Open Journal of Internal Medicine
Vol.2 No.2(2012), Article ID:19619,9 pages DOI:10.4236/ojim.2012.22020
The many faces of Crohn’s Disease: Latest concepts in etiology
![]()
Centre for Digestive Diseases, Five Dock, Australia
Email: jordana.campbell@cdd.com.au
Received 1 November 2011; revised 10 February 2012; accepted 6 March 2012
Keywords: Crohn’s Disease; etiologies; infection; classification; Syndrome; Mycobacterium avium ss paratuberculosis
ABSTRACT
The notion that Crohn’s Disease (CD) occurs as a result of an aberrant reaction to the commensal microbiota in genetically susceptible hosts is widely regarded by physicians and scientists as fact. Yet although it is undisputed that Crohn’s Disease is immune-mediated, an aberrant reaction to one’s own native flora is far from proven. The aim of the current review is to present a summary of the known infectious causes of Crohn’s Disease, whilst highlighting the limitations of using outdated methods to attempt to classify the disease as a single entity. We propose a re-classification of Crohn’s Disease, and suggest that the disease is best conceptualized as a syndrome, an “umbrella-like” term comprising a group of diseases with varying infective etiologies, which clinically, endoscopically and histologically are indistinguishable from CD.
1. INTRODUCTION
Crohn’s Disease was first described in 1904 by Polish surgeon Antoni Leśniowski [1], later by Dalziel [2] and ultimately by Crohn in 1932 [3]. Yet, after more than a century of research, the pathological processes are still poorly understood and numerous etiological questions remain. Current concepts suggest that Inflammatory Bowel Disease results from dysregulation of the mucosal immune system in genetically predisposed individuals leading to an exaggerated and ongoing activation of immunological responses to a person’s own normal microflora [4]. Previously most research has focused on autoimmune factors. Recently however, Mycobacterium avium paratuberculosis (MAP), the known pathogen responseble for the remarkably similar Johne’s Disease in livestock, has been detected in the gastrointestinal mucosa of up to 92% of CD patients [5]. With the publication of the fulfillment of Koch’s postulates [6] establishing MAP as a causal agent of CD, considerable doubt has been cast on the current “aberrant reaction” theory. Findings from the Genome Wide Association Studies implicating mutant genes involved in pathways involving the recognition and response to intracellular microbial infections, and the recognition that CD is associated with innate immune deficiency [7] add to these doubts.
These findings, strengthened by the knowledge that numerous pathogens are capable of reproducing the CD Syndrome, have led to a resurgence in theories implicating infectious etiologies of Crohn’s Disease and suggest that an etiological reevaluation is overdue. Further education, research and funding are sorely required to dramatically improve diagnostic sensitivity and specificity, particularly for MAP, as well as for other pathogens in CD. The diagnostic difficulties frequently encountered in clinical practice emphasize the need for comprehensive pathogen investigation to detect, where possible, the underlying infective cause of CD in the individual patient and move therapy away from treating inflammation alone to treating the causative agents as well as the ensuing inflammatory response.
2. VARIABLE PRESENTATIONS
The “Crohn’s Disease” label has led to much confusion in the gastroenterology field. It has provided experts with a preconceived notion of a unitary disease etiology, in spite of the fact that the name merely represents a descriptive state of the disease. This misinterpretation is perhaps best illustrated by Prantera [8] who surmised that the notion of a single pathogen etiology has been disproven given “the variable disease locations and severity” of the disease. Theoretically a single pathogen etiology for CD is possible, since it is well demonstrated that the same microbe can evoke markedly heterogenous disease manifestations. Mycobacterium tuberculosis [9] and Helicobacter pylori [10] are good examples of this phenomenon. However, with the abundant and compelling data demonstrating that various pathogens capable of causing a CD “syndrome”, and the associated diagnostic confusion, we counter that “Crohn’s Disease” is merely the clinical manifestation of any number of infective entities, some discernable and some currently indiscernible, which converge to result in a phenotypic expression of the CD Syndrome.
3. THE CROHN’S DISEASE SYNDROME (CDS)
To address the concept of Crohn’s Disease as an “umbrella-like” term or Syndrome rather than a single specific disease, it is instructive to look to an analogous inflammatory condition, caused not by one, but several pathogens, which produce an overlapping clinical and histological picture. The characteristic inflammatory presentation of pneumonia occurs as a result of infection with various bacteria, viruses, and, less often, fungi. Streptococcus pneumoniae is a common pathogen responsible for community-acquired bacterial pneumonia however other bacterial pathogens including Hemophilus influenzae, Klebsiella organisms, and Staphylococcus aureus cause a subset of pneumonia cases. These etiologic agents shift in hospital-acquired settings and are most commonly attributed to gram-negative bacilli such as E. coli, Proteus species, Klebsiella-Enterobacter, Pseudomonas, and Serratia. Mycoplasma pneumonia, Chlamydia pneumoniae, Legionella pneumophila, and Mycobacterium avium have also been reported as atypical pneumonias [11].
