Journal of Biomedical Science and Engineering
Vol.10 No.04(2017), Article ID:75779,14 pages
10.4236/jbise.2017.104011
Allele-Specific Effects on Extracellular Superoxide Dismutase Synthesis and Secretion
Sujung Jun1, Ladislav Dory2*
1Department of Physiology and Pharmacology, School of Medicine, West Virginia University Health Science Center, Morgantown, WV, USA
2Institute for Cardiovascular and Metabolic Disease, University of North Texas Health Science Center, Fort Worth, TX, USA

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: December 13, 2016; Accepted: April 25, 2017; Published: April 28, 2017
ABSTRACT
Plasma extracellular superoxide dismutase (ecSOD) concentrations are significantly (2 - 3-fold) higher in 129 mice than in the C57 strain. We reported earlier that the 129 strain carries a different allele of this enzyme, characterized by two amino acid substitutions and a 10 bp deletion in the 3’UTR of the mRNA. One of the substitutions is at the signal peptide cleavage site (position 21) while the other is within the catalytic site. Since the differences in plasma enzyme concentrations persist in congenic mice expressing the different alleles, the differences in plasma concentrations appear to be largely allele-driven and independent of the remainder of the genome. As expected, in vitro translation using rabbit reticulocyte lysate showed no differences in translational efficiency of transcripts and processing of the newly synthesized enzymes. Determinations of the rates of synthesis and secretion of the two isoforms of this enzyme in stably transfected CHO cells clearly demonstrate that the mature 129 variant is synthesized at rates nearly twice those of the wild-type isoform and has an intracellular pool size twice as large. However, both isoforms are secreted at the same fractional rate and have identical intracellular T1/2 as well as similar extent of degradation. These data explain, at least partially, the higher levels of ecSOD in congenic mice expressing the 129 variant, as well as enzyme levels in the 129 strain of mice. In silico calculations support this conclusion and indicate that the aa change at the signal peptide cleavage site (N21D) results in a substantial increase in cleavage probability at this site for the 129 variant. Our results also highlight the importance of signal peptide cleavage in determining steady-state levels of secretory proteins.
Keywords:
Extracellular Superoxide Dismutase (ecSOD), Signal Peptide Cleavage, Rates of Synthesis, Rates of Secretion, Allelic Differences

1. Introduction
Extracellular superoxide dismutase (ecSOD or SOD3) is the only enzyme that scavenges superoxide specifically in the extracellular compartment and protects components of the extracellular matrix from oxidative damage. Recent studies further strengthen the case for ecSOD and its degree of expression/tissue distribution being a significant factor in the various oxidative stress-related diseases. For instance, it has been documented that decreased ecSOD expression (possibly due to decreased copy number or promoter methylation) correlates with poor prognosis in lung and mammary carcinomas [1] [2] and pancreatic ductal adenocarcinomas [3] . Decreased ecSOD expression, which leads to increased oxidative degradation of the ECM, may thus promote cancer metastasis.
Recent studies have also shown the protective role of ecSOD on muscle integrity and congestive heart failure: it protects against cachexia, exercise intolerance, loss of mitochondria and atrophy gene expression [4] . EcSOD polymorphism has also been linked to lung function [5] . Novel polymorphisms in the non-coding 5’untranslated region (E1) and first intron (I1) are associated with reduced lung function and increased risk of clinical COPD [5] . Any polymorphisms/mutations that lead to reduced expression, activity or extracellular matrix attachment, result in increased lung tissue matrix collagen I, hyaluronan and heparin sulfate oxidative fragmentation. Such fragments are chemotactic and contribute to the recruitment of inflammatory cells [6] [7] [8] .
Clearly, the understanding of the etiology of a number of oxidative stress-re- lated diseases as well as potential therapeutic approaches for their treatment would be significantly enhanced by understanding factors that regulate the expression of this enzyme. While the involvement of several transcription factors or epigenetic regulation of ecSOD expression has been examined [1] [2] [9] - [15] , there is essentially no information regarding potential regulatory mechanisms at the posttranscriptional level [16] .
We previously reported that the 129P3/J strain of mice express an ecSOD allele which differ in several aspects from the wild-type (wt) allele expressed by the C57- BL/6J and most other strains. It contains an N21D substitution within the putative signal sequence of ecSOD, an A186S substitution in the catalytic domain and a 10 bp deletion in the 3’untranslated region (UTR) of the mRNA [17] . This genotype has a profound effect on the ecSOD phenotype: plasma ecSOD level and activity in the 129P3/J mice are 2 - 3 fold higher than those in the C57BL/6 mice, showing no apparent change in specific activity. The phenotype associated with this allele (“129”) is mostly allele-driven, as congenic mice (C57.129-sod3) expressing either ecSOD genotype on a C57BL/6 background fully recapitulate the allele-specific ecSOD phenotype [18] . The congenic mice expressing either of the ecSOD alleles are genetically 99.8% identical to C57BL/6J [18] ; taken together, these observations suggest the possibility that differences in post-transcriptional processes are responsible for observed differences in the ecSOD phenotype in mice. Moreover, one of the amino acids substitutions (N21D) is positioned at the site of signal peptide cleavage and may thus produce changes in processing efficiency.
