Journal of Cancer Therapy
Vol. 3 No. 6A (2012) , Article ID: 25168 , 15 pages DOI:10.4236/jct.2012.326119
TNFR1 Deficiency Protects Mice from Colitis-Associated Colorectal Cancer Coupled with a Decreased Level of Oxidative Damage in the Colon: Implications for Anti-TNF Therapy of Unremitting Colitis
![]()
1Department of Microbiology & Immunology, Dalhousie University, Halifax, Canada; 2Division of Anatomical Pathology, Queen Elizabeth II Health Sciences Centre, Halifax, Canada; 3Department of Pediatrics, Dalhousie University, Halifax, Canada.
Email: astadnyk@dal.ca
Received September 30th, 2012; revised October 31st, 2012; accepted November 12th, 2012
Keywords: TNF Receptor; Colitis Associated Cancer; Colorectal Cancer; Azoxymethane
ABSTRACT
It has long been appreciated that there is a direct relationship between the intensity and duration of inflammatory bowel diseases (IBD) and increasing intestinal cancer risk but which elements of the inflammatory response are responsible have not been identified. Anti-TNF drugs have been successful at treating IBD but considering the presumed anti-tumor activity of TNF, it is important to understand whether the treatment impacts on the patients’ intestinal cancer risk. We modeled this relationship by “treating mice lacking TNF receptors with a colon cancer causing combination of azoxymethane followed by repeated dextran sulphate sodium exposures (AOM + DSS regime). TNF receptor type1 gene deficient (TNFR1-/-) and TNFR2-/- mice experienced similar clinical illnesses and colonic inflammation as C57BL/6 wildtype controls during the AOM + DSS regime. Despite the inflammation, TNFR1-/- mice developed significantly fewer colon tumors than the other strains. The reduced tumor incidence was a product of the combined lack of receptor expression on hematopoietic and nonhematopoietic cells, shown using bone marrow cell chimeras of wildtype and TNFR1-/- mice. As oxidative damage is a potent contributing factor to tumorigenesis and inflammatory leukocytes make copious amounts of reactive oxygen radicals, we measured oxidative damage in the animals’ colons. TNFR1-/- mice showed less damage compared to the other strains. We subsequently examined mice deficient in their leukocyte NADPH oxidative pathway (Nox2-/-) for their cancer incidence using the AOM + DSS regime. Nox2-/- mice became inflamed but had fewer tumors than wildtype mice. We conclude that TNF promotes colon cancer including through promoting oxidative processes utilizing TNFR1 in leukocytes. Moreover, the C57BL/6 strain can be used to dissociate mechanisms of colon inflammation from tumorigenic processes. We interpret our results to mean that IBD patients on TNF antagonist therapies will potentially benefit with reduced colon cancer risk even if they do not respond with reduced inflammation.
1. Introduction
Crohn’s Disease (CD) and ulcerative colitis (UC) are remitting/relapsing inflammatory bowel diseases (IBD) associated with an increased risk of colorectal cancer (CRC) [1,2]. This risk increases with duration and severity of disease. Consequently, therapies that reduce inflammation often reduce the risk of CRC in affected individuals but exactly which element of the inflammation is mechanistically contributing to cancer risk is not clear. Those patients who fail to respond to the available antiinflammatory therapies become the susceptible population compounded by the fact that the cancer is difficult to detect on their inflamed mucosa.
The inflammation of IBD includes extensive lymphocyte and cytokine networks and pro-inflammatory TNF consequently has become a therapeutic target. TNF is a pleiotropic cytokine reportedly elevated in the serum [3,4] and inflamed mucosa. TNF signaling through its cognate receptors, TNFR1 and TNFR2, activates multiple signal transduction pathways such as NFκB, PI3K/AKT and MAPK/AP-1, all of which promote additional inflammatory mediator production [5]. Yet the precise role of TNF in driving colonic inflammation in IBD remains unresolved. In rodent models of colitis blocking TNF has yielded mixed results [6] and genetic ablation of TNF or its receptors has been shown to not significantly reduce disease measures in several models of experimental colitis [7-9]. In some cases loss of TNF exacerbated colitis, particularly in dextran sulphate sodium (DSS) colitis [10,11]. It is possible that TNF has different roles depending on the target cells. This is supported by the relative inefficacy of Etanercept, a soluble chimeric TNFR2 molecule, in IBD [12] while anti-TNF therapies targeting membrane-bound TNF on activated leukocytes are effective [13-15]. In addition to these mixed messages, the impact of anti-TNF therapies on the CRC risk of IBD patients is not known.
TNF was originally identified as the factor involved in lipopolysaccharide-induced tumor regression [16]. However, research in the past decade has challenged this concept [17], especially in models of colitis-associated cancer (CAC) [18,19]. TNF deficient mice on the C57BL/6 background experienced worse DSS-induced colitis compared to controls [10] yet had unchanged cancer development in a model of colitis-associated cancer [20] suggesting that pathways involved in inflammation differ than those involved in cancer. In contrast, mice on the BALB/c background that lacked TNFR1 showed a reduced level of inflammation and reduced colonic tumors after a combined azoxymethane (AOM) + DSS regime. A tumorigenic effect was attributed to leukocyte-specific TNFR1 as BALB/c mice irradiated and reconstituted with TNFR1-/- bone marrow cells were protected against tumor development compared to mice reconstituted with syngeneic bone marrow [18]. Studies have yet not addressed whether the TNFR2 affects CAC incidence.
In addition to the activation of multiple inflammatory signalling pathways, TNF can contribute to oxidative stress by inducing the production of reactive oxygen species (ROS) via several isoforms of NADPH oxidase [21,22]. TNF treatment can directly lead to oxidative damage and DNA mutations in cell culture [23]. ROS may contribute to tumorigenesis by damaging proteins, lipids and DNA. TNF mediated oxidative damage has not yet been evaluated in the AOM + DSS model. In our study, we sought to further clarify the contribution of TNF in CAC. We observed that mice on a C57BL/6 background that are deficient in TNFR1 are less susceptible to tumor development using the AOM + DSS regime compared to controls or TNFR2-/- mice while inflammatory measures remain similar. Additionally, we evaluated colonic oxidative damage during chronic colitis in mice lacking either TNFR, on the premise that TNFR1 may be promoting the development of tumors through oxidative damage to sensitive cells within the crypt.
2. Materials and Methods
2.1. Animals
Mice (TNFR1-/-, TNFR2-/- and gp91-phox-/- (Nox2-/-)) were obtained from Jackson Laboratories (Bar Harbor, ME) and bred as homozygous knockout pairs in our facility. C57BL/6 mice (wildtype, WT) were obtained from Jackson Laboratories. Animals were group-housed in plastic shoebox cages and food and water was available ad libitum. The rooms were maintained on a 12-hour light-dark cycle in a humidity and temperature-controlled environment. All protocols were approved by the Dalhousie University Committee for Laboratory Animals in accordance with the Canadian Council for Animal Care.
Mice (6 - 12 weeks of age) were subjected to a single intraperitoneal injection of 10 mg/kg azoxymethane (AOM, Sigma-Aldrich, St. Louis, MO) dissolved in sterile 0.9% saline. Control mice were injected with 0.9% saline diluent alone. Mice were divided into the following groups, 0.9% saline alone (no treatment, NT), AOM (AOM only), 0.9% saline followed by four cycles of DSS (DSS only) and AOM followed by 4 cycles of DSS (AOM + DSS). Each DSS cycle consisted of 3% w/v DSS (36,000 - 50,000 mw from MP Biomedicals, Solon, OH) in the animals’ drinking water for five days followed by 14 days of facility water. Each mouse’s weight and clinical score was recorded daily.
At the end of the carcinogenesis experiment the mice were anesthetized using isofluorane and exsanguinated by cardiac puncture. Their colons were removed and flushed with ice-cold PBS, weighed and the length measured. Each colon was opened longitudinally along the mesenteric axis and was first inspected under a dissecting microscope for evidence of tumors/lesions. The colon was subsequently divided in half longitudinally. One half was fixed in 10% neutral buffered formalin in a Swiss roll orientation, while the other half was weighed then snap frozen in liquid nitrogen and stored frozen at –80˚C for later evaluation. Collected blood was incubated for 10 minutes at 37˚C to coagulate then centrifuged and serum was removed and frozen for later evaluation.
2.2. Assessing Disease Severity
Clinical scores throughout the duration of the treatment periods were determined using a scale dependent on the percent weight loss, stool consistency, and the presence of occult or gross blood in the stool [9]. Formalin-fixed, paraffin-embedded samples were sectioned and stained with hematoxylin and eosin (H & E). Sections were evaluated for leukocyte infiltration (0 - 5), crypt damage (0 - 5), ulceration (0 - 3) and the presence or absence of edema (0 - 1), for a maximal score of 14 [9].
Macroscopically, tumors were counted as solid raised lesions without ulceration. Gross tumor size was determined using the average of three diameter measurements. Microscopically, the presence or absence of dysplasia was used to determine incidence. Dysplasia was described as crypts that had a well-differentiated but disorganized epithelial cell structure with nuclei that had lost their polarity, were hyperchromatic, and had a loss of goblet cells but had not penetrated the submucosa.
2.3. Immunohistochemical Staining
In general, 3 μm sectioned samples were deparaffinized and rehydrated in xylene and graded ethanol baths, respectively. Endogenous peroxidase activity was blocked using 3% H2O2 for 20 minutes, then antigen retrieval was performed in 10mM citrate buffer (pH 6) with the slides heated in a microwave for 2 minutes until boiling then maintained in a 95˚C water bath for 20 minutes. Subsequently a blocking step using 5% goat serum in PBS plus 0.1% Tween-20 for 1 hour was performed. Primary antibody was applied at concentrations as per the manufacturer’s instructions or as indicated below, overnight at 4 C diluted with blocking solution. Biotinylated secondary antibody was also diluted in blocking solution and applied to individual slides for one hour at ambient temperature. The slides were then subjected to avidin-bound horseradish-peroxidase (ABC elite, Vector Laboratories, Burlington, ON) for 30 minutes. Colour was developed using diaminobenzidine (Vector Laboratories) and slides were counterstained with Mayer’s hematoxylin (SigmaAldrich).