As in pneumonia, several pathogens can produce a syndrome clinically, colonoscopically and histologically indistinguishable from CD which, when treated correctly, can result in complete recovery. In some instances there is a clear, discernable pathogen whilst in others a pathogen has been much more difficult to identify. However due to the “single etiology” preconception, all pathogens causing a CD Syndr of CD rather than carefully considered as causal agents. Clinical experience to date, backed by the emerging discoveries of these pathogens in numerous CD cases suggests that, as in pneumonia, a single pathogen etiology of CD does not exist. Here, we present an overview of the infectious pathogens in the CD Syndrome and propose that they are not “mimickers” of the disease but actual causes of CD in subsets of patients. Furthermore, additional research is required to identify more of the infectious causes of the CD Syndrome.
4. INFECTIOUS ETIOLOGIES OF CROHN’s DISEASE SYNDROME
4.1. Mycobacterium avium ss paratuberculosis (MAP)
Although still controversial, there is a great deal of evidence implicating a causative role for Mycobacterium avium paratuberculosis in a subset of cases of Crohn’s Disease [6,12]. Hostetter’s work with Balb/C mice may provide insights into how MAP infection can cause the emergence of the Crohn’s Disease Syndrome in humans. He infected immunologically normal Balb/C mice with M. paratuberculosis and then administered controlled dosages of Dextran sodium sulfate in the drinking water and compared the resulting intestinal inflammation with controls only infected with MAP or only given DSS. MAP infection alone and DSS in the concentrations administered resulted in no or minimal inflammation. The two variables together produced extensive inflammation. His work helps unite the bodies of work implicating the enteric microbiota, genetic predisposition, dysfunctional autophagy and innate immune deficiency in the etiopathogenesis of CDS [13].
Recent technological advances, and the advent of PCR has allowed for more accurate identification of MAP via DNA amplification, identifying MAP DNA in up to 92% of CD tissues versus only 26% in healthy controls [5]. Another study of resected bowel tissues from 300 CD patients identified MAP DNA in 52% of CD patients, 2% of UC patients and 5% of controls [14], further supporting a similar study that identified MAP DNA in 6/7 (86%) resected tissue, and 4/20 (20%) biopsy specimens from CD patients, versus 2/36 (5.6%) control biopsy specimens [15]. These studies underline the high degree of MAP-positive results in CD, with higher detection rates obtained from processing of larger tissue specimens. However, an unexplained fierce resistance to the causal role of MAP in CD remains, completely at odds with the uncritical readiness to accept the current “aberrant reaction to normal colonic flora” theory [16]. Various arguments have been proffered by experts to defend preconceived ideas [16,17], with some critics claiming MAP to be a “mere bystander organism” that lodges innocuously in the intestinal mucosa of patients. This statement not only fails to explain the gross differences between MAP detection rates in CDS and controls [5,14] but also the clear pathogenicity of MAP [18].
Despite this resistance, a number of studies have been carried out reporting the success of anti-MAP therapy in CDS. A landmark study by Selby et al. (2007) using triple anti-MAP therapy in “Crohn’s Disease” reported significant remission rates of 66% at 16 weeks despite suboptimal dosing (underdosed by >30% - >50%), failure to report ITT data and incomplete capsule dissolution resulting in partial bioavailability of one drug [19,20]. Gui et al. (1997) reported on anti-MAP treatment in 52 patients with a combination of Rifabutin, Clarithromycin or Azithromycin for 6 - 35 months, documenting remission in 43 out of 52 (83%) patients [21]. A trial by Shafran et al. (2002) treating 29 CD patients with Rifabutin and Clarithromycin reported that after three months of treatment, 8 of the 29 patients (28%) were in clinical remission, 9 of the 29 patients (31%) experienced marked improvement, 8 of the 29 patients (28%) experienced minor improvement, and 4 of the 29 patients (14%) ceased medication due to intolerance [22]. Similarly, in a study by Borody et al. (2002) 67% patients achieved marked improvement with 50% colonoscopic and histological normality [23].
Despite clear evidence for the efficacy of properly chosen anti-MAP therapy in CDS trials, [24] some experts continue to request “well-designed anti-mycobacterial trials” to settle the causality dispute [25]. It should be noted that anti-MAP treatment at best currently only suppresses, but cannot yet cure this atypical Mycobacterium. To better understand anti-MAP therapy in CDS it is applicable to look to the treatment of Johne’s Disease in cattle—where there is no question as to MAP etiology. The results of which conclude that anti-MAP therapy “requires daily medication for long periods, and only effects remission and palliation of the disease instead of a definitive cure” [26]. MAP is a unique pathogen that needs to be viewed quite differently from the typical bacterial infections physicians are accustomed to. Notoriously difficult to treat, current therapy consists of multidrug regimes which can at best only suppress infection and halt disease progression. A cure is not achievable with antibiotics alone at this point. Like Johne’s Disease, anti-MAP therapy in CDS requires prolonged optimal dosing. Research and an innovative approach are needed to eradicate the dormant intracellular forms of MAP.