We now report that the relative translational efficiency of the transcripts of the two ecSOD alleles was identical in a rabbit reticulocyte translation system, in which no post-translational modification occurs. However, the rate of ecSOD mature protein synthesis in stably transfected CHO cells is significantly higher for the 129 allele, when compared to the C57 allele. These results confirm the notion that even a single amino acid substitution in the right place can have profound effects on the extent of enzyme expression.
2. Experimental Procedures
In vitro transcription and translation:
pcDNA3.1D/V5-His-TOPO plasmids with either wt or 129 ecSOD alleles (as previously described by Mirossay et al. [19] ) were linearized with Not I (New England Biolabs) at 37˚C for 2 hr. The linearized plasmids were extracted with phenol, precipitated with ethanol, and re-suspended in nuclease-free water (Ambion). In vitro transcription was performed with 5 μg linearized plasmid using the RiboMax T7 Large-Scale RNA Production kit (Promega), according to the manufacturer’s protocol, followed by purification by the phenol:chloroform method. The sequences of the template ecSOD for the in vitro transcription were confirmed in both directions. In vitro transcribed mRNAs were modified by 7-methyl G capping and polyadenylation. The capping reaction was carried out according to the manufacturer’s protocol (EPICENTRE) using 20 pmoles of mRNA/capping reaction. Capped mRNAs were polyadenylated for 30 min at 37˚C in 25 μl, containing 10 pmoles of capped mRNA and 600 U PAP, according to the manufacturer’s protocol (Ambion). Polyadenylated transcripts were purified using oligo-dT beads (Ambion). The in vitro transcribed RNAs and polyadenylated transcripts were monitored on 1% agarose-formaldehyde gels and were tested for quantity and quality by Nanodrop and Experion microcapillary electrophoresis (Bio-Rad), respectively.
In vitro translation assay reactions were performed in nuclease-treated rabbit reticulocyte lysates (RRL, Promega, cat. #L4960) at 30˚C. The translation system was charged with an equal amount of in vitro transcribed mRNA. EcSOD expression was examined by Western blotting. Semi-purified mouse plasma ecSOD, incubated with or without peptide: N-glycosidase F (PNGase F, New England Labs), was used as a reference. Post-heparin plasma ecSOD was semi-puri- fied by ConA-sepharose column as previously described [18] .
Cell culture:
CHO cells were stably transfected with either wt or 129 allele of ecSOD and established as previously published [19] . Cells were incubated with DMEM/10% FBS at 37˚C in a humidified atmosphere containing 5% CO2. To purify mRNA and genomic DNA, cells were washed with ice-cold PBS and dissolved in 1mL of TRIreagent® (Molecular Research Center) and kept at −80˚C until used.
Real-time RT-PCR:
CHO cell RNA was purified according to the manufacturer’s protocol (Molecular Research Center). EcSOD mRNA levels in stably transfected CHO cells were evaluated by real-time RT-PCR, as previously published [18] . Only cells that expressed the same amount of mRNA for either of the ecSOD alleles were used for the studies outlined below.
Continuous and pulse-chase labeling studies:
CHO cells (1.8 × 106 cells/well) were seeded in 6-well plates in DMEM/10% FBS. For continuous labeling studies (up to 8 hrs) cells were washed 2 times in methionine- and cysteine-free DMEM, followed by an incubation in the labeling medium (Met- and Cys-free DMEM, containing 60 μCi/ml [35S]-Met and Cys (Perkin Elmer, USA). Incubations were stopped at 30 min to 60 min intervals, for up to 8 hours. Total labeled protein as well as ecSOD was quantitated in cell extracts as well as medium at each time point, as described below.
Pulse-chase labeling studies were carried out in a similar manner; cells were pre-labeled with Met- and Cys-free DMEM containing 60 μCi/ml [35S]-Met and Cys for 3 hours (to label the total cellular pool of ecSOD), washed and chased in regular DMEM for indicated periods of time. To quantitate total protein and ecSOD intracellularly, as well as in the medium, cell lysates and media were collected in the presence of protease inhibitors (Sigma), containing 100 µM 4-(2- aminoethyl) benzenesulfonyl fluoride, 10 µM leupeptin and E-64, 1 µM bestatin and pepstatin-A, and 15-nM aprotinin. Prior to lysis, cells were washed twice with ice-cold PBS and collected in a final volume of 1 mL.
TCA precipitation: Total radiolabeled secreted (medium) or cellular protein synthesis was determined by trichloroacetic (TCA) acid precipitation, as previously described [20] . The results were calculated in terms of corrected cpm/mg cell protein. Cellular protein content was determined by the Lowry assay [21] .
Immunoprecipitation: Incorporation of [35S] Met and Cys into cellular and medium ecSOD was determined by immunoprecipitation with a rabbit anti ecSOD serum. Total medium ecSOD was immunoprecipitated in the presence of 0.05% Triton X-100 and a protease inhibitor cocktail (Sigma) at 4˚C for 24 h, as previously described (20). The immunoprecipitate was resolved by 12% SDS- PAGE. Slab gels processed for fluorography were soaked first in a conditioning solution (1% glycerol, 7% acetic acid and 7% methanol) and next in an enhancer solution (1 M salicylic acid) and dried. X-ray films were exposed at −80˚C and developed. The X-ray films were scanned and densitometry of ecSOD was performed using the supplied software AlphaEaseFC (Alpha Innotech). Results were expressed as “Integrated Density Value” or IDV, calculated by the software.