To measure apoptotic cells rabbit polyclonal antibodies raised against cleaved caspase-3 (Cell Signaling, Danvers, MA) was used at a 1:1000 dilution as the primary antibody. Positive stained colonic epithelial cells were counted within 100 intact crypts per section per mouse in two sections per slide. Macrophages and neutrophils were detected using a monoclonal antibodies to F4/80 (clone CI: A3-1, Serotec, Raleigh, NC) and Ly6G (Clone 1A8, BD Biosciences, San Jose, CA), respectively, at a final dilution of 1:1000 and incubated for 24 hours at 4˚C (Serotec, Raleigh, NC). Biotinylated goat anti-rat IgG (Santa Cruz Biotechnology) was used as a secondary antibody at a concentration of 1:400 and applied for 1 hour at ambient temperature. Both cell types were counted in ulcerated lesions in both the mucosa and submucosa. Five high-power fields were counted per section. To enumerate neutrophils and macrophages in tumors, cells were counted in at least 3 high power fields in tumor tissue in tumor-bearing mice. Rabbit polyclonal antibodies to TNFR1 and TNFR2 (abcam, Cambridge, MA) were used to detect the intracellular portion of the receptor within the colon sections. They were used at concentrations of 1:100 and 1:50, respectively. Oxidative damage was detected on formalin-fixed samples using a mouse antibody to 8-OHDG (clone N45.1 JaICA, Japanused at 1:100) with the following adaptations; antigen retrieval was performed using proteinase K for 20 minutes at 37˚C, mouse seroblock (Serotec) was added to the blocking solution) to prevent FcR binding, and only positive nuclei within the colonic epithelial cells were evaluated to limit counting false positives due to mouseon-mouse staining. The average number of positive cells per 20 crypts is reported.
2.4. Bone Marrow Radiation Chimeras
Six-12-week old recipient WT and TNFR1-/- female mice had 0.2% neomycin sulphate added to their drinking water for two weeks. The mice were then irradiated with two doses of 500 Rads two hours apart with a gamma-irradiator [24]. Meanwhile bone marrow cells were isolated from healthy male WT and TNFR1-/- 6 - 12-week-old donor mouse femurs and tibias and rinsed in sterile HBSS without Ca2+ Mg2+. The bone marrow cells were counted using a crystal violet stain, and recipient mice received 5 × 106 viable cells via tail vein injection following the second dose of radiation. Subsequent to the bone marrow reconstitution the mice were housed in sterile HEPA-filtered cages and fed sterile food and water containing 0.2% neomycin sulphate for two weeks. Their weights and health were monitored daily. Reportedly by 8 - 12 weeks most host immune cells are replaced [25] thus after a minimum of 8 weeks recovery the mice were subjected to the AOM + DSS regime. Chimerism was confirmed using in situ hybridization of the Y chromosome in the spleen and colon as per the manufacturer’s instructions (Star*FISH© chromosome painting, Cambio, Cambridge, UK), with an added amplication step detecting FITC. Male gut sections were used as positive controls and female gut sections were used as negative controls for the in situ hybridization.
2.5. Statistics
A two-way ANOVA was performed on most data sets using main effects strain treatment, with three levels of strain and four levels of treatment. Non-parametric data were analyzed using the Kruskal-Wallis One-way ANOVA between strains. Weight loss data was analyzed using a repeated-measures ANOVA over the treatment period. Chi square analyses were used to detect differences in the incidence between groups at a power of 0.8.
3. Results
3.1. Mouse Strain Responses to AOM + DSS
We treated WT, TNFR1-/- and TNFR2-/- mice with 10 mg/kg AOM followed by four cycles of 3% w/v DSS. Blood appeared in the stool (occult) after the second day of DSS treatment and by the fourth day, gross blood appeared in all strains treated with DSS. After the DSS treatment was discontinued in each cycle, the appearance of gross blood subsided by the end of the 14 days water period but occult blood was still detectable in the stool. There was no mortality associated with treatment in these strains. Three experiments, each using 8 - 12 mice per group, were conducted. There were no significant differences (inter-experimental variation) between experiments with respect to measures of disease severity and cancer incidence therefore they were grouped together for statistical purposes. Percent weight change during each cycle was not significantly different between strain and DSS only versus AOM + DSS treated mice while each strain lost weight during each 5 day DSS exposure (Figure 1(a)). Generally, weight lost was regained by the end of each cycle.
The severity of inflammation has been linked to the development of colon cancer [26] and there was a reduction in cancer incidence associated with reduced inflammation in BALB/c strain TNFR1-/- mice on the AOM + DSS regime [18]. We previously established that the colon inflammation did not differ between WT, TNFR1-/- and TNFR2-/- mice after a single DSS cycle [9]. In the present study, untreated mice and AOM alone mice did not have any detectable inflammation at the end of the experiment (Figure 1). In both DSS treated groups we found that most inflammation occurred in the midcolon, while little to none occurred in the proximal colon. The distal colon showed less infiltrate and less damage and ulceration than the mid-colon and was not significantly different among DSS only and DSS + AOM treated strains. Differences in the mid-colon inflammation scores were also not statistically significant between strains after four cycles of DSS in the DSS only and DSS + AOM groups (Figure 1(b)). A cellular infiltrate consisting of granulocytes (neutrophils and eosinophils), lymphocytes and macrophages was evident on hematoxylin and eosin stained sections (Figures 1(c)-(k)). Infiltrate was observed in the submucosa, mucosa and even the muscularis layers.
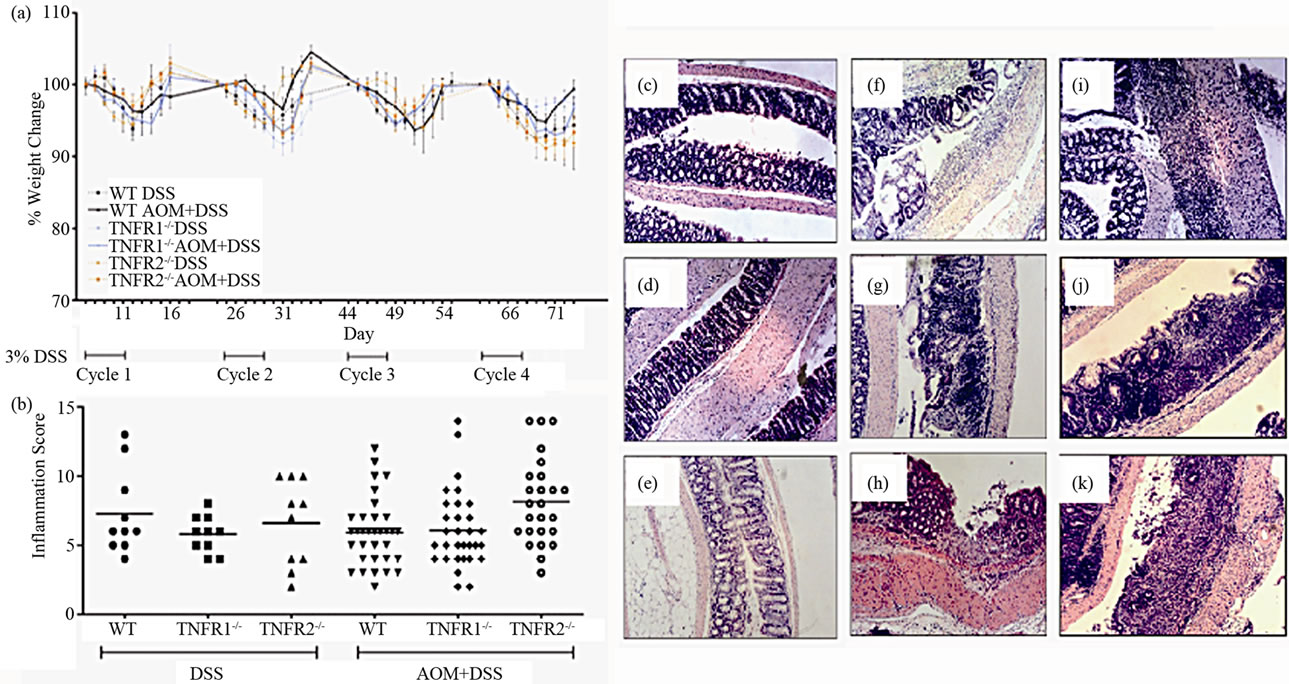
Figure 1. Illness and colon inflammation in WT, TNFR1-/- and TNFR2-/- mice treated with 10 mg/kg azoxymethane ± 4 cycles of 3% DSS. (a) Weight change during the 83-day AOM + DSS regime. Weights from days 1 - 12 of each cycle are shown. Values are the percent change in weight from the beginning of each cycle as there was a net weight gain from the beginning to the termination of the study. Data are presented as mean ± SD and statistics were performed using repeated measures ANOVA with a p < 0.05 considered significant; (b) Inflammation in hematoxylin and eosin-stained (H & E) colon sections after AOM + DSS or the DSS cycles alone. Sections were scored using a previously validated scale based on presence of edema, inflammatory cells, ulceration and crypt damage. Horizontal lines indicate the mean, and statistics were performed using non-parametric ANOVA; (c) through (k), examples of colon histology. All are H & E-stained sections using 100× original magnification; (c) WT AOM only treated mouse; (d) WT DSS only treated mouse; (e) WT AOM + DSS treated mouse; (f) TNFR1-/- AOM only treated mouse; (g) TNFR1-/- DSS only treated mouse; (h) TNFR1-/- AOM + DSS treated mouse; (i) TNFR2-/- AOM only treated mouse; (j) TNFR2-/- DSS only treated mouse; (k) TNFR2-/- AOM + DSS treated mouse.
3.2. Colon Tumor Incidence and Characteristics
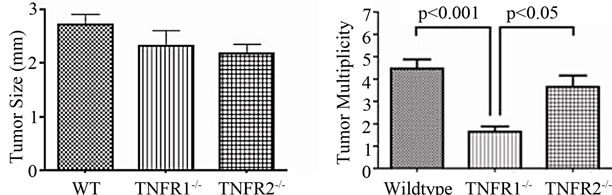
No treatment, AOM only and DSS only control mice did not develop macroscopically or microscopically detectable tumors or dysplasia, respectively. On the other hand, tumors were observed in all strains treated with DSS + AOM, both macroscopically and microscopically (Table 1). All three strains developed polyp-like tumors that were non-invasive intramucosal adenocarcinomas. Tumors were found exclusively in the mid-to distal colon. TNFR1-/- mice had a statistically significantly lower incidence of tumors and dysplasia at the end of the treatment period compared to WT and TNFR2-/- both macroscopically and microscopically (Table 1), determined by Chi-square analysis. Tumor incidence was not significantly different between WT and TNFR2-/- mice. There was no difference between the size of tumors found in the three strains (Figure 2(a)) but TNFR1-/- mice had significantly fewer tumors per mouse (Figure 2(b)). There were no statistically significant relationships between inflammation severity and tumor size, or number (not shown). When the inflammation scores between mice with and without microscopically detectable dysplasia were compared, there were no significant differences (Figure 2(c)). This is an important observation as it further suggests differences in tumor development are a function of genotype and not a difference in inflammation severity. Examples of the tumors from each strain are shown in Figures 2(d)-(f).