4.2. Mycobacterium tuberculosis (TB)
The long-recognised similarities between intestinal tuberculosis and CDS have led to this pathogen’s label as “one of the great mimics of Crohn’s disease” [27], lending further credence to the central role of mycobacteria in CDS. Both diseases are chronic, granulomatous and heterogenous in nature, with the differential diagnosis between the two diseases described as “one of the most difficult and challenging issues for the gastroenterologist, radiologist and pathologist” [28]. Like CDS, tuberculosis can affect any part of the gastrointestinal tract. One study reported that 8.5% of patients display upper gastrointestinal tract involvement, 33.8% of patients have small bowel involvement, 22.3% of patients exhibit large bowel involvement, and the remaining 44.6% of patients have extra-intestinal involvement [29]. Diagnosis of intestinal tuberculosis typically relies on the demonstration of caseating granulomas [30]. However, numerous studies report that only 19% - 25% of patients display caseating granulomas [31,32]. Further studies suggest the need for detection of acid fast bacilli and report identification in 35% to 60% of patients [33] and recent studies yield conflicting results, at times with no tubercle bacilli detected from cultured biopsy specimens [34]. Given that CDS is largely a disease of exclusion, the incredibly close resemblance between Mycobacterium tuberculosis and CDS not only confirms that mycobacteria are capable of causing the CD syndrome but also warn of the dangers of ignoring our clinical judgment to place our faith in pathology results alone.
Numerous case reports exist in the literature outlining the ability of Mycobacterium tuberculosis to present with a “Crohn’s Disease” appearance, with their shared clinical, colonoscopic and histological features often leading to misdiagnoses and mismanagement. Several cases have been summarised in Table 1.
4.3. Yersinia spp
