Journal of Biophysical Chemistry
Vol.2 No.3(2011), Article ID:6739,10 pages DOI:10.4236/jbpc.2011.23031
Prediction of a neutralizing epitope of a H5N1 virus hemagglutinin complexed with an antibody variable fragment using molecular dynamics simulation
Departemen Fisika, Fakultas MIPA, Kampus Universitas Indonesia, Depok, Indonesia; *Corresponding Author: cenmi.mulyanto@alumni.ui.ac.id
Received 19 April 2011; revised 18 May 2011; accepted 30 May 2011.
Keywords: Molecular Dynamics Simulation; Neutralizing Epitope; Antibody; H5N1 Hemagglutinin; Molecular Docking
ABSTRACT
In this present study, we predicted the neutralizing epitope of a modeled H5N1 hemagglutinin 1046T when interacted with a modeled monoclonal antibody variable fragment 8H5Fv using molecular dynamics simulation. Following the production run of the molecular dynamics simulation, we observed the average change of solvent accessible surface of the antigen alongside the formation of hydrogen bonds between the two structures during the simulation. Based on the acquired data, we predicted the neutralizing epitope of the 1046T antigen to be consisted of residues Asp 84, Glu85, Phe86, Ile87, Asn88, Val89, Pro90, Ile132, Ser136, Val147, Pro152, Tyr153, Leu154, Arg161, and Tyr268. By calculating the RMSD of the Cα backbone chain of the complex during the simulation we found the structure to be generally stable suggesting a well maintained steric hindrance, while RMSD calculation of the predicted neutralizing epitope backbone suggests the stability of the neutralizing epitope itself.
1. INTRODUCTION
Avian influenza caused by the influenza type A H5N1 virus is still one of the current major concerns of the world for causing a pandemic threat throughout the globe. Initially identified in China [1-3], the H5N1 virus has spread to the rest of Asia and also Africa. As of the time that this study is being written there have been an accumulated number of human cases reported by the World Health Organization of 543 reported human cases in total where 318 of these reported cases ended in mortality [2]. To understand the nature of the spread one might need to understand the nature of the cause at first, that is the avian influenza virus.
The H5N1 avian influenza virus, just like any other influenza virus, relies on its surface antigens to bind and interact with host cells to which they can infect. Hemagglutinin (HA) as one of the main surface glycoproteins is one of the most crucial point of study regarding the H5N1 virus. HA is not only the surface protein that directly binds to a cell receptor, but is also the protein in which humoral immune response is triggered by [3-6,10]. Another point of interest in the study of HA is due to the fact that it experiences a high mutation ratio. In light of this, vaccine developments have more often than not designed to accommodate the newest mutation of HA (or newest strain of the H5N1 virus) so that vaccines could neutralize these viruses effectively [5]. Recently, it has been suggested that structure-based vaccine design might rely on the designation of a neutralizing epitope of hemagglutinins from various strains. Through computerbased visualization and calculations of the neutralizing epitope, a common neutralizing epitope between different strains could be achieved and support aid as a target for structure-based vaccine design.
In learning more on how to prevent infection of the Influenza A virus there have been studies regarding the influenza virus specifically interacting with antibodies to identify a neutralizing epitope by experimental approach [6]. In keeping with the context of this study, a specific H5 epitope experimental study have also been conducted to learn more on the pathogenicity of the H5N1 virus [7]. Aside from experimental approaches, theoretical and computational studies have been developed using the crystallographic X-ray data and simulate a certain biological phenomena using an in silico approach [13]. Example of this is the study on identifying a common neutralizing epitope of the H5N1 virus’ main antigen, HA by introducing an antibody executed computationally [8]. In this present study, the same technique used to predict the HA neutralising epitope through computational means was executed by the molecular docking of a hemagglutinin with an antibody’s variable fragment (Fv) as a preliminary step. Then as a progression of the molecular docking, a series of molecular dynamics (MD) simulation of the Ab-HA complex was executed. This type of biomolecular simulation is frequently used as a means to understand how biomolecules move, fluctutate, interact, react, and also function [11,12]. By using MD simulation in our study, a thorough investigation of the stability of the Ab-HA complex, and more specifically the neutralizing epitope prediction, could be conducted. Hopefully, through molecular modeling and MD simulation that is shown in this study more light could be shed onto the development of structure-based vaccine design.