Intracellular ecSOD was immunoprecipitated in a similar fashion, except that 1% Triton X-100 was used and the cell lysates were pre-cleared by addition of non- immune rabbit serum, followed by protein A-agarose, to minimize background.
Statistical Analysis:
Results from experiments were reported as mean ± SEM. Statistical analyses were done using t-tests, linear regression comparisons or two-way ANOVA (GraphPad Prism). P < 0.05 was considered statistically significant.
3. Results
The in vitro translational efficiency and processing of the wt and 129 ecSOD transcript
We first examined the translation of in vitro transcribed wt or 129 ecSOD transcripts in nuclease-treated rabbit reticulocyte lysates. The translated wt and 129 ecSOD proteins appear to be the same size as the full-length, de-glycosy- lated mouse plasma ecSOD. The translation efficiency of the two transcripts appears to be similar, regardless of the transcript concentration (Figure 1), suggesting that the reticulocyte lysate was saturated even at the lowest mRNA concentrations used to allow for the detection of the product by Western blotting.
Rates of ecSOD synthesis and secretion in stably transfected CHO cells
To test the allele-dependent ecSOD phenotype in mammalian cells, we established stably transfected CHO cells, as previously described (19) with either of the ecSOD alleles. The genomic copy number and the mRNA level of ecSOD were checked by real-time qPCR. Only CHO cell clones with an equal mRNA copy number/total mRNA were used for the labeling experiments.
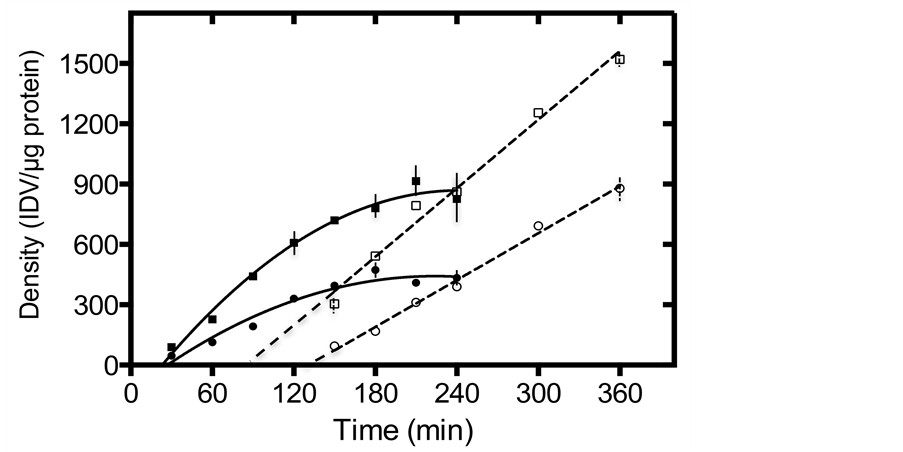
To compare the rates of synthesis, cellular pool size and secretion, we employed a continuous labeling technique. Labeled cellular ecSOD levels increased with time and both products reached plateau at about 180 min of incubation, indicating that the intracellular ecSOD pool is fully labeled within 3 hours. As shown in Figure 2(a) and Figure 2(b), the synthetic rate of mature 129 ecSOD, derived from the calculated slope of the initial rates (between times 30 - 120 min) appears to be nearly 2-fold (1.9-fold) higher than that of wt ecSOD (3.11 ± 0.39 vs. 5.90 ± 0.32 IDV/μg of protein/min), matching exactly the allele-specific differences in the cellular pool size of ecSOD (439 ± 17 vs. 850 ± 74 IDV/μg of protein, or a 1.9-fold difference). To check if the increased synthetic rates of 129 ecSOD are coupled to higher rates of secretion, ecSOD was also immunoprecipitated from the medium and is shown in Figure 2(a) & Figure 2(c). Since the
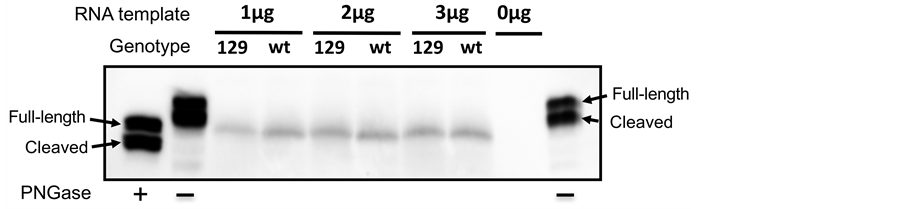
Figure 1. In vitro translation of wt and 129 ecSOD. In vitro translated ecSODs using same amounts (1 - 3 μg) of in vitro transcribed wt or 129 ecSOD mRNA were detected by Western blot to evaluate synthesis. ConA-purified mouse plasma ecSOD (first lane) and deglycosylated ecSOD by PNGase F treatment (second and last lanes) were used as a controls. Full-length subunit of mouse plasma ecSOD is 32-kDa while the heparin-binding domain-cleaved form is 30-kDa. When ecSOD is deglycosylated by PNGase, both subunits are reduced by ~3 kDa. A reaction without any RNA template (-) was used as a negative control (second to last lane). The size of wt and 129 in vitro synthesized ecSOD are similar to deglycosylated full-length plasma ecSOD.
