Neuroscience & Medicine
Vol.3 No.1(2012), Article ID:17754,4 pages DOI:10.4236/nm.2012.31010
CXB-909 Attenuates Cognitive Deficits in the mu-p-75 Saporin Mouse Model of Alzheimer’s Disease
![]()
1Field Neurosciences Institute Laboratory for Restorative Neurology, Central Michigan University, Mount Pleasant, USA; 2Program in Neuroscience, Central Michigan University, Mount Pleasant, USA; 3College of Medicine, Central Michigan University, Mount Pleasant, USA; 4Field Neurosciences Institute, Saginaw, USA.
Email: dunba1g@cmich.edu
Received December 15th, 2011; revised January 19th, 2012; accepted February 20th, 2012
Keywords: Alzheimer’s Disease; Memory Deficits; Substituted Pyrimidines; mu-p-75 Saporin; Morris Water Maze
ABSTRACT
The purpose of this study was to determine if the substituted pyrimidine, CXB-909 (formerly known as KP544) which has been shown to amplify the effects of nerve growth factor in elevating choline-acetyltransferase activity in vitro, could attenuate memory deficits in the mu-p-75 saporin injected mouse model of Alzheimer’s disease (AD). Seventyone, seven-week old C57/BL6 mice received daily oral intubation of 10, 15, or 20 mg/kg CXB-909, or vehicle (0.5% methylcellulose solution), which continued for 32 days. At postnatal week nine, mice received bilateral intra-cerebroventricular injections of mu-p-75 saporin, or sterile phosphate buffered saline. Seven days after surgery, mice were trained for two days, on a cued-platform version of the Morris water maze task, and then tested on a four-day hiddenplatform version, followed by a one-day probe version of this task. Mice injected with mu-p-75 saporin, had increased latency to find the hidden-platform compared to sham mice. Furthermore, mice treated with CXB-909 at the 10, and 15 mg/kg doses, significantly reduced their latency to reach the hidden-platform, compared to vehicle-treated mice given mu-p-75 saporin. These results suggest that CXB-909 can attenuate memory deficits in the mu-p-75 saporin injected mouse model of AD.
1. Introduction
Alzheimer’s disease (AD) is a devastating and progressive neurodegenerative disease marked by the formation of amyloid-beta plaques, neurofibrillary tangles [1], and loss of cholinergic neurons of the basal forebrain, which lead to memory loss [2]. This cholinergic deficit prompted the administration of IgG-saporin, a cholinergic toxin, to rodents in an effort to mimic memory dysfunction analogous to what is observed in AD. Murine-p-75 saporin (SAP) is an antibody-conjugated, cholinergic-specific neurotoxin, which, when injected intra-cerebroventricularly into mice, can induce memory deficits similar to those seen in AD [3,4]. SAP was first tested by Berger-Sweeney and colleagues [3], who reported that intra-cerebroventricular (ICV) injections of 3.6 µg of SAP, induced robust memory deficits as measured in the Morris Water Maze (MWM). Unfortunately this dose also resulted in a 32% mortality rate [3]. In 2008, Moreau and colleagues tested a reformulated version of SAP, which effectively induced memory deficits with doses as low as 0.4 µg with 100% survival [4]. Because most of the FDA-approved drugs for AD are cholinergic agonists with limited efficacy, other therapeutic approaches targeting cholinergic neurons are under investigation. Of particular interest is the use of neurotrophic factors, especially nerve growth factor (NGF), a protein that supports cholinergic function and survival, and which may effectively treat forebrain-related neurological decline [5-10]. Unfortunately, NGF does not readily cross the blood-brain barrier and so its clinical utility is severely limited.
Recently, the use of small molecule NGF amplifiers has captured the attention of AD researchers as a potential means of amplifying the protective effects of NGF on cholinergic neurons. One group of small molecule compounds, known as substituted pyrimidines, show considerable promise for enhancing trophic support to cells which are most vulnerable to neurodegenerative processes [11]. One of these compounds, CXB-909, formerly known as KP544 [2-amino-5-(4 chlorophenylethyl)-4-(4-trans-hydroxycyclohexyl amino) pyrimidine], is a substituted pyrimidine that has been shown to amplify the effects of NGF in vitro [12]. CXB-909 has also been shown to enhance neurite outgrowth in both rat PC12 cells [12,13], and human SH-SY5Y cells [12].