2. MATERIALS AND METHODS
2.1. Structure Preparation
The subjects of the MD simulations conducted in this study pertains two molecular structures where one complex will be made out of these two structures. The two molecular structures consist of the antigen a H5N1 virus strain (A/Indonesia/CDC1046T/2007(H5N1), which will now be referred to as 1046T for brevity and a variable region of the 8H5 antibody (or 8H5Fv). Homology modeling was implemented to generate the other two molecular structures. All unwanted water and salt molecules in the original crystal structure were removed. Following the removal of unwanted structures, a HA1 chain structure of the homotrimeric 2IBX was extracted. It has been shown in prior studies that HA1 possess the binding domain to which the sialic acid might bind to [3,26,28]. It is therefore imperative in this study to apply this structure to elucidate the binding mechanisms of an antibody variable fragment to an antigen beside it being computationally efficient in doing so.
The 1046T antigen was generated by homology modeling using the Swiss Model server [23,24]. In the Swiss Model workbench, the HA1 sequence of the 1046T antigen were uploaded to the Swiss Model server and processed by sequence alignment first then building a homology model based on the most appropriate template. The result of the generated structure from Swiss Model was then verified by using the Profile 3-D module in Discovery Studio 2.1. Aside from that, the 8H5Fv was generated by uploading its light and heavy variable sequence to the Rosetta Antibody server [25]. The Rosetta Antibody server will model each chain by homology modeling based upon the conserved framework and packed by automation in the server. The model with the most appropriate complimentary determining region (CDR) allocation was chosen for the study. Moreover, the structure was verified by using Profile-3D and compared with the structure generated by Yan et al. [8]. In this case the variable fragment of the 8H5 antibody was modeled for it has been indicated that the variable fragment of an antibody is the domain which is directly involved in triggering an immune response [14].
All structures were typed with the CHARMm force field [30] and implicitly solvated using the Generalized Born with Molecular Volume (GBMV) [22] method to create an effect of continuous dielectric environment to imitate a liquid water surrounding. Each structure was then subjected to optimization by 10,000 steps of Steepest Descent and then Conjugate Gradient.
2.2. Molecular Docking
The complex of 8H5Fv-1046T was produced through molecular docking using the Zdock module [15] in Discovery Studio 2.1. For scoring the docked complexes, Zdock uses the Pairwise Shape Complimentary (PSC) scoring function. It accounts the total number of interface atom pairs within a cutoff distance (10 Å in this study) between the receptor and ligand proteins while it penalties the number of overlapping grid point between receptor and ligand proteins. The 30 highest ranking docked poses were then subjected to refinement using the RDock module. This module attempts to achieve the best structure by removing any steric clashes in the released complex and optimizing the charge interaction of the concerning system. The best ranked pose released by RDock was then used for further study.
The complex then was implicitly solvated by GBMV and was subjected to further optimization by implementing 10,000 steps of steepest descent and conjugate gradient energy minimization under the CHARMm force field. A preliminary prediction of the neutralizing epitope was conducted. The criteria to which a residue is considered to be in the epitope is based on the criteria characterized by Yan et al. [8]. Residues of the antigen that experience decrease in solvent accessible surface of at least 7 Å2 and those that formed hydrogen bonds with antibody residues were considered to be part of the neutralizing epitope.
2.3. Molecular Dynamics Simulation of 8H5Fv-1046T Complex
Both 8H5Fv-2IBX and 8H5Fv-1046T were subjected to a 100 ps NPT molecular dynamics (MD) simulation under the CHARMm forcefield. Implicit solvation by GBMV was still applied to the system in order to simulate an aqueous environment. The NPT MD simulation [30] was preceeded by a 20 ps heating simulation involving a rise in temperature from 0 K to 300 K. The heating of the system was followed by a 50 ps equilibration process to thermally equilibrate the molecules of the systems. All simulations were executed in the simulation module of the Discovery Studio 2.1 software.
Analyses of the simulated systems will focus on the preliminary neutralizing epitope acquired from molecular docking aside from any antigen residues that exhibit any interaction with 8H5Fv, which includes:
ΔSAS: the decrease of the SAS was further observed at the final step of the simulation. Those residues that fall well below the 7 Å2 mark will be eliminated from the neutralizing epitope consideration. On the other hand, residues that were not part of the original preliminary neutralizing epitope but show decrease in SAS that is appropriate to the criterion were considered in becoming part of the neutralizing epitope.
Hydrogen Bond Analysis: formation of the hydrogen bonds during the simulation was observed with the criteria being that the distance between donor and acceptor is equal or less than 2.5 Å. Moreover, hydrogen bond occupancy with more than 50% during the simulation was considered in the study.
RMSD Analysis: observation of the overall structure backbones was observed with 2000 snapshots taken during the simulation. Apart from the backbone RMSD, the predicted neutralizing epitope backbone RMSD throughout the simulation was observed.