Yersinia preferentially invades mucosal M cells in the gastrointestinal tract overlying Peyer’s patches to cause inflammation of the ileum, right colon and appendix, indistinguishable from “CD” [35]. As in Yersinia infection, the terminal ileum in CDS is chiefly affected, with Lockhart-Mummery and Morson establishing in 1960 that ulceration of terminal ileal lymphoid follicles and Peyer’s patches are the earliest microscopic changes of “CD” [36]. The association between Yersinia and CDS is well-recognized, with both possessing remarkably similar histological and colonoscopic features, as well as a strong affinity for the ileocecal region [37]. Numerous case reports also describe the development of “CD” following Yersinia infection (Table 2) [38-40]. Despite this, only one detailed study aimed at examining the presence of Yersinia DNA in CDS tissue has been reported [35]. The study by Lamps et al. (2003) evaluated resected specimens from 52 CDS patients with confirmed “CD” for the presence of pathogenic Yersinia DNA. A total of 17 of the 54 resections (31%) contained Yersinia DNA within lesional tissues in “CD” versus none of the 120 controls (0%). Mesenteric lymph nodes were also positive for Yersinia DNA in eight of these cases. In addition, pathogenic (invasive) Yersinia DNA was detected in the appendices of two patients who later went on to develop “Crohn’s Disease”. Serological studies have also revealed significantly higher titres of IgG antibodies to Yersinia enterocolitica (Y. enterocolitica) types 3 and 9 in a series of 60 patients with “CD” and 20 with ulcerative colitis compared with controls [41].
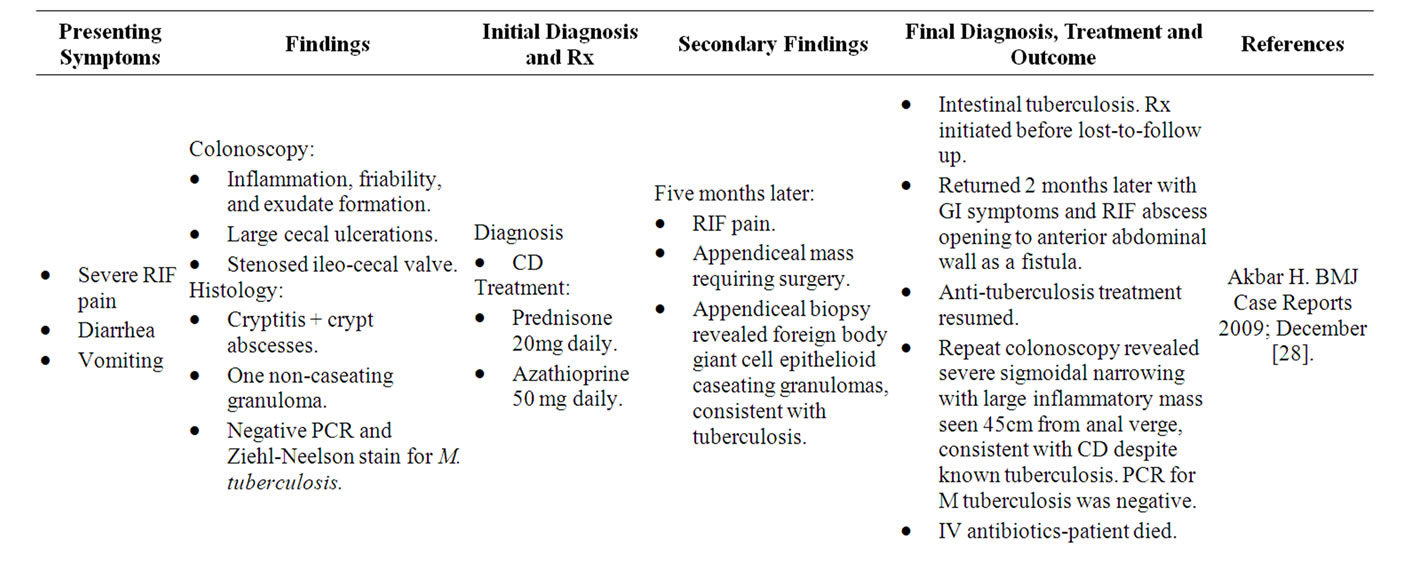
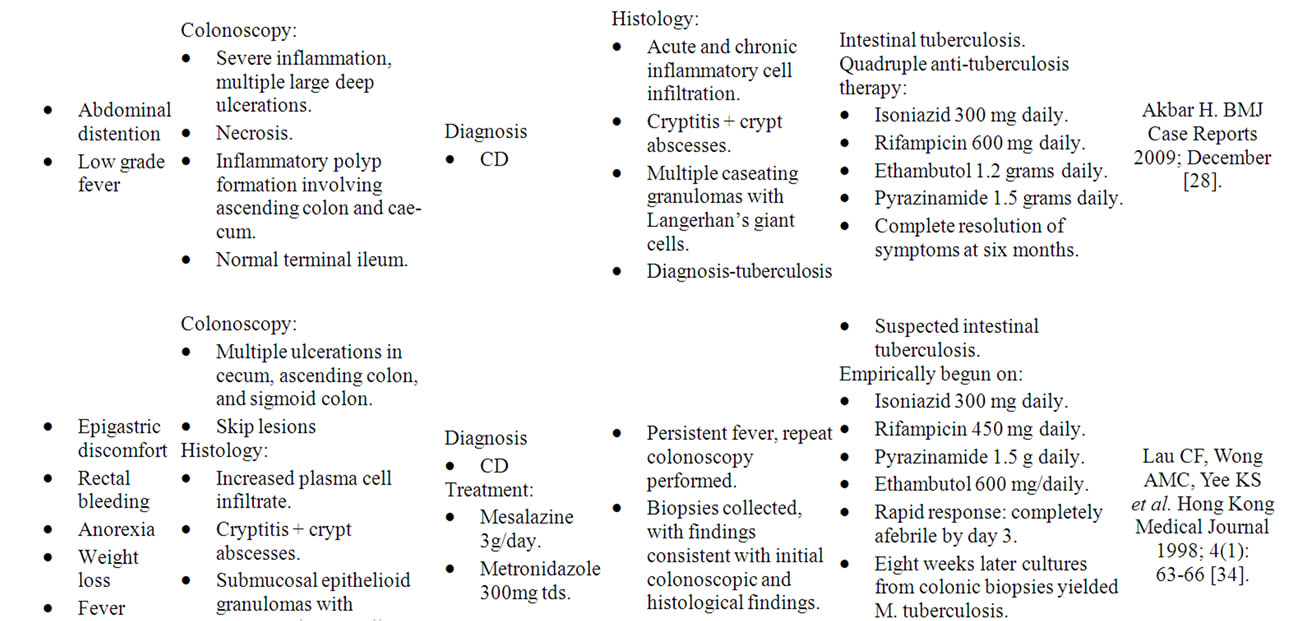

Table 1. Case reports of M. tuberculosis mimicking “CD”.
4.4. Entamoeba histolytica (E. histolytica)
Parasitic invasion of the GI mucosa by E. histolytica is made possible with the aid of cytolitic enzymes, resulting in transmural ulceration and inflammation. Diagnosis of amebiasis is established via cysts or trophozoites in faeces [42], however, as with other CDS pathogens, E. histolytica infection is also difficult to diagnose, as stool specimens, bowel biopsies, and serological studies are often negative, even in the presence of invasive amebic colitis [43]. Joos et al. (1999) reported on two patients who, following the development of chronic diarrhea, were diagnosed on colonoscopic examination with “CD”. Both were treated with immunosuppressive drugs for their “CD” which resulted in the development of amoebic liver abscesses, leading to the correct diagnosis and cure of amebic dysentery in both cases [44].