 (a)
(a)
 (b)
(b)
 (c)
(c)
Figure 2. The rate of [35S]-labeled ecSOD synthesis and secretion in stably transfected CHO cells, as determined by continuous labeling. (a) Accumulation of labeled cellular (full symbols) and medium (open symbols) ecSOD in CHO expressing the wt (circles) or 129 (squares) isoform. The slope of the linear portion of the curve (30 - 150 min) representing incorporation of [35S]-met and -cys into cellular ecSOD, which was normalized with total protein level, was used to calculate the rate of ecSOD synthesis (expressed as Integrated Density Value or IDV/μg/minute). Steady state pool sizes were calculated by averaging intensity during 180 - 240 min when the labeled ecSOD plateaud after normalization with protein. Typical fluorographs of cellular (b) or medium (c) immunoprecipitates of 129 and wt ecSOD at indicated time points. SDS-PAGE gels resolving the immunoprecipitates were exposed for 15 days in −80˚C. Densitometric analysis of labeled ecSOD was normalized by protein level.
intracellular 129 isoform of ecSOD is labeled twice as rapidly as that of the wt, it is not surprising that it appears in the medium earlier and at a more rapid rate; the time of appearance in the medium was almost 45 minutes earlier for the 129 enzyme, when compared to the wt enzyme (85 vs. 131 min of labeling). The increased rate of 129 ecSOD synthesis was matched with a significant apparent increase in the rate of secretion, relative to that of wt ecSOD. In contrast, total protein synthesis and secretion by these cells was identical (Figure 3(a) & Figure 3(b), respectively).
Pulse-chase experiments
Secretion rates derived from continuous labeling studies can be misleading and difficult to interpret, since they do not account for differences in the cellular pool size and involve partially labeled products. Moreover, such design does not provide any information regarding the potential degradation (intra- or extra- cellular) of the secreted protein. We therefore used the “pulse-chase” design, in which cells are pulsed with a label until all cellular ecSOD is labeled (3hrs, as determined in experiments shown in Figure 2) and then chased in label-free medium. Intracellular and medium ecSOD is quantitated at each time point. This allows for the calculation of secretion rates (as a portion or % of the cellular pool size at time 0 of chase) as well as the degradation (as the difference in the amounts recovered from cells and medium, relative to that found in cells at time 0 of chase). We therefore examined the half-lives and the extent of degradation of both isoforms of ecSOD. The results are shown in Figure 4. As observed in continuous labeling experiments, cellular 129 ecSOD pool size at time 0 of chase is nearly 2-fold higher (1.86-fold, 985 ± 47 vs. 1836 ± 68 IDV/μg, data not shown) than that of wt ecSOD. The apparent secretion rate is also shown to

Figure 3. The rate of [35S]-labeled total protein synthesis (a) and secretion (b) in stably transected CHO cells, as determined by continuous labeling for indicated times followed by TCA precipitation and quantitation, as described in Methods in CHO cells expressing the wt (closed symbols) or 129 isoform of ecSOD (open symbols).

Figure 4. Rates of ecSOD secretion, T1/2 and extent of degradation, as determined by pulse-chase experiments (3 independent sets of experiments). CHO cells were pre-la- beled, as described in the Methods, for 3 hours following which they were switched to normal media. The loss of labeled ecSOD in the cells and the appearance of ecSOD in the medium were followed by immunoprecipitation (as described in Methods). Data at each subsequent time point were normalized to the amount of labeled ecSOD found at time 0. The extent of degradation was calculated by subtracting the percentage of the sum of cellular and medium ecSOD at T1/2 from that found at time 0 of chase %.
be significantly higher in 129 ecSOD when compared to the wt in the media (not shown). However, when secretion (i.e., loss in cellular ecSOD and appearance of medium ecSOD) is expressed as a fraction (%) of the ecSOD labeled pool size at time 0, the fractional rates of secretion of both isoforms were identical.
Pulse-chase experiments can also reveal the intracellular half-life (T1/2) of ecSOD as well as the extent of degradation (intra- or extra-cellular). The half-life of both isoforms of ecSOD was the same (86 - 87 min). Our data indicate that once processed, the rates of ecSOD secretion are similar for both isoforms and that a similar (~20%) proportion of cellular enzyme is degraded.
4. Discussion
Our results clearly demonstrate that ecSOD polymorphism has a profound effect on several aspects of ecSOD phenotype, including rates of synthesis, cellular pool size and proteolytic cleavage, independent of the genomic context. The synthetic rate and steady state levels of intracellular 129 ecSOD are significantly higher than those of wt ecSOD in stably transfected CHO cells, despite similar levels of mRNA. These observations are analogous to the in vivo situation we reported earlier [18] . Although the apparent secretion rate is higher in CHO cells secreting 129 ecSOD compared with wt ecSOD, pulse-chase studies indicate that the fractional secretion rate is identical. These results may explain the allele-driven difference in the phenotype observed in vivo, in tissue expression vs. mRNA levels [18] .