3.3. Characterization of the Infiltrate in TNFR Deficient Mice
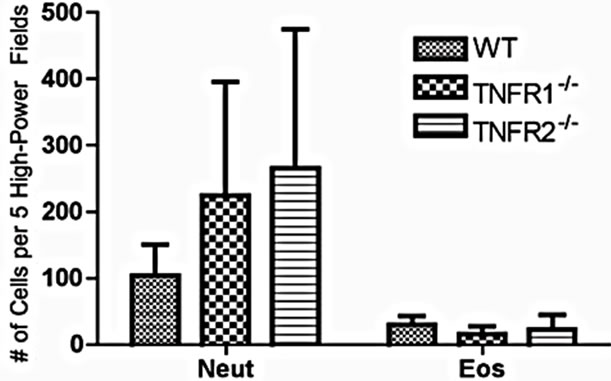
To determine if lacking either TNFR1 or TNFR2 affects leukocyte infiltration into the colon, the expression of Ly6G (neutrophils), F480 (macrophages), and congo red (eosinophils) were measured in the colon (neutrophils shown in Figure 3(a)). We determined the extent of eosinophil numbers because while there are resident eosinophils in the healthy colon, numbers rise due to DSS, and they are a source of inflammatory mediators that impact on DSS pathology [27]. Eosinophils may even play a protective role against the development of tumors [28]. No differences were found in the number of infiltrating leukocytes between strains (Figure 3).
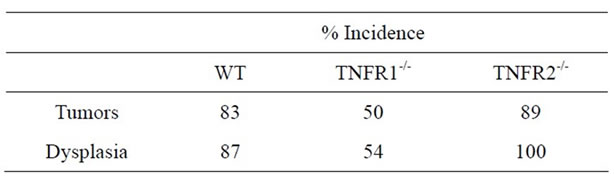
Table 1. Tumor (macroscopic) and dysplasia (microscopic) incidence (%) in WT, TNFR1-/- and TNFR2-/- all treated with AOM + DSS.
 (a)
(a)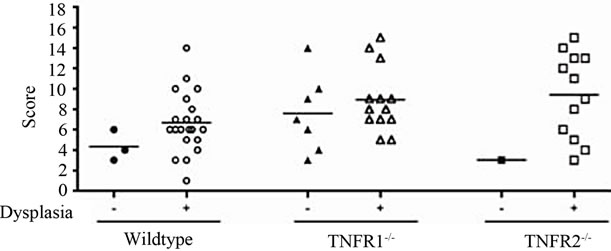 (b)
(b)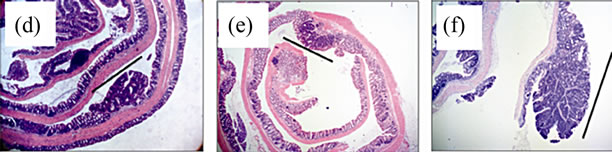 (c)
(c)
Figure 2. Tumor size and multiplicity after the AOM + DSS regime. (a) Tumor size (average of 3 diameters in mm) in WT, TNFR1-/- and TNFR2-/- mice treated with AOM + DSS. No statistical differences were found with a one-way ANOVA between strains. Data presented as mean ± SEM; (b) Tumor multiplicity (gross tumor count) in WT, TNFR1-/- and TNFR2-/- mice treated with AOM + DSS. Gross tumors were counted in the whole colon under a dissecting microscope. A one way-ANOVA found significant differences between strains, and differences between strains were determined with a Tukey’s Multiple Comparison post-test. Data presented as mean ± SEM. TNFR1-/- n = 32; WT n = 33 and TNFR2-/- n = 25 mice; (c) Inflammation scores in mice with and without dysplasia, to determine if there is a difference in the level of inflammation based on tumor presence; (d)-(f) H & E stained tissue sections depicting adenocarcinomas in each of the three strains, all at 25× magnification; (d) WT; (e) TNFR1-/-, (f) TNFR2-/-. The line denotes an adenocarcinoma.
3.4. Activated-Caspase-3 as an Indicator of Apoptosis in the Epithelium
There is a precedent for TNFR1 impacting epithelial cell homeostasis in the intestine, especially when NFκB is inhibited [29,30]. To determine the effect of treatment and genotype on colonic epithelial apoptosis, we measured activated caspase-3 by immunohistochemistry on tissue sections, as a measure of cells undergoing apoptosis. We did not find any significant effect of strain on the number of caspase 3+ colonic epithelial cells (Figure 4), although both DSS treated groups had a significantly greater number of activated caspase-3-positive cells per 100 crypts compared to untreated plus AOM only mice.
 (a)
(a)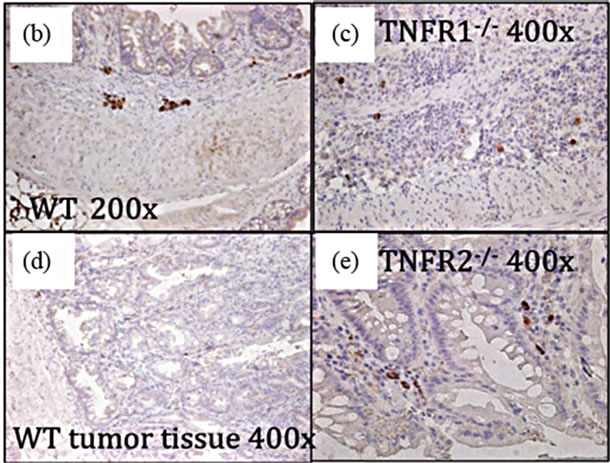
Figure 3. Cell infiltrate in the inflamed colons. (a) Cells (Neut = Ly6G+; Eos = Congo red+) were counted in 5 high power fields in AOM+DSS treated mice. Neutrophils were counted at 400× magnification in areas of inflammation with an inflammation score of at least 3 (maximum of 5 for infiltrate) within that field. Eosinophils were counted at 1000× magnification under oil immersion in inflamed areas with an inflammation score of at least 3: (b)-(e) F480+ macrophages in AOM + DSS treated mice were not different between strains; (d) Tumor tissue showing a low number of F480+ cells.
TNF and subsequent COX-2 activation and prostaglandins can induce β-catenin translocation to the nucleus. We did not observe any differences in β-catenin translocation within the epithelium between strains (not shown), suggesting it is unlikely that TNFR1 is contributing to cancer development by this mechanism.
3.5. TNF Receptor Expression in the Colon
Apart from leukocytes, TNFR1 and TNFR2 are reportedly expressed on intestinal epithelial cells [31-33]. TNFR2 expression was demonstrated in the colonic epithelium during inflammation [31]. We tested for the presence of the TNFRs in the colonic epithelium, also using immunohistochemistry, to determine if TNF might
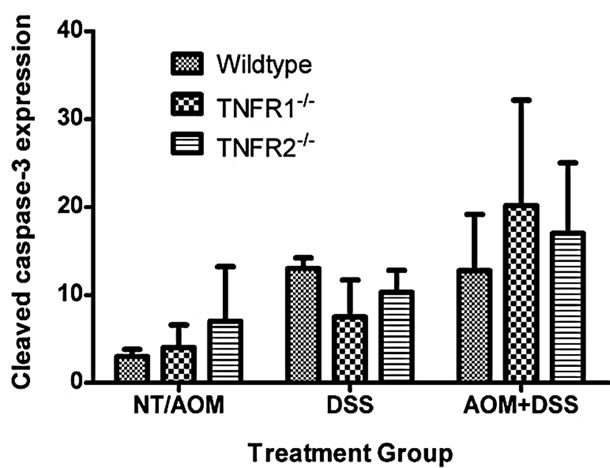
Figure 4. Apoptosis, detected by immunohistochemical detection of activated (cleaved) caspase-3, in the colon of WT, TNFR1-/- and TNFR2-/- mice. The numbers of caspase-3+ cells in the No Treatment (NT) and AOM only groups (3 NT + 3 AOM) were pooled. Caspase 3+ cells in the DSS only groups (n = 6 per strain) and AOM + DSS treated groups (n = 6 per strain) were counted within 100 intact crypts within the colon at a magnification of 400×. As a group, the mice treated with AOM + DSS had significantly greater activated Caspase-3+ cells per 100 crypts compared to NT/AOM only mice (Kruskal-Wallis One-way ANOVA for both strain and treatment).
directly affect tumor development by acting on epithelial cells (Figure 5). We found TNFR1 was expressed on the colonic epithelium in all groups and TNFR2 in both DSS treated WT groups (Figures 5(b) and (c)).
TNFR2 is readily shed during inflammation and differences in levels of shed TNFR2 between WT and TNFR1-/- mice may suggest different mechanisms are operating in these strains. We therefore measured soluble TNFR2 in the serum of mice using an ELISA. No differences were found between the two mouse strains that completed the AOM + DSS regime; however, TNFR1-/- mice treated with DSS alone had lower levels of sTNFR2 (Figure 5(e)). No sTNFR2 was detected in TNFR2-/- mice.
3.6. Colonic Cytokines after the AOM + DSS Regime
We measured cytokines present within the colon by ELISA to determine if there were any differences between strains. TNF, CXCL2 and IL-4 levels in the whole colon were similar between treatment groups and strains when corrected for both tissue weight and protein content. IL-12, however, was significantly increased within the colon of AOM + DSS treated TNFR1-/- mice compared to the other groups (Figure 6). Interferon-levels were similar in all the groups (not shown).
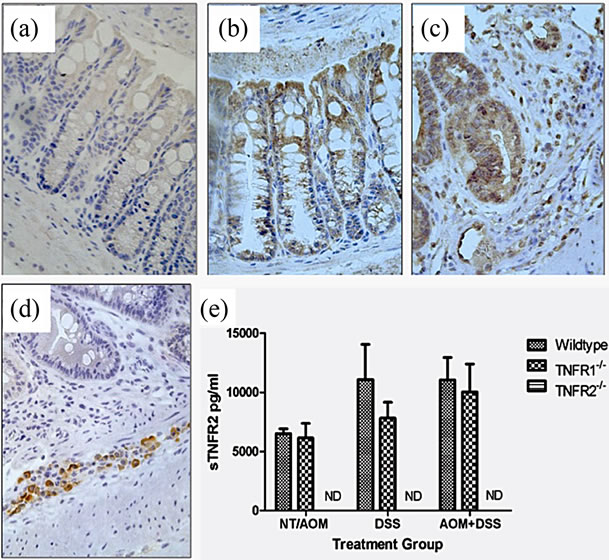
Figure 5. TNFR1 and TNFR2 expression in the colonic epithelium. TNFR1 and TNFR2 were localized by immunohistochemistry in WT mice. (a) Negative control in an untreated WT mouse section with no primary antibody application (400×); (b) TNFR1+ cells in the colon in untreated mouse colon section (400×); (c) TNFR1 expression in an AOM + DSS treated mouse section (400×); (d) TNFR2 expression in an AOM + DSS treated mouse section, with TNFR2 expression limited to cells (leukocytes) within the submucosa (400×); (e) Soluble TNFR2 levels in the serum in WT, TNFR1-/- and TNFR2-/- mice. Shown are the mean and SEM, ND = none detected.