We have also reported two patients who presented to our Centre with a classic CDS colonoscopic appearance as a result of E. histolytica infection [45]. The first patient presented with a one year history of abdominal pain, fatigue and bloody diarrhoea with colonoscopy revealing
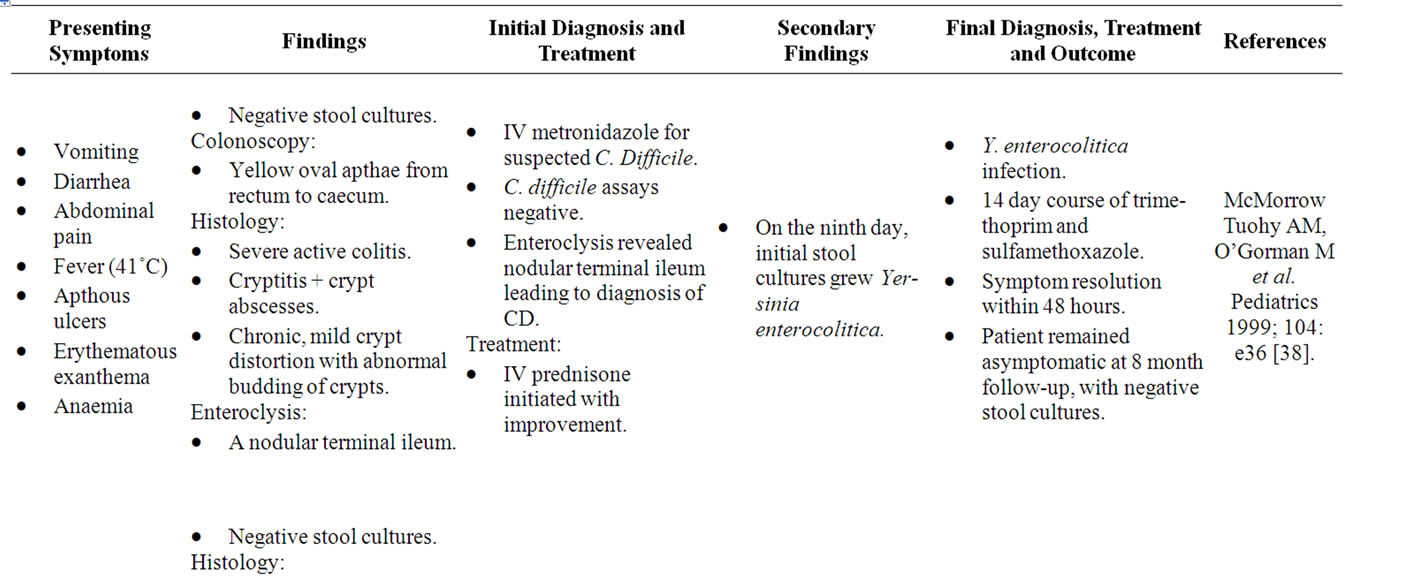


Table 2. Case reports of Yersinia mimicking “CD”.
inflammation with erosions, micro-ulcers and contact bleeding distally; and large ulcers, haustration destruction and scarring in the ascending colon. Histopathology showed moderate crypt distortion with focal cryptitis and oval foamy structures containing red blood cells, which were strongly positive for amebiasis on PAS stain. The patient was treated with an antibiotic combination and experienced a complete resolution of symptoms, with subsequent colonoscopy and histology revealing a normal mucosa. The second patient presented with a three month history of ten diarrhoeal stools daily, weight loss, abdominal pain, faecal urgency and mucus. Three years prior the patient had been treated for amebiasis, with follow-up stool results repeatedly negative for E. Histolytica. The illness “relapsed” with rectal inflammation, pseudopolyps, and large ulcerations appearing in the ascending colon and caecum. Stool culture was negative for parasites, however PAS stain revealed trophozoites of Entamoeba histolytica and the patient was successfully treated with anti-parasite treatment with amelioration of inflammation and symptoms.
The endoscopic similarities viewed with our colonoscopy case report findings of an indistinguishable disease process to “Crohn’s Disease” are similar to those reported elsewhere in the literature [46,47] and so form part of the CD Syndrome.
4.5. Adherent-Invasive Escherichia coli (AIEC)
A pathovar of Escherichia coli capable of invading and replicating within cultured intestinal epithelial cells and macrophages, inducing the secretion of large amounts of tumor necrosis factor, a key cytokine in IBD inflammation has also been isolated from “CD” lesions [48]. One study aimed at assessing the prevalence of E. coli strains in the ileal mucosa of “CD” patients isolated AIEC from 13 of 20 specimens (65%) of chronic “CD” ileal lesions and 19 (100%) “CD” patients with early ileal lesions who had endoscopic recurrence [49]. E. coli was predominant in 8 (40%) of the 20 samples from chronic ileal lesions and in 11 (57.8%) of the 19 samples from early ileal lesions. In contrast, AIEC was predominant in only 1 (9.1%) of 11 samples from healthy ileal mucosa of patients without recurrence of CD, and in 2 (15.3%) of 13 samples from controls [50]. A follow-up study examining the ileal specimens of 63 “CD” patients and 16 controls for E. coli found AIEC in 21.7% of “CD’ ileal specimens vs. 6.2% of controls. In neoterminal ileal specimens, AIEC strains were found in 36.4% of “CD” early lesions vs. 22.2% in the healthy mucosa of “CD” patients. AIEC were also found in the colonic specimens of 3.7% of CD patients, 0% of UC patients, and 1.9% of controls, with the study concluding that AIEC strains are associated specifically with the ileal mucosa in “CD” [51]. Martin et al. (2004) also reported that mucosa-associated and intramucosal E. coli were cultured more commonly in “CD” (43% and 29% respectively) than in non-inflamed controls (17% and 9% respectively) with the authors concluding that the studies support a central role for mucosal adherent E. coli in the pathogenesis of “Crohn’s Disease” [52]. Additionally the AIEC strains isolated from “CD” patients have been found to adhere to buccal cells with one study reporting that between 53% - 62% of patients with “CD” have adherent E. coli strains in their stool versus 5% - 6% of controls [53]. Furthermore, these strains are able to survive and replicate extensively within murine macrophages with a recent study reporting that the number of intracellular adherent invasive E. coli bacteria increases up to 74-fold by 48hr post-infection [54].