3. RESULTS AND DISCUSSION
3.1. Homology Modeling
To generate the 1046T model we used homology modeling to generate a 3D structure of the molecule. In order to generate the 3D structure of the 1046T antigen the FASTA sequence of the 1046T molecule was submitted to be processed in the SWISS-MODEL server. Fortunately, the 1046T protein produced a 95.4% sequence identity and 96.6% sequence similarity with its template ensuring the quality of the homology modeling. To visualize the comparison between the template, Figure 1 depicts the sequence alignment of the 1046T molecule with the 2IBX template. There were 16 residues of the template that needed to be changed to produce the 1046T and Figure 2 show the superimposed structure of the target 1046T with its template 2IBX highlighting the mutated residues.
Another task of homology modeling was also executed for the 8H5Fv 3D structure (Figure 3). For this model we submitted the amino acid sequences of the variable light chain and variable heavy chain to the Rosetta Antibody Server .The 8H5Fv consist of 229 residues with each VL and VH individually having 109 and 120 residues respectively. Each variable domain is packed together with face-to-face b-sheets and interconnected by disulfide bonds. These disulfide bonds are located at Cys23-Cys88 in VL and Cys 22-Cys 92 in VH with the average length of 2 Å which is the average disulfide bond length like other antibodies to stabilize the molecular structure. Charles Chothia onced suggested that the variable fragment of an antibody follow particular canonical structures called complimentary determining regions (CDR) [31]. In order for a structure of the Fv model to be remotely correct, the constituting amino acids must fit into these CDRs (Table 1). Another fortunate result is that the CDRs fall well in line and also resulted in the same canonical structures as did Yan et al.
To further validate the 8H5Fv structure, Profile-3D in the Discovery Studio 2.1 was used to score the compatibility of constituting residues with the surrounding environment and we found that for this size of a structure the minimum score for it to be a valid structure would have to be 46.378. The structure generated here produced a score of 103.065 guaranteeing the validity of the 8H5Fv structure that was to be used in this study. Additionally a Ramachandran plot (Figure 4) produced results showing that the generated structurese were in fact in good stereochemistry.
All structures were then structurally optimized by energy minimization. Each of the structure was subjected to 10,000 steps of steepest descent followed by 10,000

Figure 1. Sequence alignment between the template 2IBX and target 1046T.
Figure 2. The 3D structure of 1046T (purple) superimposed with template 2IBX (grey). Mutated residues are colored in blue.

Table 1. Framework and complimentary determining regions of both light and heavy variable fragments of 8H5.
of conjugate gradient minimization prior to molecular docking.
3.2. Preliminary Neutralizing Epitope Prediction by Molecular Docking
Molecular docking using the Zdock protocol in Discovery Studio 2.1 was applied to create the 8H5Fv- 1046T complex. We chose the highest ranking docking position based on pairwise shape complimentary (PSC). As a testament to Zdock’s reliability previous attempts made by CAPRI at docking HA molecules to antibody fragments have yielded positive results where native residue to residue contacts were sufficiently observed for their highest ranking complex [16,17]. In this study, we found through molecular docking that the 8H5Fv did infact interact with the regional binding domain at the hemagglutinin globular head (Figure 5).
In predicting the neutralising epitope of the antigens, the major component that was taken into consideration was the change in Solvent Accessible Surface (ΔSAS) of the HA residues at the interaction site of the 8H5Fv- 1046T complex. Conolly proposed that the solvent accessible surface of a certain molecule could in fact play a significant role [18]. The accessible molecular area denotes that area in which outside influence could trigger a biological event. In cases such as the hemagglutinin of the influenza virus it could appoint which residue of the antigen is susceptible to bind with other molecules. The ΔSAS was achieved by calculating the difference between post-molecular docking SAS and the initial premolecular docking SAS. In order for a particular residue to be included in the predicted neutralizing epitope the ΔSAS should be at least 7 Å2. With that in mind, the residues predicted to be constituents of the neutralizing epitope of 1046T are listed in Table 2 and it could be seen the result of the 1046T ΔSAS seem to fall in similar fashion with the results of Yan et al. suggesting that the 8H5Fv antibody creates a steric hindrance at the binding receptor sub-domain [8,26,28]. However, similarity that could be observed might be differed from the fact of forcefield used to characterize the structures during energy minimization. This study used the CHARMm forcefield while Yan used a Consistent Valence Forcefield (CVFF). The different approach of defining non-bonded interactions within the structure might have altered the atomic positions in the structures itself creating different antigenic sidechain atoms that interacted with the 8H5Fv atoms.