CXB-909 has shown similar promise in three rodent models of Huntington’s disease (HD) as well. Krenitsky and colleagues found that CXB-909 treatment decreased striatal lesion size, and attenuated learning deficits in the radial arm water maze (RAWM) task [14]. A similar sparing of mnemonic function was found using a 3-nitroproprionic acid (3NP) mouse model, where once again, CXB- 909 treatment attenuated 3NP-induced memory deficits in the RAWM task [11]. Finally, CXB-909 treatment attenuated motoric deficits in the R6/2 transgenic mouse model of HD [15]. In each case, CXB-909 was administered systemically, indicating that this small molecule compound is able to cross the BBB in therapeutically significant amounts [11,14]. The goal of the current study was to determine if CXB-909, which has shown great promise in HD rodent models, could provide similar therapeutic effects in the murine-p-75 saporin-injected mouse model of AD.
2. Methods
For this study, 71 C57BL/6 male (n = 36) and female (n = 35) seven-week-old mice, obtained from breeders (Jackson Laboratories, Bar Harbor, ME), were used. Beginning at postnatal week seven, mice were subdivided into treatment groups which received daily oral intubation of CXB-909 at 10, 15, or 20 mg/kg, suspended in 0.05% methylcellulose (Amend Drug and Chemical Co, Irvington, NJ) vehicle, or an equivalent volume of vehicle only, following an established protocol [15]. Dosing continued for 32 days. CXB-909 was obtained from Krenitsky Pharmaceuticals, Inc. (Durham, NC), as part of another project [15], before the compound was sold to CeNeRx BioPharma, Inc. (Cary, NC).
At postnatal week nine, mice were anesthetized with 90 mg/kg ketamine and 5 mg/kg xylazine, and placed in a stereotaxic device (David Kopf Instruments, Tujunga, CA). Anesthesia was maintained with 0.5% inhaled isoflurane (Halocarbon Products Corporation, Rivers Edge, NJ). The cranium was exposed, and burr holes drilled at –0.4 mm AP and ± 1.0 mm ML, relative to Bregma. A 10 µL syringe (Hamilton, Reno, NV) was lowered slowly through each burr hole into the ventricle at –2.6 mm DV from the cranial surface. Injections of 0.8 µg of SAP in 0.5µL sterile phosphate-buffered saline (PBS) or sterile PBS alone were made at the rate of 0.2 µL/minute. An additional sham group received a scalp incision, but no injections. Following surgery the incision was closed with 7 mm surgical wound clips. All procedures and tests followed the guidelines of the National Institutes of Health and the Institutional Animal Care and Use Committee at Central Michigan University.
One week following surgery, mice were trained on the cued-platform, hidden-platform, and probe versions of the MWM [16] task. Training and testing in the MWM followed the protocol of Moreau and colleagues [4] except that mice were tested for four, rather than five, consecutive days in the hidden-platform task.
Following the fourth day of hidden-platform testing, mice were given a single probe test as described before [4]. All swim paths in the MWM were recorded using a ceiling-mounted camera, and data were analyzed with computerized tracking software (Viewpoint, Montreal, Canada). The dependent measures on the hidden-platform task were latency to locate the platform, distance swam, and swim speed. Dependent measures for the probe test included: swim speed, distance swam, and the time spent in the target quadrant, (i.e. the quadrant of the maze where the platform had been located previously).
Data from the hidden-platform task and the probe test were analyzed using a one-way analysis of variance (ANOVA). All analyses were computed using the Statistical Package for the Social Sciences, version 17.0, with the alpha level set at 0.05. Fisher’s Protected Least Significance Difference post-hoc tests were conducted, when appropriate. No gender differences were observed on any measure, so data from male and female mice were analyzed together within each treatment group. Because the vehicle-treated mice receiving sham surgery with PBS injections (n = 8), and mice receiving sham surgery with no injections (n = 8) did not differ from each other, these mice were combined into a single SHAM + VEH group. In addition, mice receiving sham injections, and treatments of 10 (n = 8), 15 (n = 10), and 20 (n = 8) mg/kg CXB-909 did not differ from each other and were combined into a single SHAM + CXB group for analysis. These two groups were then compared with four groups of mice receiving saporin and treatments of vehicle (SAP + VEH, n = 8), 10 (SAP + CXB10, n = 8), 15 (SAP + CXB15, n = 6), and 20 (SAP + CXB20, n = 7) mg/kg CXB-909.
3. Results
A one-way ANOVA revealed that there was a significant difference between groups, in average latency to find the hidden-platform [F (5, 65) = 2.786, p = 0.024] during the four-day MWM task. Post-hoc analyses revealed that mice in the SAP + VEH group had significantly increased latency to find the hidden-platform, compared to the SHAM + VEH, SHAM + CXB, SAP + CXB10, and SAP + CXB15 groups, but not the SAP + CXB20 group. In addition, the SAP + CXB20 group took longer to find the hidden-platform than the SAP + CXB10 group, but did not differ from any other group. There were no differences between groups in total distance swum [F (5, 65) = 1.929, p = 0.101], or average swim speed [F (5, 65) = 1.726, p = 0.141] (Figure 1).