A recent study which had previously used laser capture microdissection and PCR to detect MAP DNA in granulomas of 6/15 patients with “CD”, examined archival tissue from 15 surgical cases of CD and 10 nonCrohn’s granulomatous bowel disease controls for E. coli DNA [55]. E. coli DNA was detected in microdissected granulomas in 12/15 (80%) Crohn’s disease patients and in 1/10 (10%) non-Crohn’s control granulomas (p < 0.001). E. coli DNA was also detected in 8/15 (53%) Crohn’s full-thickness sections and in 4/10 (40%) control full-thickness sections.
4.6. Campylobacter
A recent study has also detected a high prevalence of Campylobacter concisus DNA and immunoglobulin G antibodies to C. concisus in children with newly diagnosed “Crohn’s Disease”, further reinforcing the hypothesis of a persistent infection in “CD”. A study by Zhang et al. (2010) examining the fecal specimens collected from 54 children with “CD”, 27 non-inflammatory bowel disease patients and 33 healthy controls using a new PCR detected C. concisus DNA in 35/54 (65%) of “CD” samples, compared with 11/33 (33%) of healthy and 10/27 (37%) non-IBD controls [56]. The prevalence of all Campylobacter DNA using genus-specific primers in children with “CD” was 39/54 (72%), which was significantly higher than the 10/33 (30%) and 8/27 (30%) observed in healthy and non-IBD controls respectively. Additional studies have reported on an increased risk of developing “Crohn”s Disease’ following Campylobacter infection with one large study following 13,148 patients diagnosed with Campylobacter infection and 26,216 unexposed patients for a 15 year period. A follow-up diagnosis of IBD was reported in 107 infected patients (1.2%) compared with 73 uninfected patients (0.5%) with the development of CDS highest during the first year postinfection [57].
4.7. Less Common Pathogens
Apart from the common causes of CDS discussed, such as MAP, several less common bacterial, viral, fungal and parasitic candidate agents have also been reported to produce features of the CD Syndrome including Salmonella [58], Histoplasma capsulatum [59], Shigella [60], Cytomegalovirus [61], Schistosomiasis [62] and Strongyloides stercoralis [63]. Despite the obvious distinctions in etiology, the infections discussed above all share the ability to reproduce the essential features of the CD Syndrome.
5. CONCLUSION
The list of documented pathogens capable of causing “Crohn’s Disease” is extensive and one can no longer dismiss them as 'mere “mimickers” or “innocent bystanders” in the disease. Given the sheer number of infections capable of reproducing the hallmark clinical, colonoscopic and histological features of CD, it appears unlikely that a separate mechanism of an “aberrant reaction” to our own normal colonic flora is at play. It is most likely that currently unidentifiable pathogens are responsible for the remaining subset of CDS cases. Now is the time for diagnostic methods to be refined and made available for clinicians to search for one or more of the causal pathogens in their patients with CD Syndrome in order to deliver rational treatment. As with the Helicobacter saga which revealed the infective causality of ulcers, the dogmatic insistence that a single immune pathway holds the key to the pathogenesis of “Crohn’s Disease” presents a barrier to the future of CDS research and the development of effective therapies aimed at treating the etiologies of the CD Syndrome. We call for greater efforts to seek out and treat the remaining infections of CDS.
REFERENCES
- Lesniowski, A. (1904) Pamietnik towarzystwa lekarskiego warszawskiego. Polish Surgeon, 100, 630-631.
- Dalziel, T.K. (1913) Chronic Interstitial enteritis. British Medical Journal, 2, 1068-1070.
- Crohn, B.B., Ginzburg, L. and Oppenheimer, G.D. (1932) Regional Ileitis, a pathological and clinical entity. Journal of the American Medical Association, 99, 1323-1329.
- Podolsky, D.K. (2002) Inflammatory bowel disease. New England Journal of Medicine, 347, 417-429.
- Bull, T.J., McMinn, E.J., Sidi-Boumedine, K., et al. (2003) Detection and verification of Mycobacterium avium subsp. paratuberculosis in fresh ileocolonic mucosal biopsy specimens from individuals with and without Crohn’s Disease. Journal of Clinical Microbiology, 41, 2915-2923.
- Van Kruiningen, H.J., Chiodini, R.J., Thayer, W.R., et al. (1986) Experimental disease in infant goats induced by a Mycobacterium isolated from a patient with Crohn’s Disease. A preliminary report. Digestive Diseases and Sciences, 31, 1351-1360.
- Barrett, J.C., Hansoul, S., Nicolae, D.L., et al. (2008) Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s Disease. Nature Genetics, 40, 955-962.
- Prantera, C. (2007) Mycobacteria and Crohn’s Disease, The endless story. Digestive & Liver Disease, 39, 452- 454.
- Golden, M.P. (2005) Extrapulmonary tuberculosis, an overview. American Physician, 72, 1761-1768.
- Marshall, B.J. (1995) Helicobacter pylori in puptic ulcer, have Koch’s postulates been fulfilled? Annals of Medicine, 27, 565-568.
- Porter, R.S. and Kaplan, J.L. (2008) The Merck Manuals Online Medical Library. http://www.merck.com/mmpe/sec05/ch052/ch052b.html
- Chamberlin, W., Borody, T. and Naser, S. (2007) MAPassociated Crohn’s disease, MAP, Koch’s postulates, causality and Crohn’s Disease. Digestive & Liver Disease, 39, 792-794.