Additionally the hydrogen bonds that were formed between 8H5Fv and the antigen upon docking were taken into account for the only intermolecular bonds that were formed were that of hydrogen bonds. Hydrogen bonds are associated as an effort in stabilizing a protein structure [20,21] and therefore is one of the main factors in stability of the 8H5Fv-HA complexes. In this study it was observed that there were 16 hydrogen bonds formed at the interaction area of the 8H5Fv-1046T complex (Table 3). Of those 16 hydrogen bonds formed, there were seven residues from the 1046T antigen that formed hydrogen bonds with the 8H5Fv. Most of the residues observed even experienced multiple hydrogen bonds formed with different residues of the 8H5Fv. We compared our results here with the results of Yan et al. and found that the residues that formed hydrogen bonds in this study did not create similar results with Yan where

Table 2. ΔSAS of epitope candidate as a result of docking.

Table 3. Donors and acceptors of the various hydrogen bonds formed as a result of molecular docking between 1046T and 8H5Fv.
they used the 2IBX template but interestingly there is one residue that contributed in the formation of hydrogen bonds on the antigen, which are Asp76 in 2IBX and Asp 88 in 1046T. Admittedly the role of proton donor of Asp76 differs between strains which only reinforce the difference of blocking orientation of 1046T and 2IBX produced by Yan.
Figure 3. The 3D structure of 8H5Fv. Heavy variable chain is colored in red while light variable chain is colored in purple.
Derived from the results of molecular docking we could predict the initial neutralizing epitope for 1046T: of Asp59, Lys64, Asp84, Glu85, Asn88, Val89, Pro90, Ile132, Ser136, Val147, Pro152, Tyr153, Leu154, Gly155, Ser158, Arg161, Tyr268, Asn289.
3.3. Neutralizing Epitope Prediction by Molecular Dynamics Simulation
A series of molecular dynamics simulation has been performed to investigate the flexibility of the complex structure and to examine the consistency of the neutralizing epitope. The previously predicted neutralizing epitope produced by molecular docking consist of Asp59, Lys64, Asp84, Glu85, Asn88, Val89, Pro90, Ile132, Ser136, Val147, Pro152, Tyr153, Leu154, Gly155, Ser158, Arg161, Tyr268, Asn289. The complex experienced a 10 ps heating phase from 0 - 300 K followed by a 20 ps equilibration. As the final step of the MD simulation a 100 ps NPT production phase was subjected towards the complex.
As with the molecular docking step in this study, ΔSAS of the 1046T residues during the MD simulation was accounted for. In this part of the study the average ΔSAS of the antigen throughout the whole simulation was calculated. Table 4 shows the residues that are now declared as the new epitope candidate. Form the sheer number of the residues involved in the table, the residues involved this time were less than the number of residues involved prior to simulation. The dynamic nature of the atoms involved and the influence of salvation using GBMV would seem to expose some of the residues involved that were once a part of the neutralizing epitope. Residues such as Glu91, Glu97, Asn112, Ile133, Ser156,
 (a)
(a)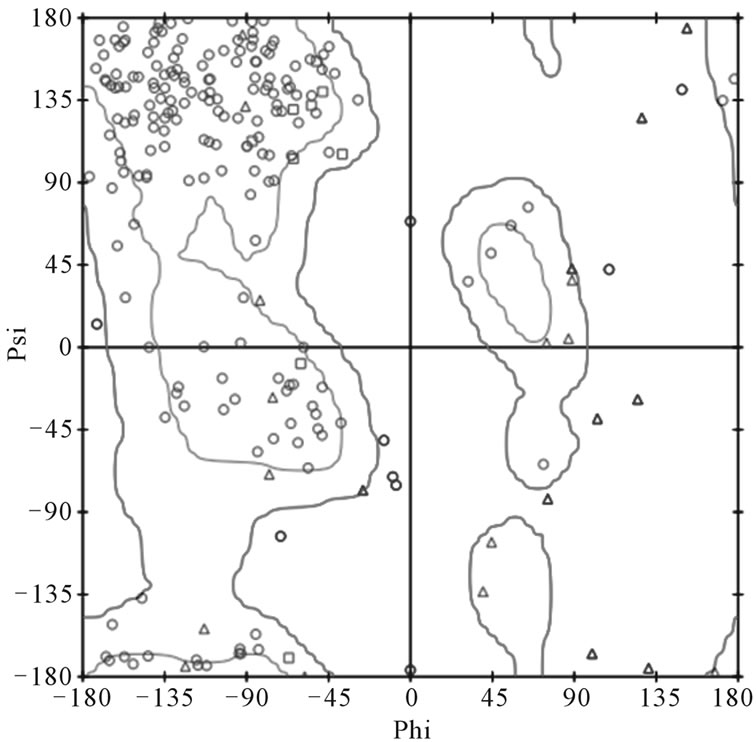 (b)
(b)
Figure 4. (a) Ramachandran plot of 1046T. Out of the 321 residues modeled 5 are in the disallowed region. Overall the 1046T is showing good stereochemistry; (b) Ramachandran plot of 8H5Fv. One residue Ser51 of the light chain is located in the disallowed region. Apart from that the structure generally shows good stereochemistry.
and Glu267 were eliminated from the epitope list due to the fact that the average ΔSAS did not reach the minimum threshold value of 7 Å2.