Using the simplest expression system, the rabbit reticulocyte lysate, the translational efficiency and processing rate were similar for both transcripts, regardless of capping or polyadenylation. These results suggest that the nuclease- treated rabbit reticulocyte lysate system may not contain the required components of the regulatory machinery responsible for the ecSOD phenotype in vivo.
The continuous labeling studies show that the synthetic rate of 129 ecSOD is 1.9-fold greater than that of wt ecSOD, leading to 1.9-fold greater steady-state intracellular levels of the enzyme. Since the detectable intracellular ecSOD is the same size as the mature glycosylated form, these results suggest that the rates of 129 ecSOD synthesis and/or processing to the mature form are greater than those of wt ecSOD. This result is supported by in silico analysis of the signal peptide cleavage likelihood or efficiency. Public domain software SignalP4.1 [22] predicts the presence of signal peptide in both sequences unequivocally, identifying residue 21 as the cleavage site. But the cleavage site score (C-score) is nearly 20% higher for the 129 allele (N21) sequence, when compared to the wild-type, C57 sequence (D21). The relative cleavage site probability at this point rises from 0.617 in the wt product to 0.774 in the 129 isoform. Although it is not clear whether the increased probability can account for the nearly 2-fold increase in the rates of synthesis, a few studies have demonstrated that sequence variation in the signal peptide can affect protein targeting and translocation, signal sequence cleavage and even post-cleavage events [23] - [28] . Importantly, the modulation of specific steps in the recognition and processing of signal sequences can have an essential role in protein biogenesis [23] . Signal sequences regulate the timing and efficiency of cleavage which can control the exit of proteins from the ER; the exceptionally slow cleavage of the native HIV gp 120 signal sequence causes prolonged retention of the protein in the ER [23] [24] . Furthermore, an amino acid substitution at the 1 position of signal peptide cleavage site of human preproapolipoprotein A-II was shown to lower the processing efficiency by signal peptidase activity [25] .
Several studies reported that signal peptidase cleavage site mutations had significant clinical effects. A substitution at the 3 position with respect to the signal peptide-processing site in Factor XSanto Domingo prevented signal peptide cleavage and the mutant protein was not secreted, leading to a bleeding disorder [26] . An Ala to Thr change at the 1 position in preprovasopressin, causing familial central diabetes insipidus, also led to failure of signal peptide processing and reduced secretion [27] . Same change at 1 position (A25T) of ADAMTS10 signal peptide cleavage site affected the secretion of full-length protein, causing recessive Weill- Marchesani syndrome (WMS) [28] . A transition mutation at position 172 of human ecSOD gene (Ala40Thr), which disrupts the tetramerization of ecSOD and prevents its secretion, leads to increased susceptibility to type 2 diabetes as well as hepatocellular carcinoma in Chinese (29). These studies clearly demonstrate that a single aa change (in the signal peptide or elsewhere) can modulate protein secretion.
It appears unlikely that the substitution in catalytic domain (A186S) of 129 ecSOD plays a role in the protein production, but we have not investigated this. It does not appear to have an effect on the specific activity, as previously shown [18] . Another major difference between the two ecSOD transcripts is a 10 bp deletion in the 3’UTR in the 129. allele. Since the 3’UTR sequence of many transcripts is subject to translational regulation by miRNA, we examined the effects of two selected miRNAs: miR-744 and miR-663. These are expected to bind to the 3’UTR of the mouse ecSOD in an area overlapping the 10 bp deletion (10 - 19 bp) region in the 129 ecSOD mRNA. The 10 bp deletion in the 129 ecSOD mRNA results in 2 (miR-744) or 3 (miR-663) mismatches of miRNA binding sites, when compared to wt allele. Although not shown, the effects of these miRNAs on the translational efficiency were examined by using a firefly luciferase assay cloned to the 3’UTR of either the wt or 129 allele of ecSOD. Neither miR-744 nor miR-663 significantly altered the rates of synthesis of the luciferase regardless of the source of 3’UTR. These results do not eliminate the possibility of other, yet to be identified miRNA(s) that may modulate translational efficiency of either transcript.
It has been suggested that ecSOD can be stored in intracellular vesicles in tissues which have the potential to be secreted upon stimulation [29] [30] [31] [32] . Previous study, which has shown localization of ecSOD in the endosomal fraction [33] , suggests that there might be some degradation of intracellular ecSOD. In this study, we observed about 21% of degradation regardless of the genotype.
In summary, our data suggest that the increased efficiency of 129 ecSOD processing contributes to the increased protein level in 129 congenic mice [18] and in the 129 strain of mice [17] ; the rates of degradation and secretion per se do not appear to play a role. These findings are in agreement with recent findings that genetic polymorphisms in ecSOD modulate protein synthesis and processing, which impact cellular and extracellular distribution of this enzyme [34] . Furthermore, these differences can profoundly impact susceptibility to multiple oxidative stress-related diseases including cardiovascular disease, cancers, lung fibrosis, even resistance to bacterial infections not only in animal models [34] [35] [36] [37] [38] but also in humans [39] [40] [41] [42] [43] .