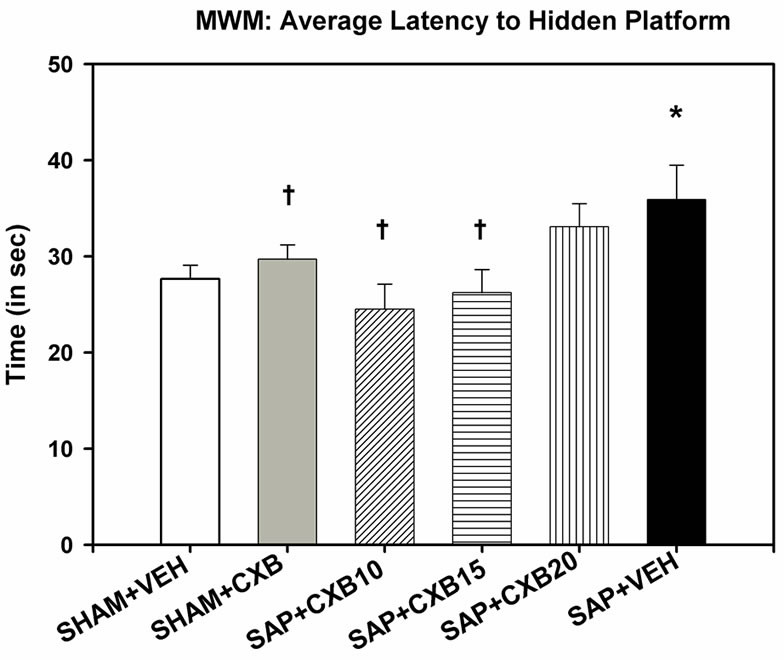
Figure 1. A one-way ANOVA showed a significant effect of average latency to find the hidden-platform during four days of testing in the MWM [F (5, 65) = 2.786, p = 0.024]. Fisher’s least significant difference post-hoc tests revealed that mice given intra-cerebroventricular injections of saporin, and vehicle treatment, took longer to find the hidden-platform, than mice receiving sham surgery, and vehicle treatment. These mice also took longer to find the hidden-platform than mice given injections of saporin, and CXB-909 treatment at 10 and 15 mg/kg (*p < 0.05 compared to SHAM + VEH group; †p < 0.05 compared to SAP + VEH group). NOTE: SHAM + VEH group includes mice receiving intracerebroventricular injections of sterile phosphate buffered saline, or a scalp incision with no injection. SHAM + CXB includes mice receiving SHAM surgery, and treatment with 10, 15, or 20 mg/kg treatments of CXB-909.
A one-way ANOVA indicated that there were no between-group differences in swim speed [F (5, 65) =1.150, p = 0.344], or distance swum [F (5, 65) = 1.377, p = 0.245] in the probe task. However, there was a significant difference of time spent swimming in the target quadrant [F (5, 65) = 2.393, p = 0.047]. Post-hoc analysis revealed that the SAP + VEH and SAP + CXB20 mice were the only groups who spent less time than SHAM + VEH mice in the target quadrant, which previously contained the hidden-platform (Figure 2).