A lower than 7 Å2 DSAS value indicate that the corresponding residue with that SAS value is exposed to the external environment which could result in the binding of a solvent or, even in this case, a cell receptor. This
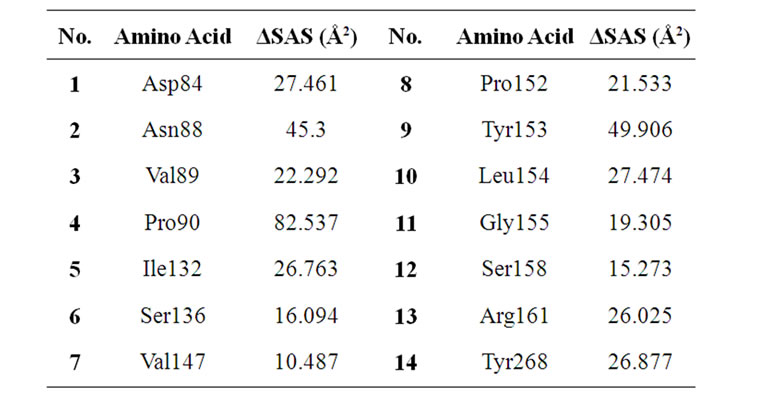
Table 4. Average ΔSAS of epitope candidates after MD simulation.
threshold value of course was the value that was used for selecting the amino acid that made up the predicted neutralizing epitope and also used in the study conducted by Yan et al. Out of the 20 amino acid in the predicted neutralizing epitope there were 4 residues that had a negative value. These residues were Glu91, Ile133, Ser156, and Glu267. However, the 1046T residues that were calculated to have that lower than 7 Å2 value were still buried in the complex and surrounded by other residues that interact with and/or affected by the 8H5Fv. Hence, even though these residues with less than 7 Å2 values are still part of the neutralizing epitope, these calculations suggest that the 8h5Fv neutralize the 2IBX antigen more effectively than the 1046T antigen.
Another criteria for a residue to be included as an epitope candidate is the formation hydrogen atoms between residues of 8H5Fv with 1046T. We observed the formation of hydrogen bonds during the production MD simulation and we found the occupancy of the hydrogen bonds during the 100 ps simulation that is shown in Figure 6. The residues that contributed towards the formation of hydrogen bonds were added from seven 1046T residues becoming eight residues during the production simulation. A surprising turn of event ensued when a residue that was not included in Table 3 as a result of molecular docking existed in the occupancy chart in Figure 6. The residue Ile87 did not show any sign of formation of a hydrogen bond with a 8H5Fv residue after molecular docking. The contribution of Ile87 to the formation of hydrogen bonds suggests that MD simulation influenced the movement of the Ile87 residue toward the residues of 8H5Fv. The influence of the dynamics would have to be great because by the end of the simulation run the Ile87 exhibited the highest occupancy compared to the rest of the residues.
Based on hydrogen bond analysis and DSAS calculations from the MD simulation, residues that are included in the predicted could be identified. The residues that are part of the predicted epitope are Asp 84, Glu85, Phe86,
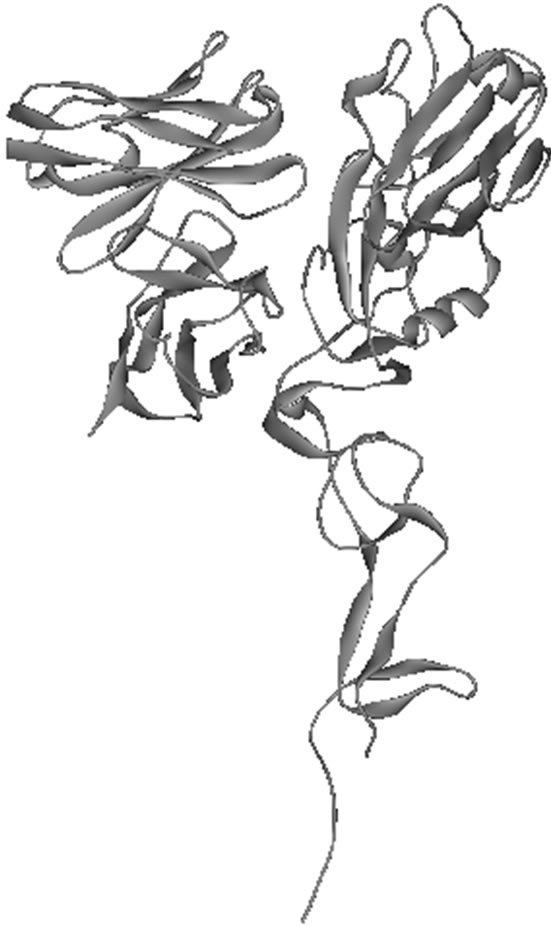 (a)
(a) (b)
(b)
Figure 5. The docked structure of the 8H5Fv- 1046T complex displayed in (a) ribbon style and (b) solvent accessible surface. The 8H5Fv is colored green while 1046T is purple.