It appears likely that additional, yet to be discovered mutations in the ecSOD gene lead to variations in gene expression at both, transcriptional and translational level and disease susceptibilty. Considering the important role ecSOD plays in maintaining the integrity of the extracellular matrix and the extent of oxidative stress in that compartment, it is clear that a good understanding of the factors that regulate the expression of this enzyme is of high clinical relevance and merits further studies. As our study indicates, even small, single amino acid substitutions, while having little or no effect on the enzyme activity may profoundly affect the extent of secretion and thus the extent of protection of the ECM.
Cite this paper
Jun, S.J. and Dory, L. (2017) Allele-Specific Effects on Extracellular Superoxide Dismutase Synthesis and Secretion. J. Biomedical Science and Engineering, 10, 135-148. https://doi.org/10.4236/jbise.2017.104011
References
- 1. Teoh-Fitzgerald, M.L., Fitzgerald, M.P., Jensen, T.J., Futscher, B.W. and Domann, F.E. (2012) Genetic and Epigenetic Inactivation of Extracellular Superoxide Dismutase Promotes an Invasive Phenotype in Human Lung Cancer by Disrupting ECM Homeostasis. Molecular Cancer Research, 10, 40-51.
https://doi.org/10.1158/1541-7786.MCR-11-0501 - 2. Teoh-Fitzgerald, M.L., Fitzgerald, M.P., Zhong, W., Askeland, R.W. and Domann, F.E. (2014) Epigenetic Reprogramming Governs EcSOD Expression during Human Mammary Epithelial Cell Differentiation, Tumorigenesis and Metastasis. Oncogene, 33, 358-368.
https://doi.org/10.1038/onc.2012.582 - 3. O,Leary, B.R., Fath, M.A., Bellizzi, A.M., Hrabe, J.E., Button, A.M., Allen, B.G., et al. (2015) Loss of SOD3 (EcSOD) Expression Promotes an Aggressive Phenotype in Human Pancreatic Ductal Adenocarcinoma. Clinical Cancer Research, 21, 1741-1751.
https://doi.org/10.1158/1078-0432.CCR-14-1959 - 4. Okutsu, M., Call, J.A., Lira, V.A., Zhang, M., Donet, J.A., French, B.A., et al. (2014) Extracellular Superoxide Dismutase Ameliorates Skeletal Muscle Abnormalities, Cachexia, and Exercise Intolerance in Mice with Congestive Heart Failure. Circulation: Heart Failure, 7, 519-530.
https://doi.org/10.1161/CIRCHEARTFAILURE.113.000841 - 5. Dahl, M., Bowler, R.P., Juul, K., Crapo, J.D., Levy, S. and Nordestgaard, B.G. (2008) Superoxide Dismutase 3 Polymorphism Associated with Reduced Lung Function in Two Large Populations. American Journal of Respiratory and Critical Care Medicine, 178, 906-912.
https://doi.org/10.1164/rccm.200804-549OC - 6. Gao, F., Koenitzer, J.R., Tobolewski, J.M., Jiang, D., Liang, J., Noble, P.W., et al. (2008) Extracellular Superoxide Dismutase Inhibits Inflammation by Preventing Oxidative Fragmentation of Hyaluronan. The Journal of Biological Chemistry, 283, 6058-6066.
https://doi.org/10.1074/jbc.M709273200 - 7. Kliment, C.R., Tobolewski, J.M., Manni, M.L., Tan, R.J., Enghild, J. and Oury, T.D. (2008) Extracellular Superoxide Dismutase Protects against Matrix Degradation of Heparan Sulfate in the Lung. Antioxidants & Redox Signaling, 10, 261-268.
https://doi.org/10.1089/ars.2007.1906 - 8. Petersen, S.V., Oury, T.D., Ostergaard, L., Valnickova, Z., Wegrzyn, J., Thogersen, I.B., et al. (2004) Extracellular Superoxide Dismutase (EC-SOD) Binds to Type I Collagen and Protects against Oxidative Fragmentation. The Journal of Biological Chemistry, 279, 13705-13710.
https://doi.org/10.1074/jbc.M310217200 - 9. Zelko, I.N. and Folz, R.J. (2003) Myeloid Zinc Finger (MZF)-Like, Kruppel-Like and ETS Families of Transcription Factors Determine the Cell-Specific Expression of Mouse Extracellular Superoxide Dismutase. Biochemical Journal, 369, 375-386.
https://doi.org/10.1042/bj20021431 - 10. Zelko, I.N. and Folz, R.J. (2004) Sp1 and Sp3 Transcription Factors Mediate Trichostatin A—Induced and Basal Expression of Extracellular Superoxide Dismutase. Free Radical Biology & Medicine, 37, 1256-1271.
- 11. Zelko, I.N., Mueller, M.R. and Folz, R.J. (2008) Transcription Factors sp1 and sp3 Regulate Expression of Human Extracellular Superoxide Dismutase in Lung Fibroblasts. American Journal of Respiratory Cell and Molecular Biology, 39, 243-251.
https://doi.org/10.1165/rcmb.2007-0378OC - 12. Itoh, S., Ozumi, K., Kim, H.W., Nakagawa, O., McKinney, R.D., Folz, R.J., et al. (2009) Novel Mechanism for Regulation of Extracellular SOD Transcription and Activity by Copper: Role of Antioxidant-1. Free Radical Biology & Medicine, 46, 95-104.