- Johnson, C., Wannemuehler, M.J. and Hostetter, J.M. (2011) Persistent enteric mycobacterial infection enhances sensitivity to acute mucosal injury. Experimental Biology and Research, 236, 36-43.
- Autschbach, F., Eisold, S., Hinz, U., et al. (2005) High prevalence of Mycobacterium avium subspecies paratuberculosis IS900 DNA in gut tissues from individuals with Crohn’s Disease. Gut, 54, 944-949.
- Schwartz, D., Shafran, I., Romero, C., et al. (2000) Use of short term culture for identification of Mycobacterium avium subsp. paratuberculosis in tissue from Crohn’s Disease patients. Clinical Microbiology & Infection, 6, 303-307.
- Sartor, C. (2005) Does Mycobacterium avium subspecies paratuberculosis cause Crohn’s Disease? Gut, 54, 898- 898.
- Prantera, C. (2007) Mycobacteria and Crohn’s Disease, The endless story. Digestive & Liver Disease, 39, 452- 454.
- Hermon-Taylor, J. (2009) Mycobacterium avium subspecies paratuberculosis, Crohn’s Disease and the doomsday scenario. Gut Pathogens, 1, 15.
- Selby, W., Pavli, P., Crotty, B., et al. (2007). Two-year combination antibiotic therapy with clarithromycin, rifabutin and clofazamine for Crohn’s Disease. Gastroenterology, 132, 2313-2319.
- Behr, M.A. and Hanley, J. (2008) Antimycobacterial therapy for Crohn’s Disease, a reanalysis. The Lancet Infectious Diseases, 8, 344.
- Gui, G.P, Thomas, P.R., Tizard, M.L., et al. (1997) Two-year outcomes analysis of Crohn’s Disease treated with rifabutin and macrolide antibiotics. Journal of Antimicrobial Chemotherapy, 30, 393-400.
- Shafran, I., Kugler, L., El-Zaatari, F.A., et al. (2002) Open clinical trial of rifabutin and clarithromycin therapy in Crohn’s Disease. Digestive & Liver Disease, 34, 22- 28.
- Borody, T.J., Leis, S., Warren, E.F., et al. (2002) Treatment of severe Crohn’s Disease using antimycobacterial triple therapy—Approaching a cure? Digestive & Liver Disease, 34, 29-38.
- Hanauer, S.B., Feagan, B.G., Lichtenstein, G.R., et al. (2002) Maintenance infliximab for Crohn’s Disease, the ACCENT I randomised trial. Lancet, 359, 1541-1549.
- Hanauer, S.B. (2007) More likely than not. Nature Clinical Practice Gastroenterology & Hepatology, 4, 469.
- St Jean, G. (1996) Treatment of clinical paratuberculosis in cattle. Veterinary Clinics of North America, Food Animal Practice, 12, 417-430.
- Sibartie, V., Kirwan, W.O., O’Mahony, S., et al. (2007). Intestinal tuberculosis mimicking Crohn’s Disease, lessons relearned in a new era. European Journal of Gastroenterology & Hepatology, 19, 347-349.
- Akbar, I. (2009) Intestinal tuberculosis and Crohn’s Disease, the dilemma of similarities and misdiagnosis. BMJ Case Reports.
- Al Kawari, M.A., Mohamed, A.E., Yasawy, M.l., et al. (1995) Protean manifestation of gastrointestinal tuberculosis, report on 130 patients. Journal of Clinical Gastroenterology, 20, 225-232.
- Bhargava, D., Kushwaha, A.K., Dasarathy, S., et al. (1992) Endoscopic diagnosis of segmental colonic tuberculosis. Gastrointestinal Endoscopy, 38, 571-574.
- Shah, S., Thomas, V., Mathan, M., et al. (1995) Colonoscopic stuy of 50 patients with colonic tuberculosis. Gut, 33, 347-351.
- Singh, V., Kumar, P., Kamal, J., et al. (1995) Clinicocolonoscopic profile of colonic tuberculosis. American Journal of Gastroenterology, 20, 225-232.
- Marshall, J.B. (1993) Tuberculosis of the gastrointestinal tract and peritoneum. American Journal of Gastroenterology, 88, 989-999.
- Lau, C.F., Wong, A.M.C., Yee, K.S., et al. (1998) A case of colonic tuberculosis mimicking Crohn’s Disease. Hong Kong Medical Journal, 4, 63-66.
- Lamps, L.W., Madhusudhan, K.T., Havens, J.M., et al. (2003) Pathogenic Yersinia DNA is detected in bowel and mesenteric lymph nodes from patients with Crohn’s Disease. The American Journal of Surgical Pathology, 27, 220-227
- Lockhart-Mummery, H.E. and Morson, B.C. (1960) Crohn’s Disease (regional enteritis) of the large intestine and its distinction from ulcerative colitis. Gut, 1, 87-105.
- Payne, M., Girdwood, A.H., Roost, R.W., et al. (1987) Yersinia enterocolitica and Crohn’s Disease, a case report. South African Medical Journal, 472, 53-55.
- McMorrow Tuohy, A.M., O’Gorman, M., Byington, C., et al. (1999) Yersinia Enterocolitis mimicking Crohn’s Disease in a toddler. Pediatrics, 104, 1-4.