3.7. TNFR1 Bone Marrow Chimeras and Tumor Incidence
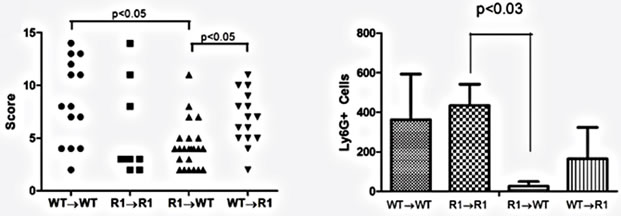
Having established that epithelial cells possess the TNFR1 we next sought to determine whether the tumor-promoting actions of TNF are due to activities directly on mesenchymal and stromal cells, an indirect effect due to activation of hematopoietic cells or possibly both. We created bone marrow chimeras using WT and TNFR1-/- mice and treated them with the AOM + DSS regime. After the final DSS cycle all the mice exhibited inflammation in their colons. The histopathological scores were similar between WT→WT mice and R1→R1 control mice, which suggests there was no confounding influence of the irradiation and reconstitution on the inflammatory response (Figure 7(a)). WT mice had similar histological inflammation to the two control groups; however, R1→WT mice had a significantly decreased level of colonic inflammation compared to both WT→WT and WT→R1 mice. R1→WT mice also showed reduced Ly6G+ cell counts compared to R1→R1 mice but infiltration was similar among the other strains (Figure 7(b)). Examples of infiltrated ulcers found in mice of each strain are shown in Figures 7(c)-(f).
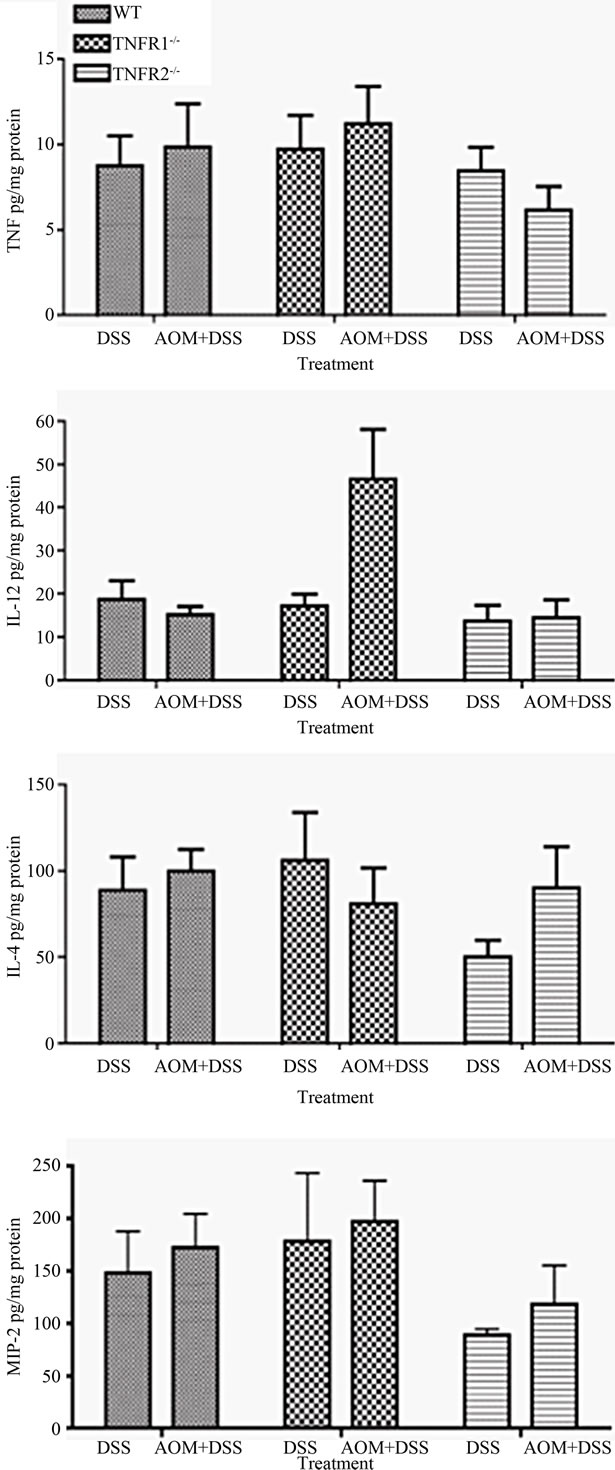
Figure 6. Selected colonic cytokine levels in colitis-associated colon cancer. Cytokine concentrations were measured in whole colon extracts from DSS only (n = 6 - 8) and DSS + AOM treated (n = 10 - 15) mice using an ELISA. Data are presented as mean ± SEM and expressed per mg of protein. Statistics were determined by Kruskal-Wallis ANOVA for both strain and treatment. Significant differences denoted with a star (p < 0.05).
 (a)
(a)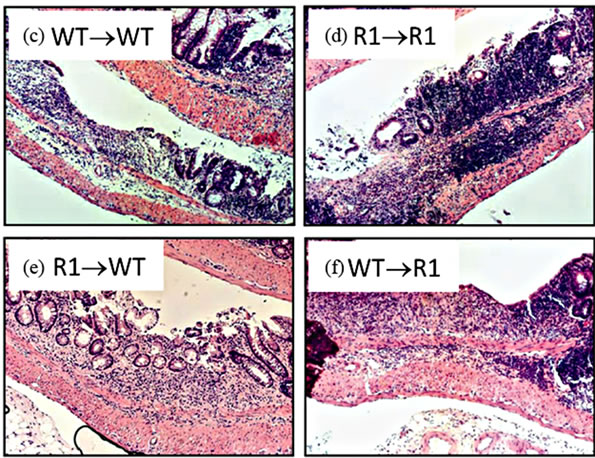 (b)
(b)
Figure 7. TNFR1 on bone-marrow-derived cells contributes to chronic intestinal inflammation. (a) Colon inflammation scores of chimeric mice treated with the AOM + DSS regime. Data are plotted as individual mice. Differences were assessed using Kruskal-Wallis One-way ANOVA with a Dunn’s post-test (p < 0.05). Number of animals per group: WT→WT n = 14, R1→R1 n = 8, R1→WT n = 23, WT→R1 n = 15; (b) Ly6G+ cells within inflamed areas in 5 highpowered fields of colon sections from chimeric mice. Comparisons were measured by Kruskal-Wallis One-way ANOVA; (c)-(f) examples of H & E stained sections from chimeric mice, at 100× magnification, showing areas of inflammation in each group.
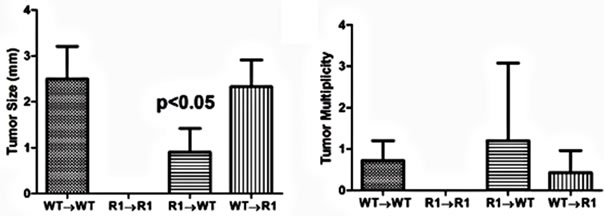
With regard to cancer development, WT→WT mice had an incidence similar to non-chimeric mice (87%). Strikingly, we failed to observe any tumors or dysplasia in R1→R1 mice (incidence of 0%, Table 2). Mice lacking either stromal/mesenchymal (WT→R1) or bonemarrow-derived TNFR1 (R1→WT) had significantly reduced incidences of tumors, roughly half the number compared to WT→WT though higher than R1→R1 mice (Table 2). The average tumor size was significantly reduced in R1→WT mice compared to other groups with tumors (Figure 8(a)), while tumor multiplicity was not affected by chimerism in tumor-bearing reconstituted mice (Figure 8(b)). These chimeric data suggest that in the C57BL/6 strain, leukocyte-selective TNFR1 deficiency can affect inflammation but both hematopoietic and stromal TNFR1 expression can contribute to tumorigenesis. This has to be interpreted in the context of R1→R1 mice not developing any tumors, which implies
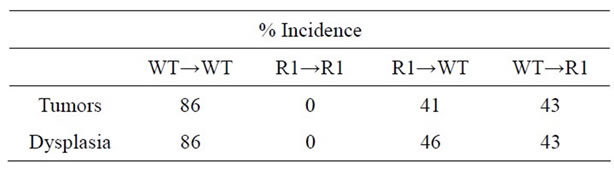
Table 2. Incidence of tumors and dysplasia in chimeric mice treated with AOM + DSS regime.
 (a)
(a)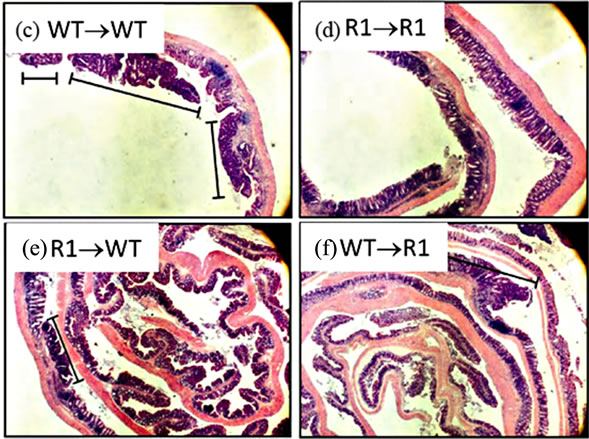 (b)
(b)
Figure 8. Tumor characteristics in TNFR1-/-, wild type chimeric mice. (a) Tumor size from tumor-bearing mice. A one-way ANOVA with a Tukey’s multiple comparison post-test was used to determine differences between groups; (b) Tumor multiplicity in tumor-bearing mice determined by gross examination, with the exception of R1→R1 mice, which did not develop tumors. A one-way ANOVA was used to determine statistically significant differences between groups in tumor-bearing mice; (c)-(f) Representative H & E stained sections from (c) WT→WT mouse; (d) R1→R1 mouse; (e) R1→WT mouse; (f) WT→R1 mouse. All images are 25× original magnification and the lines denote tumors.
that the treatments, while not impacting on inflammation did confound the cancer mechanisms since non-irradiated TNFR1-/- mice did get tumors.
