Ethyl Acetate with Glycerol Using Homogeneous or Heterogeneous Acid Catalysts. Journal of the Brazilian Chemical Society, 24, 17-25.
http://dx.doi.org/10.1590/S0103-50532013000100004 - 28. Ma, M., DiLollo, A., Mercuri, R., Lee, T., Bundang, M. and Kwong, E. (2002) HPLC and LC-MS Studies of the Transesterification Reaction of Methylparaben with Twelve 3- to 6-carbon Sugar Alcohols and Propylene Glycol and the Isomerization of the Reaction Products by Acyl Migration. Journal of Chromatographic Science. 40, 170-177.
http://dx.doi.org/10.1093/chromsci/40.3.170 - 29. Thompson, M.J., Fell, A.F., Clark, B.J. and Robinson, M.L. (1993) LC Studies on the Potential Interaction of Paraben Preservatives with Sorbitol and Glycerol. Journal of Pharmaceutical and Biomedical Analysis, 11, 233-240.
http://dx.doi.org/10.1016/0731-7085(93)80202-C - 30. Hensel, A., Leisenheimer, S., Muller, A., Busker, E., Wolf-Heuss, E. and Engel, J. (1995) Transesterification Reactions of Parabens(alkyl 4-Hydroxy Benzoates) with Polyols in Aqueous Solutions. Journal of Pharmaceutical Sciences, 84, 115-118.
http://dx.doi.org/10.1002/jps.2600840126 - 31. Larsen, J., Staerk, D., Cornett, C., Hansen, S.H. and Jaroszewski, J.W. (2009) Identification of Reaction Products between Drug Substances and Excipients by HPLC-SPE-NMR: Ester and Amide Formation between Citric Acid and 5-Aminosalicylic Acid. Journal of Pharmaceutical and Biomedical Analysis, 49, 839-842.
http://dx.doi.org/10.1016/j.jpba.2008.12.028 - 32. Schou-Pedersen, A.M., Cornett, C., Nyberg, N., Ostergaard, J. and Hansen, S.H. (2015) Structure Elucidation and Quantification of Impurities Formed between 6-Aminocaproic Acid and the Excipients Citric Acid and Sorbitol in an Oral Solution Using High-Resolution Mass Spectrometry and Nuclear Magnetic Resonance Spectroscopy. Journal of Pharmaceutical and Biomedical Analysis, 107, 333-340.
http://dx.doi.org/10.1016/j.jpba.2015.01.022 - 33. Yu, H., Cornett, C., Larsen, J. and Hansen, S.H. (2010) Reaction between Drug Substances and Pharmaceutical Excipients: Formation of Esters between Cetirizine and Polyols. Journal of Pharmaceutical and Biomedical Analysis, 53, 745-750.
http://dx.doi.org/10.1016/j.jpba.2010.05.001 - 34. Hotha, K.K., Roychowdhury, S. and Subramanian, V. (2015) Drug-Excipient Interaction of Methylphenidate with Glycerin in Methylphenidate Oral Solution and Identification of Its Transesterification Products by UPLC-MS/MS. American Journal of Analytical Chemistry (Communicated).
- 35. Nebel, B., Mittelbach, M. and Uray, G. (2008) Determination of the Composition of Acetyl Glycerol Mixtures by 1H NMR Followed by GC Investigation. Analytical Chemistry, 80, 8712-8716.
http://dx.doi.org/10.1021/ac800706s - 36. Sutter, M., Dayoub, W., Métay, E., Raou, Y. and Lemaire, M. (2013) 1-O-Alkyl (di) Glycerol Ethers Synthesis from Methyl Esters and Triglycerides by Two Pathways: Catalytic Reductive Alkylation and Transesterification/Reduction. Green Chemistry, 15, 786-797.
http://dx.doi.org/10.1039/c3gc36907b - 37. Larsen, J., Cornett, C., Jaroszewski, J.W. and Hansen, S.H. (2009) Reaction between Drug Substances and Pharmaceutical Excipients: Formation of Citric Acid Esters and Amides of Carvedilol in the Solid State. Journal of Pharmaceutical and Biomedical Analysis, 49, 11-17.