- MacFarlane, P.I. (1986) Yersinia enterocolitica mimicking Crohn’s Disease. Journal of Pediatric Gastroenterology & Nutrition, 5, 671-672.
- Treacher, D.F. and Jewell, D.P. (1985) Yersinia colitis associated with Crohn’s Disease. Postgraduate Medical Journal, 61, 173-174.
- Ibbotson, J.P., Pease, P.E. and Allan, R.N. (1987) Serological studies in Crohn’s Disease. Eur Journal of Clinical Microbiology, 6, 286-290.
- Kaya, M., Aydin, F. and Buyukbayram, H. (2005) A rare cause of colonic stricture, amebiasis. Turkish Journal of Gastroenterology, 16, 236-239.
- Merino, E., Glender, W., Del Muro, R., et al. (1990) Evaluation of the ELISA test for detection of Entamoeba histolytica in feces. Journal of Clinical Laboratory Analysis, 4, 39-42.
- Joos, L., Loosli, J., Spichtin, P., et al. (1999) Am- Öebenleberabszess bei chronischer Kolitis: Revision der diagnose “Morbus Crohn”. Schweiz Med Wochenschr, 129, 1656-1659.
- Borody, T.J., Campbell, J., Torres, M., et al. (2009). Entamoeba histolytica, another cause of Crohn’s Disease. American Journal of Gastroenterology, 104, 990.
- Pai, S. (2009) Amebic colitis can mimic Tuberculosis and Inflammatory Bowel Disease on endoscopy and biopsy. International Journal of Surgical Pathology, 17, 116-121.
- Purnomo, H.J., Tarius, A., Simadibrata, M., et al. (2007). Chronic diarrhea caused by amebic colitis and Inflammatory Bowel Disease. Indonesian Journal of Gastroenterology, Hepatology & Digestive Endoscopy, 8.
- Croxen, M.A. and Finlay, B. (2010) Molecular mechanisms of Escherichia coli pathogenicity. Nature Reviews Microbiology, 8, 26-38.
- Barnich, N. (2007) Adherent invasive Escherichia coli and Crohn’s Disease. Current Opinion in Gastroenterology, 23, 16-20.
- Darfeuille-Michaud, A., Neut, C., Barnich, N., et al. (1998) Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s Disease. Gastroenterology, 115, 1405-1413.
- Darfeuille-Michaud, A., Boudeau, J., Bulois, P., et al. (2004) High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s Disease. Gastroenterology, 127, 412-421.
- Martin, H., Campbell, B.J., Hart, A., et al. (2004). Enhanced Escherichia coli adherence and invasion in Crohn’s Disease and colon cancer. Gastroenterology, 127, 80-93.
- Giaffer, M.H., Holdsworth, C.D. and Duerden, B.I. (1992) Virulence properties of E. coli strains isolated from patients with inflammatory bowel disease. Gut, 33, 646- 650.
- Glasser, A.L., Boudeau, J., Barnich, N., et al. (2001) Adherent invasive Escherichia coli strains from patients with Crohn’s Disease survive and replicate within macrophages without inducing host cell death. Infection and Immunity, 69, 5529-5537.
- Kelly, R.P., Lee, G., Collins, J.K., et al. (2004) Bacterial DNA within granulomas of patients with Crohn’s Disease-detection by laser capture microdissection and PCR. American Journal of Gastroenterology, 99, 1539-1543.
- Man, S.M., Zhang, L., Day, A.S., et al. (2010) Campylobacter concisus and other Campylobacter species in children with newly diagnosed Crohn’s Disease. Inflammatory Bowel Disease, 16, 1008-1016.
- Gradel, K.O., Nielson, H.L., Schønheyder, H.C., et al. (2009) Increased short and long-term risk of Inflammatory Bowel Disease after Salmonella or Campylobacter gastroenteritis. Gastroenterology, 137, 495-501.
- Dionisio, D., Esperti, F., Vivarelli, A., et al. (2001) Acute terminal ileitis mimicking Crohn’s Disease caused by Salmonella venezian. International Journal of Infectious Diseases, 5, 225-227.
- Alberti-Flor, J. and Granda, A. (1986) Ileocaecal histoplasmosis mimicking Crohn’s Disease in a patient with Job’s syndrome. Digestion, 33, 176-180.
- [61] DiLauro, S. and Crum-CIanflone, N.F. (2010) Ileitis, when it is not Crohn’s Disease. Current Gastroenterology Reports, 12, 249-258.
- [62] Khan, F.N., Prasad, V. and Klein, M.D. (2009) Cytomegalovirus enteritis mimicking Crohn’s Disease in a lupus nephritis patient, a case report. World Journal of Gastroenterology, 15, 4327-4330.
- [63] Sheikh, M.A. and Ashraf, S.A. (2007) Terminal ileum schistosomiasis with perianal fistula mimicking Crohn’s Disease. Saudi Medical Journal, 28, 1449-1452.
- [64] Gutierrez, Y., Bhatia, P., Garbadawala, S., et al. (1996). Strongyloides stercoralis eosinophilic granulomatous enterocolitis. American Journal of Surgical Pathology, 20, 603-612.
NOTES
*Thomas J. Borody has a pecuniary interest in both the Centre for Digestive Diseases and Giaconda Ltd., the licensor of MyocondaTM (an anti-MAP treatment).