3.8. Oxidative Damage in the Inflamed Colon
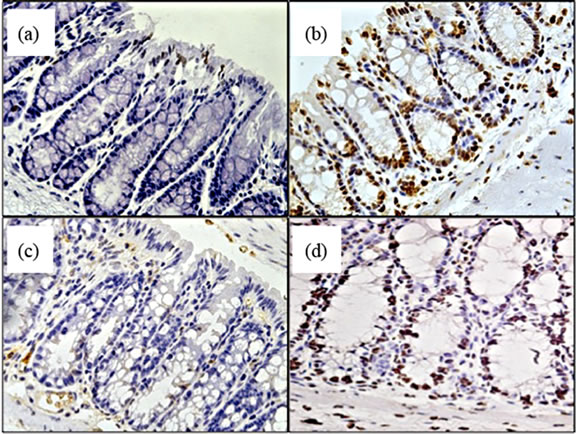
Beginning to investigate the mechanisms behind TNF promoting carcinogenesis, we sought to determine whether TNF differentially mediated oxidative damage in the colon through the two receptor types. Reactive oxygen species (ROS) are produced as byproducts from a range of metabolic processes, including energy metabolism, but are also produced by leukocytes during inflammation to kill bacteria [34]. Normally, excessive ROS are scavenged and neutralized by antioxidant enzymes. Failure of these scavenger systems leads to tissue and cell damage by ROS attack. ROS damage a wide range of cellular processes including cell cycle control and DNA repair, leading to mutations [34]. We assessed whether there were detectable differences in oxidative damage in the colons of TNFR deficient mice by measuring epithelial nuclear 8-OHDG (Figure 9(a)), a DNA adduct formed after exposure to oxygen radicals that promotes DNA mutations favourable to tumorigenesis [35]. We found a significant difference between strains treated with AOM + DSS in the number of epithelial cells exhibiting oxidative damage, with TNFR1-/- mice showing reduced numbers (Figures 9(c) and (e)) compared to WT (Figures 9(b),
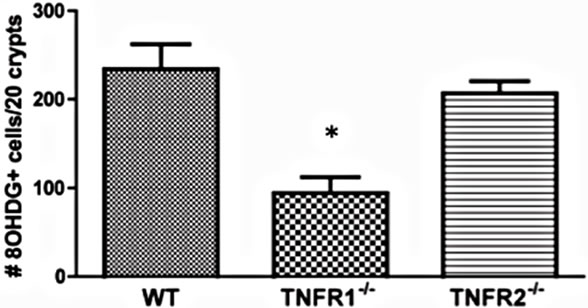 (e)
(e)
Figure 9. Oxidative damage detected by 8-OHDG immunohistochemical staining of colonic epithelial nuclei. (a)-(d) photomicrograph examples of 8-OHDG+ staining within the colonic crypt epithelium of, (a) AOM only treated WT mouse; (b) AOM + DSS treated WT mouse; (c) AOM + DSS treated TNFR1-/- mouse; and (d) AOM + DSS treated TNFR2-/- mouse. Original magnification in all images was 400× and positive nuclei stain brown; (e) The average number of 8-OHDG+ cells within 20 intact colonic crypts from AOM + DSS treated mice. Shown are the mean and SEM. * denotes statistically significant difference, p < 0.05, determined by a Kruskal-Wallis one-way ANOVA with a Dunn’s multiple comparison post-test.
(e)) and TNFR2-/- mice (Figures 9(d) and (e)). Untreated mice had very few 8-OHDG+ nuclei/20 crypts (Figure 9(a)).
3.9. Role of Oxidative Radicals in Colitis-Associated Cancer
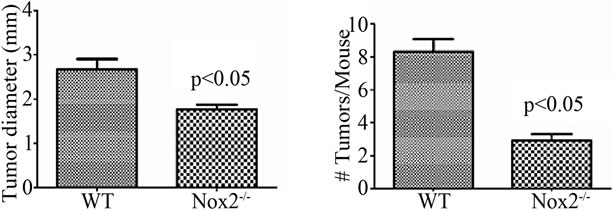
With evidence that TNFR1-/- mice express less oxidative damage, we next determined whether mice deficient in leukocyte ROS production might also experience a lower tumor incidence than WT mice. NADPH oxidase 2 (Nox2) is an enzyme complex critical in the formation of ROS involved in phagocyte function [36]. The contribution of Nox2 has been investigated in acute DSS colitis and mice deficient in the p47-phox subunit are as susceptible to inflammation and illness as WT mice on the C57BL/6 background [37]. We first exposed mice deficient in the Nox2 component, gp91-phox (Nox2-/-), to a single DSS cycle, and confirmed they experienced clinical illness scores and colon inflammation scores similar to C57BL/6 control mice (data not shown). We subsequently treated a cohort of control and Nox2-/- mice to DSS only or the AOM + DSS regime. As with the acute DSS exposure, Nox2-/- mice experienced a pattern of weight loss similar to C57BL/6 mice, only some of the control mice in this series of experiments suffered from a precipitous drop in weight and health during the fourth DSS cycles (Figure 10(a)). An autopsy showed the mice had colonic tumors occluding the lumen. Despite these mice having cancer, only mice that completed the 83 day experiment were used in further analyses. All groups of mice had similar levels of colon inflammation (Figures 10(b) and (c)), including the extent of leukocyte infiltration (Figure 10(e)). All C57BL/6 mice developed colonic tumors while significantly fewer, 58% of the Nox2-/- mice, were observed with tumors (Table 3). The number of mice with dysplasia, while higher at 71%, was still significantly lower than the control strain mouse incidence (Table 3). The numbers of tumors per mouse and size of the tumors were also greater in C57BL/6 mice (Figures 11(a) and (b)). Thus mice with a deficient capacity to produce ROS in leukocytes are less susceptible to develop CRC in the AOM + DSS regime.
4. Discussion
Anti-TNF drugs are being used in increasing numbers of IBD patients and it is highly desirable that we understand how the drugs may change the risk of these patients to develop CRC. Pre-clinical studies in rodents will provide some clues, particularly with regard to predicting changes in humans. Indeed, a precedent has been published reporting that TNFR1-/- mice on the BALB/c genetic background experience fewer tumors than the WT strain, associated with less inflammation, at the conclusion of the
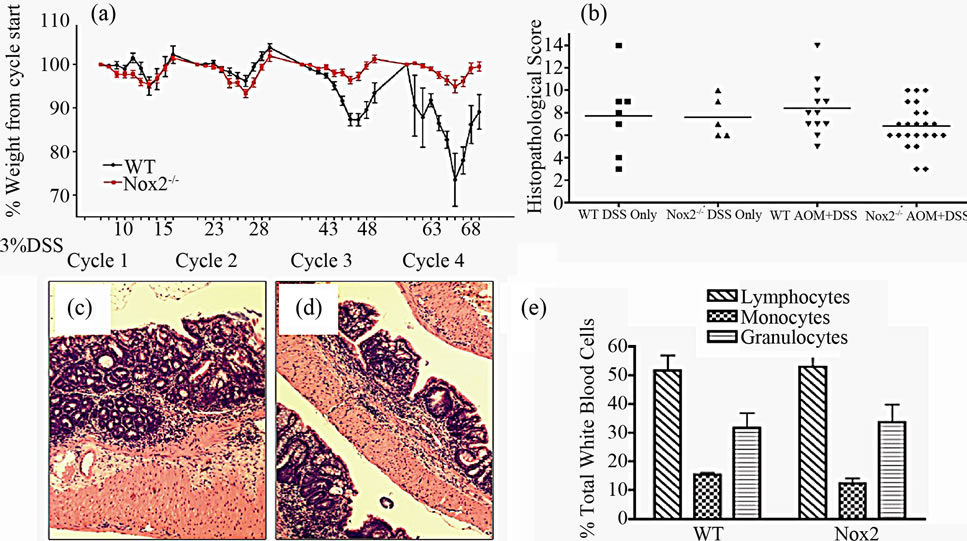
Figure 10. Characteristics of chronic colitis in Nox2-/- mice. (a) Pattern of weight loss during the 83-day experiment in WT (n = 22) and Nox2-/- (n = 24) mice during the AOM + DSS regime. Despite some C57BL/6 mice in this series of experiments experiencing precipitous weight loss during the 4th DSS cycle (requiring euthanasia), there is no significant difference between strains over the course of the experiment assessed by a repeated measures ANOVA (p < 0.05); (b) Colon inflammation scores are not significantly different between WT (n = 12) and Nox2-/- (n = 24) mice after treatment. Only those mice that were present at the end of the experiment were included in scoring. Representative H & E stained section showing inflammation within the mid-colon in (c) WT and (d) Nox2-/- mice (100× original magnification); (e) Blood leukocyte differential in WT and Nox2-/- mice following treatment with the AOM + DSS regime.
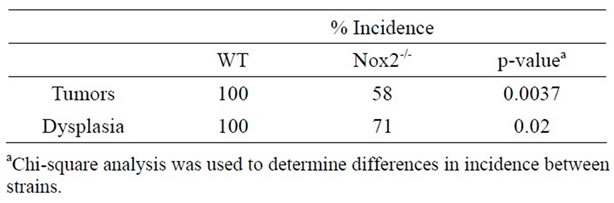
Table 3. Incidence of colon tumors and dysplasia WT and Nox2-/- mice treated with the AOM + DSS regime.
AOM + DSS regime [18,19]. The reduced tumor incidence was linked to hematopoietic cell loss of the receptor, shown using receptor chimeric animals. So our finding that TNFR1-/- mice on the C57BL/6 genetic background have reduced tumor incidence and dysplasia incidence and tumor load may not be unexpected. However, our results build on this observation by showing that in the C57BL/6 strain the reduction in tumors is not associated with reduced colon inflammation and that both hematopoietic and nonhematopoietic cell loss of the TNFR1 contributed to this phenotype. The differences in our outcomes may be influenced by the fact that BALB/c mice are less sensitive to DSS treatment compared to C57BL/6 mice [38] and that BALB/c mice are much more sensitive to AOM when compared to C57BL/6 mice [39]. Finally, we also show that the TNFR2 does not play a role in CAC, either cell-bound or as a shed, soluble antagonist. Our results identify that the mechanism behind tumorigenesis is separate from the mecha-
 (a)
(a)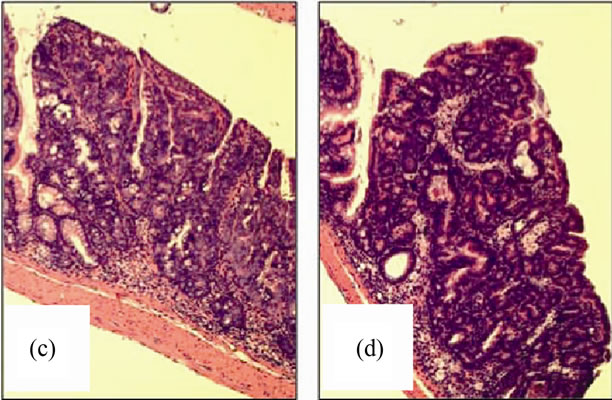 (b)
(b)
Figure 11. Nox2-/- mice are protected from AOM + DSS tumor development. (a) The average tumor diameter was significantly lower in Nox2-/- mice. Significance was determined using a two-tailed t-test; (b) Tumor multiplicity was significantly reduced in Nox2-/- mice. Significance was determined using a two-tailed t-test. H & E stained sections showing a non-invasive adenocarcinoma in (c) WT and (d) Nox2-/- mice (100× original magnification for both images).
nisms that drive the inflammation. This finding supports studies using drugs instead of receptor deficient animals, as both Etanercept and Infliximab have been shown to prevent CAC in a DSS mouse model, also without affecting inflammation [18,19]. Together these observations have profound implications for treating chronic IBD. While possibly ineffective at reducing inflammation due to IBD, continued use of TNF antagonists may still reduce the risk of CRC. This understanding from pre-clinical research should lead to discussions over whether to discontinue treatments in the non-responsive patients, who of course are at the greatest risk of CRC.