http://dx.doi.org/10.1016/j.jpba.2008.09.039 - 38. Mostafavi, A., Abedi, G., Jamshidi, A., Afzali, D. and Talebi, M. (2009) Development and Validation of a HPLC Method for the Determination of Buprenorphine Hydrochloride, Naloxone Hydrochloride and Noroxymorphone in a Tablet Formulation. Talanta, 77, 1415-1419.
http://dx.doi.org/10.1016/j.talanta.2008.09.024 - 39. Cao, X., Jia, F. and Tao, Q.F. (2013) Characterization of Impurity-I in Dexamethasone Sodium Phosphate Injection. Chinese Journal of Pharmaceutical Analysis, 33, 1016-1020.
- 40. Smith, G.B., Weinstock, L.M., Roberts Jr, F.E., Brenner, G.S., Hoinowski, A.M., Arison, B.H. and Douglas, A.W. (1972) Kinetics of Equilibration of Bisulfite and Dexamethasone-21-Phosphate in Aqueous Solution. Journal of Pharmaceutical Sciences, 61, 708-716.
http://dx.doi.org/10.1002/jps.2600610508 - 41. Rozman Peterka, T., Grahek, R., Hren, J., Bastarda, A., Bergles, J. and Urleb, U. (2015) Solid State Compatibility Study and Characterization of a Novel Degradation Product of Tacrolimus in Formulation. Journal of Pharmaceutical and Biomedical Analysis, 110, 67-75.
http://dx.doi.org/10.1016/j.jpba.2015.02.047 - 42. Rangaiah, K.V., Chattaraj, S.V. and Das, S.K. (1994) Effects of Process Variables and Excipients on Tabletting Parameters of Norfloxacin Tablets. Drug Development and Industrial Pharmacy, 20, 2175-2182.
http://dx.doi.org/10.3109/03639049409050231 - 43. Oliveira, P.R., Bernardi, L.S., Murakami, F.S., Mendes, C. and Silva, M.A.S. (2009) Thermal Characterization and Compatibility Studies of Norfloxacin for Development of Extended Release Tablets. Journal of Thermal Analysis and Calorimetry, 91, 741-745.
http://dx.doi.org/10.1007/s10973-009-0347-2 - 44. Serajuddin, A.T., Thakur, A.B., Ghoshal, R.N., Fakes, M.G., Ranadive, S.A., Morris, K.R. and Varia, S.A. (1999) Selection of Solid Dosage Form Composition through Drug-Excipient Compatibility Testing. Journal of Pharmaceutical Science, 88, 696-704.
http://dx.doi.org/10.1021/js980434g - 45. Jansen, P.J., Oren, P.L., Kemp, C.A., Maple, S.R. and Baertschi, S.W. (1998) Characterization of Impurities Formed by Interaction of Duloxetine HCl with Enteric Polymers Hydroxypropyl Methylcellulose Acetate Succinate and Hydroxypropyl Methylcellulose Phthalate. Journal of Pharmaceutical Science, 87, 81-85.
http://dx.doi.org/10.1021/js970133r - 46. Wirth, D.D., Baertschi, S.W., Johnson, R.A., Maple, S.R., Miller, M.S. and Hallenback, D.K. (1998) Maillard Reaction of Lactose and Fluoxetine Hydrochloride, A Secondary Amine. Journal of Pharmaceutical Sciences, 87, 31-39.
http://dx.doi.org/10.1021/js9702067 - 47. Yaylayan, V.A. and Huyghues-Despointes, A. (1994) Chemistry of Amadori Rearrangement Products: Analysis, Synthesis, Kinetics, Reactions, and Spectroscopic Properties. Critical Reviews in Food Science and Nutrition, 34, 321-369.
http://dx.doi.org/10.1080/10408399409527667 - 48. Ellis, G.P. (1959) The Maillard Reaction. Advances in Carbohydrate Chemistry, 14, 63-134.
http://dx.doi.org/10.1016/s0096-5332(08)60223-4 - 49. Devani, M.B., Shishoo, C.J., Doshi, K.J. and Patel, H.B. (1985) Kinetic Studies of the Interaction between Isoniazid and Reducing Sugars. Journal of Pharmaceutical Sciences, 74, 427-432.
http://dx.doi.org/10.1002/jps.2600740412 - 50. Hodge, J.E. (1955) The Amadori Rearrangement. Advances in Carbohydrate Chemistry, 10, 169-205.
http://dx.doi.org/10.1016/s0096-5332(08)60392-6 - 51. Colaco, C.A.L.S., Collett, M. and Roser, B.J. (1996) Pharmaceutical Formulation Instability and the Maillard Reaction. ChemOggi, 14, 32-37.