Ile87, Asn88, Val89, Pro90, Ile132, Ser136, Val147, Pro152, Tyr153, Leu154, Arg161, and Tyr268.
As an approach to examine the flexibility and stability of the complex, we calculated the RMSD of the complex at every 1 ps of the simulation. The result is Figure 7 where it shows that during simulation the RMSD fluctuated with increased value. The RMSD for the structure in the starting state was at around 1.4 Å and ended at 1.5 Å. During the 75 ps, the RMSD began to increase until the 2.7 Å mark around 90 ps. The regular fluctuating RMSD graph suggests that the complex tend to be stable and flexible throughout the 100 ps MD simulation.
Previously, the global structure was the focus of observation. In this section the most significant area of the study will be observed and reported, i.e. the predicted neutralizing epitope. In this section RMSD observation of the neutralizing epitope based on DSAS analysis and

Figure 6. Hydrogen bond occupancy of the residues contributing in the formation of hydrogen bonds during the production run of the MD simulation.
Figure 7. Root mean square deviation of the (red) global structure and the predicted neutralizing epitope of 1046T (black).
hydrogen bond occupancy will be accounted for the affectivity of the neutralizing activity triggered by 8H5Fv against the hemagglutinin.
The RMSD of the 8H5Fv-1046T neutralizing epitope could be seen in Figure 7. The RMSD of the neutralizing epitope fluctuated at an average RMSD of 1.09 Å. Comparing the neutralizing epitope RMSD plot with the overall complex RMSD yield in a similar pattern at the last 25 ps of the simulation. The rise in RMSD at the 75 ps mark was apparent for the predicted neutralizing epitope RMSD and it also showed a similar downward slope at the end of the graph. The range of the RMSD spanning from 0.3 Å to 2 Å experiencing a regular fluctuating pattern in between the values indicate that the stability is well maintained during the simulation. Not only that, the stability of the predicted epitope could be caused by the hydrogen bonds formed during the simulation. Having observed five out of eight residues that formed hydrogen bonds creating occupancy above 50% show that structural stability of the neutralizing epitope was influenced by the formation of hydrogen bonds during the simulation.
From the data gathered during simulation, we were able to visualize the predicted neutralizing epitope (Figure 8). The predicted epitope displayed here agrees with epitopes defined by Ndifon et al. [32] In their study Ndifon defined various neutralizing epitopes at the hemagglutinin of a H3N2 influenza virus. There were five epitopes that surround the receptor binding site. All five epitopes are located at the globular head of the hemagglutinin. Our predicted epitope here correspond to one of the epitopes that surround the receptor binding site of H3N2. In this sense it seems that there is a conserved epitope between different hemagglutinin subtypes. Experimentally, this has been proven to be able to occur by Okuno et al. [9]. In the study produced by Okuno conserved neutralizing epitope were found between H1 and
Figure 8. The pattern of the predicted neutralizing epitope (red) of 1046T shown in molecular surface.
H2 subtypes. If epitope conservation could happen then there is a chance that the H5N1 epitope in this study could be found in the H3N2 strain virus used by Ndifon. Moreover, it has been inferred that the residue Glu91 of the 1046T (Glu79 in 2IBX) implicated in ligand-binding specifity [26,27]. In our study that particular residue is well located at the predicted neutralizing epitope. The inclusion of Glu91 in the predicted neutralizing epitope could enforce the point that the 8H5Fv could neutralize the 1046T antigen effectively.