Contrasting this literal and optimistic translation of ours and others’ findings, other models of CAC have reported no effect of TNF antagonism on cancer outcomes. Sakai et al. showed that TNF deficiency in an Apcmin+/− strain on the C57BL/6 background did not lead to a reduction in chronic colitis or tumor development in mice also treated with DSS [20]. Apcmin+/− mice spontaneously develop tumors throughout the small and large intestine and DSS further promotes the development of colonic tumors [40,41]. In agreement with our study, the level of colonic inflammation after DSS treatment was similar between TNF−/− and TNF+/+ mice; however, DSS was only given once, not accounting for the remitting recurring course of the disease [20]. Small intestinal tumor development was also not affected by the presence or absence of TNF. It is possible that the Apcmin+/− mutation ablated the protection that TNF deficiency might have otherwise provided in this model, as previous studies suggested that TNF deficiency was fatal in DSS colitis on a C57BL/6 background, the same background used in the study by Sakai et al. [10].
In our study, despite a similar level of inflammation tumor severity was reduced when TNFR1 was absent. This suggests that an element of TNF signaling that does not directly affect inflammation is responsible for the reduction in cancer susceptibility. It also suggests that inflammation is not necessarily directly related to tumorigenesis. Indirectly supporting this finding is our observation that most of the tumors were found in the mid to distal colon, with the majority of tumors located close to the anus, while inflammation is typically more proximal. This suggests that the anatomical location of the inflammation may not be as important as systemic inflammation in the development of colonic tumors. This also has support from past observations in IBD where microscopic colitis can influence tumor development as opposed to the presence of gross inflammation [42].
Beginning to dissect the mechanisms behind the protumorigenic activity of TNF, we generated irradiated bone marrow chimeras to determine whether the reduction in cancer incidence between WT and TNFR1-/- mice was due to the receptor deficiency on leukocytes versus on epithelial and other stromal/mesenchymal (e.g. nonhematopoietic) cells. Our data for the cancer incidence of these chimeras, at roughly half the incidence of control mice, supports the interpretation that TNFR1 expression on both cell sources is important for TNF led tumor development. Exactly which cell type(s) expressing TNFR1 contribute to this pathway to tumor development remains to be determined. Our finding that nonhematopoietic TNFR1 deficiency results in a significant reduction in tumor incidence differs from the results of Papivanova et al., who reported that TNFR1 on leukocytes alone was important [18]. Noteworthy in their study, BALB/c mice did not show colonic epithelial TNFR1 expression therefore it is possible that pro-tumorigenic signaling through TNFR1 did not occur in epithelial cells in the BALB/c strain.
Our conclusion that the reduced colon cancer incidence in mice lacking TNFR1 expression in bone marrowderived cells has a caveat since the mice also had a significantly decreased colon inflammation score compared to WT mice reconstituted with WT bone-marrow. This means the chimera’s response better resembles the response seen in BALB/c mice. We do not know the mechanism behind the reduced inflammation but if TNFR1 is pathogenic acting through the bone marrow derived cells, it is possible that it is protective when expressed in the stromal/epithelial cells, and that this protective mechanism manifests as less inflammation in the chimeras. TNFR1 potently activates NFκB and while NFκB drives inflammatory mediator production in most cell types, NFκB activation can be beneficial in promoting epithelial barrier properties, which would presumably act to limit inflammation [32,30]. Nevertheless, we believe the mechanism for the reduced inflammation is not linked to the carcinogenic mechanisms since this association was already lost in non-chimeric TNFR1-/- animals.
Further to a possible direct effect of TNF, it may play role in homeostasis of the colonic epithelium. TNF in some circumstances can lead to epithelial cell apoptosis [43,44] and treatment with anti-TNF therapy may reduce colonic epithelial cell apoptosis [45]. In this case we would predict changes in the number of apoptotic cells and whether an alteration in this cause of apoptosis by TNFR deficiency would affect cancer incidence is unknown. However, this idea was not supported by our measure of activated caspase-3+ cells. There were no differences between strains in the number of activated caspase-3+ cells within the colon, suggesting that TNFRs likely do not play a significant role in controlling the rate of colonic epithelial apoptosis, at least in the AOM + DSS model.
TNF is a potent stimulus for multiple inflammatory cytokines and one might predict that the lack of TNF receptors would be reflected in other cytokine levels. While we did not conduct a comprehensive survey of cytokines our finding that representative pro-inflammatory cytokines, TNF and CXCL2, were not different between treatments or strains is consistent with the lack of difference in inflammation between the strains. While it has been reported that serum and colonic TNF are elevated during intestinal inflammation [3], this seem to not always be the case [46,47]. CXCL2, signaling through CXCR2, is chemotactic for neutrophils [48]. This chemokine is one of a larger family any number of which may be important in DSS-colitis, as treating mice with a CXCR2 blocking antiserum or a genetic deficiency for CXCR2 reduces DSS colitis [49,50]. IL-4 is regarded as an anti-inflammatory mediator although it is reported elevated in the DSS inflamed colon [51]. Again, there were no significant differences between levels among the strains. The one cytokine that we did find changed happened to be elevated IL-12 limited to TNFR1-/- mice treated with AOM + DSS. IL-12 is produced predominantly by dendritic cells inducing T cell maturation and differentiation [52], and is also involved in the cytotoxic response by CD8+ T cells and natural killer cells. Increased IL-12 could therefore be indicative of an increase in anti-cancer immunity, partially explaining a protective effect of TNFR1 deficiency in this model [53]. To be more confident in this possibility it would be important to know when in the course of the AOM + DSS regime levels may have become elevated.
Finally, with regard to possible mechanisms, oxidative stress, which is increased in colitis [54], has not previously been linked to intestinal TNFR1 in vivo. On the other hand, TNFR1 has been shown to contribute to oxidative stress in an animal model of lung injury [52]. We observed that TNFR1-/- mice had significantly fewer 8-OHDG+ colonic epithelial nuclei compared to WT and TNFR2-/- mice. 8-OHDG is a DNA adduct formed after oxidative damage that can lead to translocations and alterations in gene expression, and is normally cleaved by a DNA repair enzyme. High levels of 8-OHDG are associated with tumorigenesis [55] and is found elevated in UC patients with dysplasia [56]. As TNFR1 directly associates with Nox enzymes [21], it is possible that TNFR1 deficiency reduces oxidative stress, potentially reducing CAC. While our data showing reduced oxidative damage implicate oxygen radicals in the tumorigenic mechanism, we could not conclude that the reduction in oxidative damage within the epithelium of TNFR1-/- mice is a result of reduced ROS production by infiltrating leukocytes. It is possible that internal production of ROS by colonic epithelial cells, mediated by the NADPH oxidase isoform Nox1, is responsible for the difference in 8-OHDG levels. Nox1 can be directly activated [57] and upregulated by TNF [22], and Nox1 has been associated with CAC in the colon [58]. To distinguish between the contributions of these two possible cell sources of ROS we chose to examine mice deficient in leukocyte Nox2 for their cancer incidence. We indeed observed that the mice experienced fewer colon tumors than wildtype mice, despite similar levels of inflammation. In this series of experiments the high tumor incidence in the wildtype strain was unexpected but may be due to the azoxymethane oxidizing and becoming more potent during storage but at least the same batch was used in both strains of mice, so we are confident in the conclusion that Nox2-/- mice experienced less cancer.
In conclusion, TNFR1 deficiency is protective against the development of CAC and this protective effect is independent of the severity of inflammation. This means TNF promotes cancer development through TNFR1, which we show is dependent on both leukocytic and nonhematopoietic cell TNFR1 expression. Finally, a reduction in oxidative damage by leukocytes partially accounts for the protective effect of TNFR1 deficiency. The implications are that TNF antagonist treatments ought to continue to be used in the non-responsive IBD patient as the treatments may reduce the patient’s CRC risk.
5. Acknowledgements
We wish to thank Hana James for her technical expertise throughout the study. The research was funded by a Cancer Research Training Program (CRTP) studentship to RMS, and grants from the CRTP and Canadian Cancer Society (Nova Scotia Division) to AWS.