- 52. Qiu, Z., Stowell, J.G., Cao, W, Morris, K.R., Byrn, S.R. and Carvajal, M.T. (2005) Effect of Milling and Compression on the Solid-State Maillard Reaction. Journal of Pharmaceutical Science, 94, 2568-2580.
http://dx.doi.org/10.1002/jps.20448 - 53. Sharma, N., Rao, S.S., Ullas, G., Kumar, N.D.A. and Reddy, A.M. (2011) Optimization and Validation of Reverse Phase Liquid Chromatographic Method for Estimation of Cetirizine Mannitol Ester Impurity in Cetirizine Hydrochloride Chewable Tablet. Journal of Liquid Chromatography & Related Technologies, 34, 2157-2168.
http://dx.doi.org/10.1080/10826076.2011.585485 - 54. Djordjevic Filijovic, N., Antonijevic, M.D., Pavlovic, A., Vuckovic, I., Nikolic, K. and Agbaba, D. (2015) The stress Stability of Olanzapine: Studies of Interactions with Excipients in Solid State Pharmaceutical Formulations. Drug Development and Industrial Pharmacy, 41, 502-514.
http://dx.doi.org/10.3109/03639045.2014.884114 - 55. Hiriyanna, S.G., Basavaiah, K., Goud, P.S.K., Dhayanithi, V., Raju, K. and Pati, H.N. (2008) Identification and Characterization of Olanzapine Degradation Products under Oxidative Stress Conditions. Acta Chromatogrphica, 20, 81-93.
http://dx.doi.org/10.1556/AChrom.20.2008.1.7 - 56. Harmon, P.A., Yin, W., Bowen, W.E., Tyrrell, R.J. and Reed, R.A. (2000) Liquid Chromatography-Mass Spectrometry and Proton Nuclear Magnetic Resonance Characterization of Trace Level Condensation Products Formed between Lactose and the Amine-Containing Diuretic Hydrochlorothiazide. Journal of Pharmaceutical Science, 89, 920-929.
http://dx.doi.org/10.1002/1520-6017(200007)89:7<920::AID-JPS9>3.0.CO;2-W - 57. Deppeler, H.P. (1981) Hydrochlorothiazide. Analytical Profiles Drug Substances, 10, 405-441.
http://dx.doi.org/10.1016/S0099-5428(08)60646-2 - 58. Lovdahl, M.J., Hurley, T.R., Tobias, B. and Priebe, S.R. (2002) Synthesis and Characterization of Pregabalin Lactose Conjugate Degradation Products. Journal of Pharmaceutical and Biomedical Analysis, 28, 917-924.
http://dx.doi.org/10.1016/S0731-7085(01)00710-5 - 59. Monajjemzadeh, F., Hassanzadeh, D., Valizadeh, H., Siahi-Shadbad, M.R., Mojarrad, J.S., Robertson, T.A. and Roberts, M.S. (2009) Compatibility Studies of Acyclovir and Lactose in Physical Mixtures and Commercial Tablets. European Journal of Pharmaceutics and Biopharmaceutics, 73, 404-413.
http://dx.doi.org/10.1016/j.ejpb.2009.06.012 - 60. Yu, X., Buevich, A.V., Li, M., Wang, X. and Rustum, A.M. (2013) A Compatibility Study of a Secondary Amine Active Pharmaceutical Ingredient with Starch: Identification of a Novel Degradant Formed between Desloratadine and a Starch Impurity Using LC-MS (n) and NMR Spectroscopy. Journal of Pharmaceutical Sciences, 102, 717-731.