4. CONCLUSIONS
In this present study, we have examined the interacttion between a modeled hemagglutinin structure with a modeled monoclonal antibody variable fragment using molecular dynamics simulation. To generate the aforementioned structures the antigen 3D structure was generated through homology modeling based on a recent X-ray crystallographic structure of an H5 subtype of avian influenza 2IBX as a template to build the 1046T model of A/Indonesia/CDC1046T/2007(H5N1) based on its amino acid sequence. Apart from the antigen, the variable fragment of 8H5Fv was built from multiple templates that were decided in the Rosetta Antibody Server. Evaluation was subsequently executed towards the generated structure and the results of the structure evaluation were strong enough to lend credibility of the homology modeling of both structures.
Through molecular docking of the antigen and the antibody variable fragment using the Zdock protocol in the Discovery Studio 2.1 software we were able to make a preliminary prediction of the neutralizing epitope based on the change of solvent accessible surface and the hydrogen bonds formed between the two structures. Then with a series of molecular dynamics simulations we were able to provide a prediction of the neutralizing epitope based on the production run of the MD simulation. By calculating the average ΔSAS and the formation of hydrogen bonds during the production run we predicted the neutralizing epitope of 1046T to consist of residues Asp 84, Glu85, Phe86, Ile87, Asn88, Val89, Pro90, Ile132, Ser136, Val147, Pro152, Tyr153, Leu154, Arg161, and Tyr268. By observing the RMSD of the predicted neutralizing epitope, we gathered that the structure of the predicted epitope to be stable during the MD simulation, suggesting that possible neutralization efforts of the 8H5Fv upon the 1046T antigen to be effective. To further investigate the stability and consistency of the predicted neutralizing epitope, an MD simulation with longer time is much needed. Hopefully, the role of MD simulation in predicting neutralizing epitope of the 1046T structure will shed light onto further signify the role of MD simulations in structure based vaccine design.
5. ACKNOWLEDGEMENTS
We would like to give the utmost gratitude towards Ding Ming Chee of Accelrys for giving us assistance and the opportunity to try the Discovery Studio 2.1 software.
REFERENCES
- The Writing Committee of the World Health Organization (WHO) Consultation on Human Influenza A/H5 (2005) Avian influenza A (H5N1) infection in humans. The New England Journal of Medicine, 353, 1374-1385. doi:10.1056/NEJMra052211
- World Health Organzation (2009) Cumulative number of confirmed human cases of avian influenza A/(H5N1) reported to WHO. http://www.who.int
- De Jong, M.D. and Hien, T.T. (2006) Avian influenza A (H5N1). Oxford University Clinical Research Unit, Hospital for Tropical Diseases, Ho Chi Minh City, Viet Nam.
- Kamps, B.S., Hoffman, C. and Preiser, W. (2006) Influenza Report 2006, Flying Publisher, Paris. http://www.infleunzareport.com
- Swayne, D.E., (2008) Avian influenza. Blackwell Publishing, Ames.
- Lim, A., Wong, S., Chan, A., Chan, C., Ooi, E. and Hanson, B. (2008) Epitope characterization of the protective monoclonal antibody VN04-2 shows broadly neutralizing against highly pathogenic H5N1. Virology Journal, 5, 80. doi:10.1186/1743-422X-5-80
- Philpot M., Easterday B.C. and Hinshaw V.S. (1989) Neutralizing epitopes of the H5 hemagglutinin from a virulent avian influenza virus and their relationship to pathogenicity. The Journal of Virology, 63, 3453-3458.
- Yan, Y.Q., Li, S., Yang, C.Y., Luo, W.X., Wang, M.Q., Chen, Y.X., Luo, H.F., Wu, T.Z. and Xia, N.S. (2008) Prediction of a common neutralizing epitope of H5N1 avian influenza virus by in silico molecular docking. Chinese Science Bulletin, 53, 868-877. doi:10.1007/s11434-008-0161-4
- Okuno,Y., Isegawa, Y., Sasao, F. and Dan Ueda, S. (1999) A common neutralizing epitope conserved between the haemagglutinins influenza virus a H1 and H2 strains. Journal of Virology, 67, 2552-2558.
- Yen, H.L. and Peiris, J.S.M. (2009) Mapping antibody epitopes of the avian H5N1 influenza virus. PloS Medicine, 6, e1000064. doi.org/10.1371/journal.pmed.1000064
- Jensen, F. (2007) Introduction to computational chemistry. 2nd Edition, John Wiley & Sons, Ltd., Chichester.
- Mulholland, A.J. (2008) Introduction: Biomolecular simulation. Journal of the Royal Society Interface, 5, S169-S172. doi:10.1098/rsif.2008.0385.focus
- Leach, A. (2001) Molecular modeling: Principles and application. Prentice Hall, Upper Saddle River.