REFERENCES
- C. Canavan, K. R. Abrams and J. Mayberry, “MetaAnalysis: Colorectal and Small Bowel Cancer Risk in Patients with Crohn’s Disease,” Alimentary Pharmacology & Therapeutics, Vol. 23, No. 8, 2006, pp. 1097-1104. doi:10.1111/j.1365-2036.2006.02854.x
- J. A. Eaden, K. R. Abrams and J. F. Mayberry, “The Risk of Colorectal Cancer in Ulcerative Colitis: A MetaAnalysis,” Gut, Vol. 48, No. 4, 2001, pp. 526-535. doi:10.1136/gut.48.4.526
- S. H. Murch, C. P. Braegger, J. A. Walker-Smith and T. T. MacDonald, “Location of Tumour Necrosis Factor a by Immunohistochemistry in Chronic Inflammatory Bowel Disease,” Gut, Vol. 34, No. 12, 1993, pp. 1705-1709. doi:10.1136/gut.34.12.1705
- S. H. Murch, V. A. Lamkin, M. O. Savage, J. A. Walker-Smith and T. T. MacDonald, “Serum Concentrations of Tumour Necrosis Factor a in Childhood Chronic Inflammatory Bowel Disease,” Gut, Vol. 32, No. 8, 1991, pp. 913-917. doi:10.1136/gut.32.8.913
- M. Karin and E. Gallagher, “TNFR Signaling: Ubiquitin-Conjugated Traffic Signals Control Stop-and-Go for MAPK Signaling Complexes,” Immunological Reviews, Vol. 228, No. 1, 2009, pp. 225-240. doi:10.1111/j.1600-065X.2008.00755.x
- G. Kojouharoff, W. Hans, F. Obermeier, D. N. Männel, T. Andus, J. Schölmerich, V. Gross and W. Falk, “Neutralization of Tumour Necrosis Factor (TNF) But Not of IL-1 Reduces Inflammation in Chronic Dextran Sulphate Sodium-Induced Colitis in Mice,” Clinical & Experimental Immunology, Vol. 107, No. 2, 1997, pp. 353-358. doi:10.1111/j.1365-2249.1997.291-ce1184.x
- K. Kinoshita, M. Hori, M. Fujisawa, K. Sato, T. Ohama, E. Momotani and H. Ozaki, “Role of TNF-Alpha in Muscularis Inflammation and Motility Disorder in a TNBSInduced Colitis Model: Clues from TNF-Alpha-Deficient Mice,” Neurogastroenterology & Motility, Vol. 18, No. 7, 2006, pp. 578-588. doi:10.1111/j.1365-2982.2006.00784.x
- M. Nakai, K. Sudo, Y. Yamada, Y. Kojima, T. Kato, K. Saito, H. Moriwaki and M. Seishima, “The Role of the Tumor Necrosis Factor Receptor in 2,4,6-Trinitrobenzene Sulfonic Acid (TNBS)-Induced Colitis in Mice,” Digestive Diseases and Sciences, Vol. 50, No. 9, 2005, pp. 1669-1676. doi:10.1007/s10620-005-2913-1
- R. M. Stillie and A. W. Stadnyk, “Role of TNF Receptors, TNFR1 and TNFR2, in Dextran Sodium Sulfate-Induced Colitis,” Inflammatory Bowel Diseases, Vol. 15, No. 10, 2009, pp. 1515-1525. doi:10.1002/ibd.20951
- Y. Naito, T. Takagi, O. Handa, T. Ishikawa, S. Nakagawa, T. Yamaguchi, N. Yoshida, M. Minami, M. Kita, J. Imanishi and T. Yoshikawa, “Enhanced Intestinal Inflammation Induced by Dextran Sulfate Sodium in Tumor Necrosis Factor-Alpha Deficient Mice,” Journal of Gastroenterology and Hepatology, Vol. 18, No. 5, 2003, pp. 560-569. doi:10.1046/j.1440-1746.2003.03034.x
- E. Mizoguchi, Y. Hachiya, M. Kawada, K. Nagatani, A. Ogawa, K. Sugimoto, A. Mizoguchi and D. K. Podolsky, “TNF Receptor Type I-Dependent Activation of Innate Responses to Reduce Intestinal Damage-Associated Mortality,” Gastroenterology, Vol. 134, No. 2, 2008, pp. 470- 480. doi:10.1053/j.gastro.2007.11.055
- W. J. Sandborn, S. B. Hanauer, S. Katz, M. A. Safdi, D. G. Wolf, R. D. Baerg, W. J. Tremaine, T. Johnson, N. N. Diehl and A. R. Zinsmeister, “Etanercept for Active Crohn’s Disease: A Randomized, Double-Blind, Placebo-Controlled Trial,” Gastroenterology, Vol. 121, No. 5, 2001, pp. 1088-1094. doi:10.1053/gast.2001.28674
- B. E. Sands, W. J. Tremaine, W. J. Sandborn, P. J. Rutgeerts, S. B. Hanauer, L. Mayer, S. R. Targan and D. K. Podolsky, “Infliximab in the Treatment of Severe, Steroid-Refractory Ulcerative Colitis: A Pilot Study,” Inflammatory Bowel Diseases, Vol. 7, No. 2, 2001, pp. 83- 88. doi:10.1097/00054725-200105000-00001
- S. B. Hanauer, W. J. Sandborn, P. J. Rutgeerts, R. N. Fedorak, M. Lukas, D. MacIntosh, R. Panaccione, D. Wolf and P. Pollack, “Human Anti-Tumor Necrosis Factor Monoclonal Antibody (Adalimumab) in Crohn’s Disease: the Classic-I Trial,” Gastroenterology, Vol. 130, No. 2, 2006, pp. 323-333. doi:10.1053/j.gastro.2005.11.030
- J. F. Colombel, W. J. Sandborn, P. Rutgeerts, R. Enns, S. B. Hanauer, R. Panaccione, S. Schreiber, D. Byczkowski, J. Li, J. D. Kent and P. F. Pollack, “Adalimumab for Maintenance of Clinical Response and Remission in Patients with Crohn’s Disease: The CHARM Trial,” Gastroenterology, Vol. 132, No. 1, 2007, pp. 52-65. doi:10.1053/j.gastro.2006.11.041
- E. A. Carswell, L. J. Old, R. L. Kassel, S. Green, N. Fiore and B. Williamson, “An Endotoxin-Induced Serum Factor That Causes Necrosis of Tumors,” Proceedings of the National Academy of Science of the Unite States of America, Vol. 72, No. 9, 1975, pp. 3666-3670. doi:10.1073/pnas.72.9.3666
- F. Balkwill and C. Joffroy, “TNF: A Tumor-Suppressing Factor or a Tumor-Promoting Factor?” Future Oncology, Vol. 6, No. 12, 2010, pp. 1833-1836. doi:10.2217/fon.10.155
- B. K. Popivanova, M. Kitamura, Y. Wu, T. Kondo, T. Kagaya, S. Kaneko, M. Oshima, C. Fujii and N. Mukaida, “Blocking TNF-Alpha in Mice Reduces Colorectal Car- cinogenesis Associated with Chronic Colitis,” Journal of Clinical Investigation, Vol. 118, No. 2, 2008, pp. 560- 570.
- M. Onizawa, T. Nagaishi, T. Kanai, K. Nagano, S. Oshima, Y. Nemoto, A. Yoshioka, T. Totsuka, R. Okamoto, T. Nakamura, N. Sakamoto, K. Tsuchiya, K. Aoki, K. Ohya, H. Yagita and M. Watanabe, “Signaling Pathway via TNF-Alpha/NF-kappaB in Intestinal Epithelial Cells May Be Directly Involved in Colitis-Associated Carcinogenesis,” American Journal of Physiology Gastrointestinal and Liver Physiology, Vol. 296, No. 4, 2009, pp. G850-G859. doi:10.1152/ajpgi.00071.2008
- H. Sakai, Y. Yamada, M. Shimizu, K. Saito, H. Moriwaki and A. Hara, “Genetic Ablation of Tnfá Demonstrates No Detectable Suppressive Effect on Inflammation-Related Mousecolon Tumorigenesis,” Chemico-Biological Interactions, Vol. 184, No. 3, 2010, pp. 423-430. doi:10.1016/j.cbi.2010.01.014
- B. Yazdanpanah, K. Wiegmann, V. Tchikov, O. Krut, C. Pongratz, M. Schramm, A. Kleinridders, T. Wunderlich, H. Kashkar, O. Utermohlen, J. C. Bruning, S. Schutze and M. Kronke, “Riboflavin Kinase Couples TNF Receptor 1 to NADPH Oxidase,” Nature, Vol. 460, No. 7259, 2009, pp. 1159-1163. doi:10.1038/nature08206
- Y. Kuwano, K. Tominaga, T. Kawahara, H. Sasaki, K. Takeo, K. Nishida, K. Masuda, T. Kawai, S. TeshimaKondo and K. Rokutan, “Tumor Necrosis Factor Alpha Activates Transcription of the NADPH Oxidase Organizer 1 (NOXO1) Gene and Upregulates Superoxide Production in Colon Epithelial Cells,” Free Radical Biology and Medicine, Vol. 45, No. 12, 2008, pp. 1642-1652. doi:10.1016/j.freeradbiomed.2008.08.033
- B. Yan, H. Wang, Z. N. Rabbani, Y. Zhao, W. Li, Y. Yuan, F. Li, M. W. Dewhirst and C. Y. Li, “Tumor Necrosis Factor-Alpha Is a Potent Endogenous Mutagen That Promotes Cellular Transformation,” Cancer Research, Vol. 66, No. 24, 2006, pp. 11565-11570. doi:10.1158/0008-5472.CAN-06-2540
- R. vanOs, H. D. Thames, A. W. Konings and J. D. Down, “Radiation Dose-Fractionation and Dose-Rate Relationships for Long-Term Repopulating Hemopoietic Stem Cells in a Murine Bone Marrow Transplant Model,” Radiation Research, Vol. 136, No. 1, 1993, pp. 118-125. doi:10.2307/3578648
- M. Aparicio-Vergara, R. Shiri-Sverdlov, G. de Haan and M. H. Hofker, “Bone Marrow Transplantation in Mice as a Tool for Studying the Role of Hematopoietic Cells in Metabolic and Cardiovascular Diseases,” Atherosclerosis, Vol. 213, No. 2, 2010, pp. 335-344. doi:10.1016/j.atherosclerosis.2010.05.030
- S. H. Itzkowitz and X. Yio, “Inflammation and Cancer. IV. Colorectal Cancer in Inflammatory Bowel Disease: The Role of Inflammation,” American Journal of Physiology Gastrointestinal and Liver Physiology, Vol. 287, No. 1, 2004, pp. G7-G17. doi:10.1152/ajpgi.00079.2004
- E. Forbes, T. Murase, M. Yang, K. I. Matthaei, J. J. Lee, N. A. Lee, P. S. Foster and S. P. Hogan, “Immunopathogenesis of Experimental Ulcerative Colitis Is Mediated by Eosinophil Peroxidase,” The Journal of Immunology, Vol. 172, No. 9, 2004, pp. 5664-5675.
- F. Legrand, V. Driss, M. Delbeke, S. Loiseau, E. Hermann, D. Dombrowicz and M. Capron, “Human Eosinophils Exert TNF-á and Granzyme A-Mediated Tumoricidal Activity toward Colon Carcinoma Cells,” The Journal of Immunology, Vol. 185, No. 12, 2010, pp. 7443-7451. doi:10.4049/jimmunol.1000446
- A. Nenci, C. Becker, A. Wullaert, R. Gareus, G. van Loo, S. Danese, M. Huth, A. Nikolaev, C. Neufert, B. Madison, D. Gumucio and M. F. Neurath, “Epithelial NEMO Links Innate Immunity to Chronic Intestinal Inflammation,” Nature, Vol. 446, No. 7135, 2007, pp. 557-561. doi:10.1038/nature05698
- R. Kajino-Sakamoto, M. Inagaki, E. Lippert, S. Akira, S. Robine, K. Matsumoto, C. Jobin and J. Ninomiya-Tsuji, “Enterocyte-Derived TAK1 Signaling Prevents Epithelium Apoptosis and the Development of Ileitis and Colitis,” The Journal of Immunology, Vol. 181, No. 2, 2008, pp. 1143-1152.