http://dx.doi.org/10.1002/jps.23416 - 61. Abdoh, A., Al-Omari, M.M., Badwan, A.A. and Jaber, A.M. (2004) Amlodipine Besylate-Excipients Interaction in Solid Dosage Form. Pharmaceutical Development and Technology, 9, 15-24.
http://dx.doi.org/10.1081/PDT-120027414 - 62. Hartauer, K.J., Arbuthnot, G.N., Baertschi, S.W., Johnson, R.A., Luke, W.D., Pearson, N.G., Rickard, E.C., Tingle, C.A., Tsang, P.K. and Wiens, R.E. (2000) Influence of Peroxide Impurities in Povidone and Crospovidone on the Stability of Raloxifene Hydrochloride in Tablets: Identification and Control of an Oxidative Degradation Product. Pharmaceutical Development Technology, 5, 303-310.
http://dx.doi.org/10.1081/PDT-100100545 - 63. Yarkala, S., Amaravadi, S., Rao, V.U., Vijaykumar, V., Navalgund, S.G. and Jagdish, B. (2009) Role of Excipients on N-Oxide Raloxifene Generation from Raloxifene-Excipients Binary Mixtures. Chemical and Pharmaceutical Bulletin, 57, 1174-1177.
http://dx.doi.org/10.1248/cpb.57.1174 - 64. Heyes, W.F., Salmon, J.R. and Marlow, W. (1980) High-Performance Liquid Chromatography Separation of the N- and S-Oxides of Fluphenazine and Fluphenazine Decanoate. Journal of Chromatography, 194, 416-420.
http://dx.doi.org/10.1016/S0021-9673(00)81433-8 - 65. Wasylaschuk, W.R., Harmon, P.A., Wagner, G., Harman, A.B., Templeton, A.C., Xu, H. and Reed, R.A. (2007) Evaluation of Hydro Peroxides in Common Pharmaceutical Excipients. Journal of Pharmaceutical Sciences, 96, 106-116.
http://dx.doi.org/10.1002/jps.20726 - 66. Buhler, V. (2008) Kollidon: Polyvinylpyrrolidone Excipients for the Pharmaceuticals. Springer-Verlag Berlin Heidelberg, Germany, 118.
- 67. Narang, A.S., Rao, V.M. and Desai, D.S. (2012) Effect of Antioxidants and Silicates on Peroxides in Povidone. Journal of Pharmaceutical Science, 101, 127-139.
http://dx.doi.org/10.1002/jps.22729 - 68. Kumar, V. and Kalonia Devendra, S. (2006) Removal of Peroxides in Polyethylene Glycols by Vacuum Drying: Implications in the Stability of Biotech and Pharmaceutical Formulations. AAPS PharmSciTech, 7, 62.
http://dx.doi.org/10.1208/pt070362 - 69. Tuleu, C., Khela, M.K., Evans, D.F., Jones, B.E., Nagata, S. and Basit, A.W. (2007) A Scintigraphic Investigation of the disintegration Behavior of Capsules in Fasting Subjects: A Comparison of Hypromellose Capsules Containing Carrageenan as a Gelling Agent and Standard Gelatin Capsules. European Journal of Pharmaceutical Sciences, 30, 251-255.
http://dx.doi.org/10.1016/j.ejps.2006.11.008 - 70. Sims, J.L., Carreira, J.A., Carrier, D.J., Crabtree, S.R., Easton, L., Hancock, S.A. and Simcox, C.E. (2003) A New Approach to Accelerated Drug-Excipient Compatibility Testing. A New Approach to Accelerated Drug-Excipient Compatibility Testing. Pharmaceutical Development and Technology, 8, 119-126.
http://dx.doi.org/10.1081/PDT-120018476 - 71. Hotha, K.K., Patel, T., Roychowdhury, S. and Subramanian, V. (2014) Development of Better-Quality Assay Method for the Citric Acid and Sodium Citrate in Ophthalmic/Oral Solutions and Their Application to Deformulation Studies. American Journal of Analytical Chemistry, 5, 1249-1260.
http://dx.doi.org/10.4236/ajac.2014.517131
NOTES

*Corresponding author.