- Morea, V., Lesk, A.M. and Tramontano, A. (2009) Antibody modeling: Implications for engineering and design. Methods, 20, 267-279. doi:10.1006/meth.1999.0921
- Chen, R. and Weng, Z. (2003) A novel shape complimentarity scoring function for protein—Protein docking. Poteins, 5, 397-408.
- Chen, R., Tong, W., Mintseris, J., Li, L. and Weng, Z. (2003) ZDOCK predictions for the CAPRI challenge. PROTEINS: Structure, Function, and Genetics, 52, 68-73. doi:10.1002/prot.10388
- Wiehe, K., Pierce, B., Mintseris, J., Tong, W., Anderson, R., Chen, R. and Weng, Z. (2005) ZDOCK and RDOCK performance in CAPRI rounds 3, 4, and 5. Proteins, 60, 207-213. doi:10.1002/prot.20559
- Connolly, M.L. (1983) Solvent accessible surfaces of proteins and nucleic acids. Science, 221, 709-713. doi:10.1126/science.6879170
- Durham, E., Dorr, B., Woetzel, N., Staritzbichler, R. and Meiler, J. (2009) Solvent accessible surface area approximations for rapid and accurate protein structure prediction. Journal of Molecular Modeling, 15, 1093- 1108. doi:10.1007/s00894-009-0454-9
- Rose, G.D. (1993) Hydrogen bonding, hydrophobicity, packing, and protein folding. Annual Review of Biophysics and Biomolecular Structure, 22, 381-415. doi:10.1146/annurev.bb.22.060193.002121
- Wong, S., Bernacki, K. and Jacobson, M. (2005) Competition between intramolecular hydrogen bonds and salvation in phosphorylated peptides: Simulations with explicit and implicit solvent. The Journal of Physical Chemistry B, 109, 5249-5258. doi:10.1021/jp046333q
- Feig, M., Onufriev, A., Lee, M.S., Im, W., Case, D.A. and Brooks, C.L. (2004) Performance comparison of generalized born and poisson methods in the calculation of electrostatic solvation energies for protein structures. Journal of Computational Chemistry, 25, 265-284. do:10.1002/jcc.10378
- Arnold, K., Bordoli, L., Kopp, J. and Schwede, T. (2006) The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics, 22, 195-201. doi:10.1093/bioinformatics/bti770
- Peitsch, M.C. (1995) Protein modeling by E-mail. BioTechnology, 13, 658-660. doi:10.1038/nbt0795-658
- Sircar, A., Kim, E. and Gray, J.J. (2009) Rosetta antibody: Antibody variable region homology modeling server. Nucleic Acids Research, 37, W474-W479.
- Yamada, S., Suzuki, Y., Suzuki, T., Le, M., Sakai-Tagawa, Y., Muramoto, Y., Ito, M., Kiso, M., Horimoto, T., Shinya, K., Sawada, T., Kiso, M., Usui, T., Murata, T., Lin, Y., Hay, A., Haire, L., Stevens, D., Russell, R., Gamblin, S., Skehel, J. and Kawaoka, Y. (2006) Haemagglutinin mutations responsible for the binding of H5N1 influenza A viruses to human-type receptors. Nature, 444, 378-382. doi:10.1038/nature05264
- Kasson, P., Ensign, D. and Pande, V. (2009) Combining molecular dynamics with Bayesian analysis to predict evaluate ligand-binding mutations in influenza hemagglutinin. Journal of the American Chemical Society, 131, 11339-11340. doi:10.1021/ja904557w
- Stevens, J., Blixt, O., Tumpey, T., Taubenberger, J.K., Paulson, J. and Wilson, I. (2006) Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science, 312, 404-410.
- Brooks, B., Brooks, C.L. III, Mackerell, A., Nilsson, L., Peterealla, R., Roux, B., Won, Y. and Archontis, G., et al. (2009) CHARMM: The biomolecular simulation program. Journal of Computational Chemistry, 30, 1545- 1614. doi:10.1002/jcc.21287
- Rapaport, D.C. (2004) The art of molecular dynamics simulation. 2nd Edition, Cambridge University Press, Cambridge. doi:10.1017/CBO9780511816581
- Chothia, C. and Lesk, A.M. (1987) Canonical structures for the hypervariable regions of immunoglobulins. Journal of Molecular Modeling, 196, 901-917. doi:10.1016/0022-2836(87)90412-8
- Ndifon, W., Wingreen, N.S., Levin, S.A. (2009) Differential neutralization efficiency of hemagglutinin epitopes, antibody interference, and the design of influenza vaccines. Proceedings of the National Academy of Sciences, 106, 8701-8706.