- 13. Zelko, I.N., Mueller, M.R. and Folz, R.J. (2010) CpG Methylation Attenuates Sp1 and Sp3 Binding to the Human Extracellular Superoxide Dismutase Promoter and Regulates Its Cell-Specific Expression. Free Radical Biology & Medicine, 48, 895-904.
- 14. Laukkanen, M.O., Mannermaa, S., Hiltunen, M.O., Aittomaki, S., Airenne, K., Janne, J., et al. (1999) Local Hypomethylation in Atherosclerosis Found in Rabbit ec-sod Gene. Arteriosclerosis, Thrombosis, and Vascular Biology, 19, 2171-2178.
https://doi.org/10.1161/01.ATV.19.9.2171 - 15. Zelko, I.N., Stepp, M.W., Vorst, A.L. and Folz, R.J. (2011) Histone Acetylation Regulates the Cell-Specific and Interferon-Gamma-Inducible Expression of Extracellular Superoxide Dismutase in Human Pulmonary Arteries. American Journal of Respiratory Cell and Molecular Biology, 45, 953-961.
https://doi.org/10.1165/rcmb.2011-0012OC - 16. Miao, L. and St Clair, D.K. (2009) Regulation of Superoxide Dismutase Genes: Implications in Disease. Free Radical Biology & Medicine, 47, 344-356.
- 17. Pierce, A., Whitlark, J. and Dory, L. (2003) Extracellular Superoxide Dismutase Polymorphism in Mice. Arteriosclerosis, Thrombosis, and Vascular Biology, 23, 1820-1825.
https://doi.org/10.1161/01.ATV.0000089011.51491.A6 - 18. Jun, S., Pierce, A. and Dory, L. (2010) Extracellular Superoxide Dismutase Polymorphism in Mice: Allele-Specific Effects on Phenotype. Free Radical Biology & Medicine, 48, 590-596.
- 19. Mirossay, A., Jun, S. and Dory, L. (2007) Cloning and Characterization of Two Alleles of the Murine Extracellular Superoxide Dismutase Gene. Biochemical and Biophysical Research Communications, 352, 739-743.
- 20. Dory, L. (1989) Synthesis and Secretion of apoE in Thioglycolate-Elicited Mouse Peritoneal Macrophages: Effect of Cholesterol Efflux. The Journal of Lipid Research, 30, 809-816.
- 21. Lowry, O.H., Rosebrough, N.J., Farr, A.L. and Randall, R.J. (1951) Protein Measurement with the Folin Phenol Reagent. The Journal of Biological Chemistry, 193, 265-275.
- 22. Petersen, T.N., Brunak, S., von Heijne, G. and Nielsen, H. (2011) Signal P 4.0: Discriminating Signal Peptides from Transmembrane Regions. Nature Methods, 8, 785-786.
https://doi.org/10.1038/nmeth.1701 - 23. Hegde, R.S. and Bernstein, H.D. (2006) The Surprising Complexity of Signal Sequences. Trends in Biochemical Sciences, 31, 563-571.
- 24. Li, Y., Bergeron, J.J., Luo, L., Ou, W.J., Thomas, D.Y. and Kang, C.Y. (1996) Effects of Inefficient Cleavage of the Signal Sequence of HIV-1 gp 120 on Its Association with Calnexin, Folding, and Intracellular Transport. Proceedings of the National Academy of Sciences, 93, 9606-9611.
https://doi.org/10.1073/pnas.93.18.9606 - 25. Folz, R.J., Nothwehr, S.F. and Gordon, J.I. (1988) Substrate Specificity of Eukaryotic Signal Peptidase. Site-Saturation Mutagenesis at Position-1 Regulates Cleavage between Multiple Sites in Human Pre (Delta Pro) Apolipoprotein A-II. The Journal of Biological Chemistry, 263, 2070-2078.
- 26. Racchi, M., Watzke, H.H., High, K.A. and Lively, M.O. (1993) Human Coagulation Factor X Deficiency Caused by a Mutant Signal Peptide That Blocks Cleavage by Signal Peptidase But Not Targeting and Translocation to the Endoplasmic Reticulum. The Journal of Biological Chemistry, 268, 5735-5740.
- 27. Ito, M., Oiso, Y., Murase, T., Kondo, K., Saito, H., Chinzei, T., et al. (1993) Possible Involvement of Inefficient Cleavage of Preprovasopressin by Signal Peptidase as a Cause for Familial Central Diabetes Insipidus. The Journal of Clinical Investigation, 91, 2565-2571.
https://doi.org/10.1172/JCI116494 - 28. Kutz, W.E., Wang, L.W., Dagoneau, N., Odrcic, K.J., Cormier-Daire, V., Traboulsi, E.I., et al. (2008) Functional Analysis of an ADAMTS10 Signal Peptide Mutation in Weill-Marchesani Syndrome Demonstrates A Long-Range Effect on Secretion of the Full-Length Enzyme. Human Mutation, 29, 1425-1434.
https://doi.org/10.1002/humu.20797 - 29. Boggess, K.A., Kay, H.H., Crapo, J.D., Moore, W.F., Suliman, H.B. and Oury, T.D. (2000) Differential Localization of Placental Extracellular Superoxide Dismutase as Pregnancy Progresses. American Journal of Obstetrics & Gynecology, 183, 199-205.