4. Discussion
The major finding of this study is that SAP induced a significant memory deficit in mice as measured by the MWM hidden-platform and probe tests, which was alleviated by 10 and 15 mg/kg treatments of CXB-909. No differences were detected between groups in swim speed in either test, suggesting that motor function was not impaired in these mice. Surprisingly, deficits in both MWM tests were unexpectedly pronounced in the SAP + CXB20 treatment group. A parsimonious explanation for this phenomenon is that the 20 mg/kg dose may have greatly exceeded the optimal effective dose. This is the highest reported dose
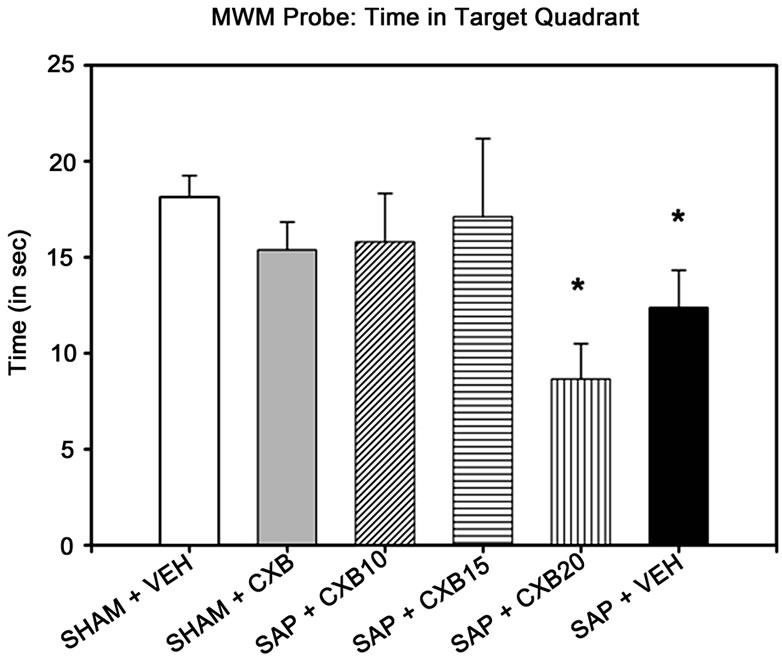
Figure 2. Morris water maze probe test results. Only mice given saporin and daily treatments of either vehicle (methylcellulose) or 20 mg/kg CXB-909 spent less time swimming in the quadrant that previously contained the escape platform in the Morris water maze probe task than vehicle-treated SHAM controls (*p < 0.05 compared to SHAM + VEH). NOTE: SHAM + VEH group includes mice receiving intra-cerebroventricular injections of sterile phosphate buffered saline, or a scalp incision with no injection. SHAM + CXB includes mice receiving SHAM surgery, and treatment with 10, 15, or 20 mg/kg treatments of CXB-909.
of CXB-909 ever administered to rodents, and more importantly twice the 10 mg/kg dose reported previously for reducing behavioral deficits in the R6/2 mouse model of HD [15].
It also seems to be the case that overexpression of neurotrophic factors, such as brain derived neurotrophic factor (BDNF) results in memory deficits [17], Furthermore, there is recent evidence that the unprocessed precursor to NGF, pro-NGF, seems to enhance deterioration of basal forebrain cholinergic neurons, itself, by initiating alternative signaling not directly related to the role of NGF [18]. It is not entirely clear how the substituted pyrimidine promotes NGF activity, but it is conceivable that it may also increase pro-NGF, which may accumulate to more toxic levels at higher doses. Nonetheless, mice receiving the lowand medium-doses of CXB-909 treatment showed significant cognitive improvements, indicating, for the first time, that CXB-909 can attenuate memory deficits in a rodent model of AD.
These results support the work of Moreau and colleagues, who also found significant memory deficits following SAP administration [4]. The SAP model is particularly well suited for early-stage drug discovery, because memory deficits occur very quickly following administration, which provides an efficient model for screening possible treatments. The present findings also support previous work that has shown that CXB-909 treatment can attenuate cognitive deficits in rodent models of HD [11,14].
Although further work is needed to delineate its precise mechanism of action, CXB-909 has been shown to amplify NGF in vitro [12] and has neuroprotective effects in vivo [14], which suggests that the behavioral sparing observed in the present study may be due to neurotrophic-like effects, such as amplification of NGF. The present finding underscores the fact that CXB-909 treatment has repeatedly led to behavioral sparing in several rodent models of neurodegeneration and suggests that this compound confers significant neuroprotective properties and warrants further consideration as a potential treatment for AD.
5. Acknowledgements
This project was supported by funds from the Field Neurosciences Institute and the John G. Kulhavi Endowed Professorship in Neuroscience to (GLD), and the Central Michigan University Program in Neuroscience. The authors thank Ming Lu for his technical assistance in completing this project.
REFERENCES
- Z. S. Khachaturian, “Diagnosis of Alzheimer’s Disease,” Archives of Neurology, Vol. 42, No. 11, 1985, pp. 1097- 1105. doi:10.1001/archneur.1985.04060100083029
- R. T. Bartus, “On Neurodegenerative Diseases, Models, and Treatment Strategies: Lessons Learned and Lessons Forgot-Ten a Generation Following the Cholinergic Hypothesis,” Experimental Neurology, Vol. 163, No. 2, 2000, pp. 495-529. doi:10.1006/exnr.2000.7397
- J. Berger-Sweeney, N. A. Stearns, S. L. Murg, et al., “Selective Immunolesions of Cholinergic Neurons in Mice: Effects on Neuroanatomy, Neurochemistry, and Behavior,” Journal of Neuroscience, Vol. 21, No. 20, 2001, pp. 8164-8173.