- E. Mizoguchi, A. Mizoguchi, H. Takedatsu, E. Cario, Y. P. De Jong, C. J. Ooi, R. J. Xavier, C. Terhorst, D. K. Podolsky and A. K. Bhan, “Role of Tumor Necrosis Factor Receptor 2 (TNFR2) in Colonic Epithelial Hyperplasia and Chronic Intestinal Inflammation in Mice,” Gastroenterology, Vol. 122, No. 1, 2002, pp. 134-144. doi:10.1053/gast.2002.30347
- F. Wang, B. T. Schwarz, W. V. Graham, Y. Wang, L. Su, D. R. Clayburgh, C. Abraham and J. R. Turner, “IFNGamma-Induced TNFR2 Expression Is Required for TNF-Dependent Intestinal Epithelial Barrier Dysfunction,” Gastroenterology, Vol. 131, No. 4, 2006, pp. 1153- 1163. doi:10.1053/j.gastro.2006.08.022
- K. L. Edelblum, J. A. Goettel, T. Koyama, S. J. McElroy, F. Yan and D. B. Polk, “TNFR1 Promotes Tumor Necrosis Factor-Mediated Mouse Colon Epithelial Cell Survival through RAF Activation of NF-kappaB,” The Journal of Biological Chemistry, Vol. 283, No. 43, 2008, pp. 29485-29494. doi:10.1074/jbc.M801269200
- S. P. Hussain, L. J. Hofseth and C. C. Harris, “Radical Causes of Cancer,” Nature Reviews Cancer, Vol. 3, No. 4, 2003, pp. 276-285. doi:10.1038/nrc1046
- H. Kasai, “Analysis of a Form of Oxidative DNA Damage, 8-Hydroxy-2’-Deoxyguanosine, as a Marker of Cellular Oxidative Stress during Carcinogenesis,” Mutation Research, Vol. 387, No. 3, 1997, pp. 157-163.
- B. Rada and T. L. Leto, “Oxidative Innate Immune Defenses by Nox/Duox Family NADPH Oxidases,” Contributions to Microbiology, Vol. 15, 2008, pp. 164-187. doi:10.1159/000136357
- C. F. Krieglstein, W. H. Cerwinka, F. S. Laroux, J. W. Salter, J. M. Russell, G. Schuermann, M. B. Grisham, C. R. Ross and D. N. Granger, “Regulation of Murine Intestinal Inflammation by Reactive Metabolites of Oxygen and Nitrogen: Divergent Roles of Superoxide and Nitric Oxide,” The Journal of Cell Biology, Vol. 194, No. 9, 2001, pp. 1207-1218. doi:10.1084/jem.194.9.1207
- S. Melgar, A. Karlsson and E. Michaëlsson, “Acute Colitis Induced by Dextran Sulfate Sodium Progresses to Chronicity in C57Bl/6 but Not in BALB/c Mice: Correlation between Symptoms and Inflammation,” American Journal of Physiology Gastrointestinal and Liver Physiology, Vol. 288, No. 6, 2005, pp. G1328-G1338. doi:10.1152/ajpgi.00467.2004
- R. Suzuki, H. Kohno, S. Sugie, H. Nakagama and T. Tanaka, “Strain Differences in the Susceptibility to Azoxymethane and Dextran Sodium Sulfate-Induced Colon Carcinogenesis in Mice,” Carcinogenesis, Vol. 27, No. 1, 2010, pp. 162-169. doi:10.1093/carcin/bgi205
- T. Tanaka, H. Kohno, R. Suzuki, K. Hata, S. Sugie, N. Niho, K. Sakano, M. Takahashi and K. Wakabaysahi, “Dextran sodium Sulfate Strongly Promotes Colorectal Carcinogenesis in Apc(Min/+) Mice: Inflammatory Stimuli by Dextran Sodium Sulfate Results in Development of Multiple Colonic Neoplasms,” International Journal of Cancer, Vol. 118, No. 1, 2006, pp. 25-34. doi:10.1002/ijc.21282
- K. Yoshimi, T. Tanaka, A. Takizawa, M. Kato, M. Hirabayashi, T. Mashimo, T. Serikawa and T. Kuramoto, “Enhanced Colitis-Associated Colon Carcinogenesis in a Novel Apc Mutant Rat,” Cancer Science, Vol. 100, No. 11, 2009, pp. 2022-2027. doi:10.1111/j.1349-7006.2009.01287.x
- C. Mathy, K. Schneider, Y. Y. Chen, M. Varma, J. P. Terdiman and U. Mahadevan, “Gross versus Microscopic Pancolitis and the Occurrence of Neoplasia in Ulcerative Colitis,” Inflammatory Bowel Diseases, Vol. 9, No. 6, 2003, pp. 351-355. doi:10.1097/00054725-200311000-00002
- P. F. Piguet, C. Vesin, Y. Donati and C. Barazzone, “TNF-Induced Enterocyte Apoptosis and Detachment in Mice: Induction of Caspases and Prevention by a Caspase Inhibitor, ZVAD-Fmk,” Laboratory Investigation, Vol. 79, No. 4, 1999, pp. 495-500.
- P. F. Piguet, C. Vesin, J. Guo, Y. Donati and C. Barazzone, “TNF-Induced Enterocyte Apoptosis in Mice Is Mediated by the TNF Receptor 1 and Does Not Require p53,” European Journal of Immunology, Vol. 28, No. 11, 1998, pp. 3499-3505. doi:10.1002/(SICI)1521-4141(199811)28:11<3499::AID-IMMU3499>3.0.CO;2-Q
- S. Zeissig, C. Bojarski, N. Buergel, J. Mankertz, M. Zeitz, M. Fromm and J.-D. Schulzke, “Downregulation of Epithelial Apoptosis and Barrier Repair in Active Crohn’s Disease by Tumour Necrosis Factor Alpha Antibody Treatment,” Gut, Vol. 53, No. 9, 2004, pp. 1295-1302. doi:10.1136/gut.2003.036632
- J. S. Hyams, W. R. Treem, E. Eddy, N. Wyzga and R. E. Moore, “Tumor Necrosis Factor-A Is Not Elevated in Children with Inflammatory Bowel Disease,” Journal of Pediatric Gastroenterology & Nutrition, Vol. 12, No. 2, 1991, pp. 233-236. doi:10.1097/00005176-199102000-00016
- O. H. Nielsen, J. Brynskov and K. Bendtzen, “Circulating and Mucosal Concentrations of Tumour Necrosis Factor and Inhibitor(s) in Chronic Inflammatory Bowel Disease,” Danish Medical Bulletin, Vol. 40, No. 2, 1993, pp. 247-249.
- M. E. Spehlmann, S. M. Dann, P. Hruz, E. Hanson, D. F. McCole and L. Eckmann, “CXCR2-Dependent Mucosal Neutrophil Influx Protects Against Colitis-Associated Diarrhea Caused by an Attaching/Effacing Lesion-Forming Bacterial Pathogen,” The Journal of Immunology, Vol. 183, No. 5, 2009, pp. 3332-3343. doi:10.4049/jimmunol.0900600
- S. M. Farooq, R. M. Stillie, C. Svanborg, M. Svensson, R. M. Strieter and A. W. Stadnyk, “Therapeutic Effect of Antagonizing CXCR2 on Neutrophil Infiltration and DSS Induced Colitis,” The Journal of Pharmacology Experimental Therapeutics, Vol. 329, No. 1, 2009, pp. 123-129. doi:10.1124/jpet.108.145862
- P. Buanne, E. Di Carlo, L. Caputi, L. Brandolini, M. Mosca, F. Cattani, L. Pellegrini, L. Biordi, G. Coletti, C. Sorrentino, G. Fedele, F. Colotta, G. Melillo and R. Bertini, “Crucial Pathophysiological Role of CXCR2 in Experimental Ulcerative Colitis in Mice,” Journal of Leukocyte Biology, Vol. 82, No. 5, 2007, pp. 1239-1246. doi:10.1189/jlb.0207118
- P. Alex, N. C. Zachos, T. Nguyen, L. Gonzales, T. -E. Chen, L. S. Conklin, M. Centola and X. Li, “Distinct Cytokine Patterns Identified From Multiplex Profiles of Murine DSS and TNBS-Induced Colitis,” Inflammatory Bowel Diseases, Vol. 15, No. 3, 2009, pp. 341-352. doi:10.1002/ibd.20753
- T. S. Lee, H. C. Yen, C. C. Pan and L. Y. Chau, “The Role of Interleukin 12 in the Development of Atherosclerosis in ApoE-Deficient Mice,” Arteriosclerosis, Thrombosis and Vascular Biology, Vol. 19, No. 3, 1999, pp. 734-742. doi:10.1161/01.ATV.19.3.734
- M. A. EngelandM. F. Neurath, “Anticancer Properties of the IL-12 Family—Focus on Colorectal Cancer,” Current Medicinal Chemistry, Vol. 17, No. 29, 2010, pp. 3303- 3308.
- Y. Dincer, Y. Erzin, S. Himmetoglu, K. N. Gunes, K. Bal and T. Akcay, “Oxidative DNA Damage and Antioxidant Activity in Patients with Inflammatory Bowel Disease,” Digestive Diseases and Sciences, Vol. 52, No. 7, 2007, pp. 1636-1641. doi:10.1007/s10620-006-9386-8
- J. Liao, D. N. Seril, G. G. Lu, M. Zhang, S. Toyokuni, A. L. Yang and G. Y. Yang, “Increased Susceptibility of Chronic Ulcerative Colitis-Induced Carcinoma Develop- ment in DNA Repair Enzyme Ogg1 Deficient Mice,” Molecular Carcinogenesis, Vol. 47, No. 8, 2008, pp. 638- 646.
- R. D’Inca, R. Cardin, L. Benazzato, I. Angriman, D. Martines and G. C. Sturniolo, “Oxidative DNA Damage in the Mucosa of Ulcerative Colitis Increases with Disease Duration and Dysplasia,” Inflammatory Bowel Diseases, Vol. 10, No. 1, 2004, pp. 23-27. doi:10.1097/00054725-200401000-00003
- Y. S. Kim, M. J. Morgan, S. Choksi and Z. G. Liu, “TNF-Induced Activation of the Nox1 NADPH Oxidase and Its Role in the Induction of Necrotic Cell Death,” Molecular Cell, Vol. 26, No. 5, 2007, pp. 675-687. doi:10.1016/j.molcel.2007.04.021
- K. Rokutan, T. Kawahara, Y. Kuwano, K. Tominaga, A. Sekiyama and S. Teshima-Kondo, “NADPH Oxidases in the Gastrointestinal Tract: A Potential Role of Nox1 in Innate Immune Response and Carcinogenesis,” Antioxidants & Redox Signaling, Vol. 8, No. 9-10, 2006, pp. 1573-1582. doi:10.1089/ars.2006.8.1573