- 30. Nozik-Grayck, E., Dieterle, C.S., Piantadosi, C.A., Enghild, J.J. and Oury, T.D. (2000) Secretion of Extracellular Superoxide Dismutase in Neonatal Lungs. The American Journal of Physiology-Lung Cellular and Molecular Physiology, 279, L977-L984.
- 31. Oury, T.D., Card, J.P. and Klann, E. (1999) Localization of Extracellular Superoxide Dismutase in Adult Mouse Brain. Brain Research, 850, 96-103.
- 32. Iversen, M.B., Gottfredsen, R.H., Larsen, U.G., Enghild, J.J., Praetorius, J., Borregaard, N., et al. (2016) Extracellular Superoxide Dismutase Is Present in Secretory Vesicles of Human Neutrophils and Released upon Stimulation. Free Radical Biology & Medicine, 97, 478-488.
- 33. Chu, Y., Piper, R., Richardson, S., Watanabe, Y., Patel, P. and Heistad, D.D. (2006) Endocytosis of Extracellular Superoxide Dismutase into Endothelial Cells: Role of the Heparin-Binding Domain. Arteriosclerosis, Thrombosis, and Vascular Biology, 26, 1985-1990.
https://doi.org/10.1161/01.ATV.0000234921.88489.5c - 34. Hartney, J.M., Stidham, T., Goldstrohm, D.A., Oberley-Deegan, R.E., Weaver, M.R., Valnickova-Hansen, Z., et al. (2014) A Common Polymorphism in Extracellular Superoxide Dismutase Affects Cardiopulmonary Disease Risk by Altering Protein Distribution. Circulation: Cardiovascular Genetics, 7, 659-666.
https://doi.org/10.1161/CIRCGENETICS.113.000504 - 35. Jun, S., Fattman, C.L., Kim, B.J., Jones, H. and Dory, L. (2011) Allele-Specific Effects of ecSOD on Asbestos-Induced Fibro Proliferative Lung Disease in Mice. Free Radical Biology & Medicine, 50, 1288-1296.
- 36. Break, T.J., Jun, S., Indramohan, M., Carr, K.D., Sieve, A.N., Dory, L., et al. (2012) Extracellular Superoxide Dismutase Inhibits Innate Immune Responses and Clearance of an Intracellular Bacterial Infection. The Journal of Immunology, 188, 3342-3350. https://doi.org/10.4049/jimmunol.1102341
- 37. Break, T.J., Witter, A.R., Indramohan, M., Mummert, M.E., Dory, L. and Berg, R.E. (2016) Extracellular Superoxide Dismutase Enhances Recruitment of Immature Neutrophils to the Liver. Infection and Immunity, 84, 3302-3312.
https://doi.org/10.1128/IAI.00603-16 - 38. Mouradian, G.C., Gaurav, R., Pugliese, S., El Kasmi, K., Hartman, B., Hernandez-Lagunas, L., et al. (2017) Superoxide Dismutase 3 R213G Single-Nucleotide Polymorphism Blocks Murine Bleomycin-Induced Fibrosis and Promotes Resolution of Inflammation. American Journal of Respiratory Cell and Molecular Biology, 56, 362-371.
https://doi.org/10.1165/rcmb.2016-0153OC - 39. Su, S., He, K., Li, J., Wu, J., Zhang, M., Feng, C., et al. (2015) Genetic Polymorphisms in Antioxidant Enzyme Genes and Susceptibility to Hepatocellular Carcinoma in Chinese Population: A Case-Control Study. Tumor Biology, 36, 4627-4632.
https://doi.org/10.1007/s13277-015-3110-2 - 40. Yang, Y.M., Xie, X.R. and Jin, A.L. (2016) Genetic Polymorphisms in Extracellular Superoxide Dismutase Leu53Leu, Arg213Gly, and Ala40Thr and Susceptibility to Type 2 Diabetes Mellitus. Genetics and Molecular Research, 15, gmr15048418.
https://doi.org/10.4238/gmr15048418 - 41. Zhang, C., Guo, L., Qin, Y. and Li, G. (2016) Correlation between Helicobacter Pylori Infection and Polymorphism of Adiponectin Gene Promoter-11391G/A, Superoxide Dismutase Gene Innonalcoholic Fatty Liver Disease. Journal of Central South University. Medical Sciences, 41, 359-366.
- 42. Oberley-Deegan, R.E., Regan, E.A., Kinnula, V.L. and Crapo, J.D. (2009) Extracellular Superoxide Dismutase and Risk of COPD. COPD, 6, 307-312.
https://doi.org/10.1080/15412550903085193 - 43. Mohammedi, K., Bellili-Munoz, N., Marklund, S.L., Driss, F., Le Nagard, H., Patente, T.A., et al. (2015) Plasma Extracellular Superoxide Dismutase Concentration, Allelic Variations in the SOD3 Gene and Risk of Myocardial Infarction and All-Cause Mortality in People with Type 1 and Type 2 Diabetes. Cardiovascular Diabetology, 14, 845.
https://doi.org/10.1186/s12933-014-0163-2