- P. H. Moreau, B. Cosquer, H. Jeltsch, et al., “Neuroanatomical and Behavioral Effects of a Novel Version of the Cholinergic Immunotoxin mu-p-75-Saporin in Mice,” Hippocampus, Vol. 18, No. 6, 2008, pp. 610-622. doi:10.1002/hipo.20422
- M. H. Tuszynski, “Intraparenchymal NGF Infusions Rescue Degenerating Cholinergic Neurons,” Cell Transplant, Vol. 9, No. 9, 2000, pp. 629-636.
- A. C. Cuello, M. A. Bruno and K. F. Bell, “NGF-Cholinergic Dependency in Brain Aging, MCI and Alzheimer’s Disease,” Current Alzheimers Research, Vol. 4, No. 4, 2007, pp. 351-358. doi:10.2174/156720507781788774
- V. Di Fausto, M. Fiore, P. Tirassa, A. Lambiase and L. Aloe, “Eye Drop NGF Administration Promotes the Recovery of Chemically Injured Cholinergic Neurons of Adult Mouse forebrain,” European Journal of Neuroscience, Vol. 26, No. 9, 2007, pp. 2473-2480. doi:10.1111/j.1460-9568.2007.05883.x
- H. Kewitz, K. L. Rost, O. Pleul and A. Handke, “DoseRelated Effects of Nerve Growth Factor (NGF) on Choline Acetyltransferase (ChAT), Acetylcholine (ACh) Content and ACh Turnover in the Brain of Newborn Rats,” Neurochemistry International, Vol. 17, No. 2, 1990, pp. 239-244. doi:10.1016/0197-0186(90)90146-K
- A. H. Nagahara, T. Bernot, R. Moseanko, et al., “LongTerm Reversal of Cholinergic Neuronal Decline in Aged Non-Human Primates by Lentiviral NGF Gene Delivery,” Experimental Neurology, Vol 215, No. 1, 2009, pp. 153- 159. doi:10.1016/j.expneurol.2008.10.004
- A. Salehi, J. D. Delcroix and D. F. Swaab, “Alzheimer’s Disease and NGF Signaling,” Journal of Neural Transmission, Vol. 111, No. 3, 2004, pp. 323-345. doi:10.1007/s00702-003-0091-x
- G. L. Dunbar, M. I. Sandstrom, J. Rossignol and L. Lescaudron, “Eurotrophic Enhancers as Therapy for Behavioral Deficits in Rodent Models of Huntington’s Disease: Use of Gangliosides, Substituted Pyrimidines, and Mesenchymal Stem Cells,” Behavioral Cognitive Neuroscience Reviews, Vol. 5, No. 2, 2006, pp. 63-79. doi:10.1177/1534582306289367
- J. A. Fyfe, L. M. Beauchamp and A. O. Caggiano, “P544 Amplifies the Effects of Nerve Growth Factor on Cell Differentiation and Is Neuroprotective,” Drug Development Research, Vol. 62, No. 1, 2004, pp. 49-59. doi:10.1002/ddr.10385
- M. A. Geist, C. Volbracht, J. Podhorna, et al., “Wide Spectrum Modulation by KP-544 in Models Relevant for Neuronal Survival,” Neuroreport, Vol. 16, No. 6, 2007, pp. 571-575.
- T. A. Krenitsky, J. Dillberger, E. Zotova, et al., “KP544, a Nerve Growth Factor Amplifier: Pharmacokinetics, Safety, and Efficacy in the Rat,” Pharmacology & Pharmaceutical Medicine, Vol. 62, No. 1, 2004, pp. 50-60.
- N. D. Dey, A. J. Boersen, R. A. Myers, et al., “The Novel Sub-Stituted Pyrimidine, KP544, Reduces Motor Deficits in the R6/2 Transgenic Mouse Model of Huntington’s Disease,” Restorative Neurology and Neuroscience, Vol. 25, No. 5-6, 2007, pp. 485-492.
- R. Morris, “Developments of a Water-Maze Procedure for Studying Spatial Learning in the Rat,” Journal of Neuroscience Methods, Vol. 11, No. 1, 1984, pp. 47-60. doi:10.1016/0165-0270(84)90007-4
- F. Papaleo, J. L. Silverman, J. Aney, et al., “Working Memory Deficits Increased Anxiety-Like Traits and Seizure Susceptibility in BDNF Overexpressing Mice,” Learning & Memory, Vol. 18, No. 8, 2011, pp. 534-544.
- A. M. Fortress, M. Buhusi, K. L. Helke and A. C. Granholm, “Cholinergic Degeneration and Alterations in the TrkA and p75NTR Balance as a Result of Pro-NGF Injection into Aged Rats,” Journal of Aging Research, Vol. 2011, 2011, p. 1-10. doi:10.4061/2011/460543