Journal of Cancer Therapy
Vol.4 No.6A1(2013), Article ID:33923,6 pages DOI:10.4236/jct.2013.46A1001
Human Multipotent Stem Cell Proteins Induce Apoptosis in Skin Cancer Cells
![]()
1Department of Dermatology, University of California, Mt. Zion Cancer Research Center, San Francisco, USA; 2Histogen, Inc., San Diego, USA.
Email: gnaughton@histogeninc.com
Copyright © 2013 Christian Posch et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received May 30th, 2013; revised June 27th, 2013; accepted July 4th, 2013
Keywords: Human Extracellular Matrix; Cell Conditioned Media; Melanoma; Caspase; Apoptosis
ABSTRACT
Unique characteristics in fetal development include scar-less wound healing and the paucity of tumor formation. Recent studies have demonstrated that the embryonic microenvironment can reverse melanoma cells to a benign melanocyte phenotype. We bioengineered embryonic-like compositions and tested the anti-cancer activity of this material on a panel of skin cancer lines. To simulate the embryonic environment, neonatal fibroblasts were grown in hypoxic suspension cultures. The cells reverted back into multipotent stem cells as evidenced by the upregulation of SOX2, Oct4, NANOG, and KLF4 genes, and by the expression of stem cell-associated proteins including Nodal, Brachyury, Nestin, and Oct4. Cell Conditioned Media (CCM) and human Extracellular Matrix Proteins (hECM) produced by these cells were tested for their ability to reduce cell viability in skin cancer cell lines. In vitro studies with CCM and hECM show reduction in Squamous Cell Carcinoma (SCC), Basal Cell Carcinoma (BCC) and melanoma cell number through upregulation of caspases and induction of apoptosis. In the chick allantoic membrane assay, melanoma load was reduced by up to 80% with hECM treatment compared to vehicle treated controls (p < 0.05). Similar inhibition was seen with SCC cells. In a xenograft mouse model of subcutaneous melanoma, tumor growth was inhibited by 70% - 90%. These data suggest that CCM and hECM have anti tumor potential and might offer a new treatment strategy in skin cancer.
1. Introduction
Research has elucidated key roles that the tumor extracellular matrix (ECM) plays in promoting tumor growth and metastasis, as well as the anti-tumor properties of the innate host ECM. The ability of cancer cells to expand locally and develop regional as well as distant metastasis relies on their biomechanical properties and their microenvironment which consists of collagens, laminins, a variety of growth factors and several matrix proteins [1]. Continuous interaction of the malignant cells with, as well as the conditioning of, the microenvironment allows cells to invade the connective tissue, basement membranes and endothelial layers. The embryonic microenvironment, however, seems to bear anticancerogenic characteristics.
There is growing evidence, that secretory products of fetal mesenchyme can reverse many aspects of the cancer phenotype. Metastatic melanoma cells, when placed within the neural crest of the developing chicken egg, integrate and differentiate toward the resident cells’ phenotype, displaying features of normal, benign tissue [2,3]. When LS1474T human colon cells and Dunning prostatic adenocarcinoma were co-cultured with rat fetal mesenchyme and seminal vesicle mesenchyme, LS1474T cells formed a polarized layer, contact inhibited with tight junctions resembling intestinal epithelium and displayed features of less aggressive prostate cancer [4,5]. Rat basal cell carcinoma cells cultured in primed uterine tissue lost their malignant growth behavior [6]. Further evidence of the ability of mesenchymal products to influence cell phenotype was shown with melanocytes and melanoma cells; when normal primary melanocytes were cultured in contact with ECM isolated from melanoma tumors they differentiated into melanoma cells. Conversely, melanoma cells cultured in fetal ECM reverted into benign melanocytes [7].
In addition to whole mesenchyme and ECM, individual biomolecules found in the embryonic environment have been demonstrated to slow or reverse neoplastic behavior. The proteoglycan decorin strongly inhibits tumor growth and metastasis by inhibiting ErbB2 receptor tyrosine kinase thereby reducing EGF receptor expression [8,9]. Lack of the proteoglycan dystrolycan in the basement membrane has been strongly linked to breast and prostate cancer progression [10,11]. Fractions of the noncollagenous 1 (NC-1) domain of collagen VII, the tumstatins, inhibit attachment and proliferation of cancer cells by binding intergrin αvβ3 and endostatin collagen 18 by binding integrin α5β1 [12]. Both of these fragments are being explored as anti-cancer therapies. Many secreted proteins found in the embryonic microenvironment are present in cell conditioned media (CCM). The embryonic environment is hypoxic, and is characterized by rapid cell growth, scar-less healing and the ability to control tumor growth. Unlike adult tissue, it contains predominantly collagen III and IV and low levels of TGFβ [13]. Although traditional mammalian cell culture has involved normoxic conditions of 21% oxygen and 5% CO2, Ezashi et al. have shown that mimicking the embryonic condition of hypoxia enhances the growth of human embryonic stem cells (hESCs). Furthermore, hESCs and adult stem cells show improved maintenance of their undifferentiated state under low oxygen conditions [14-16]. We have studied the effect of hypoxia on neonatal fibroblasts and discovered that hypoxic conditions of 1% - 5% oxygen resulted in the cells differentially expressing over 5000 genes as compared to cells grown in otherwise identical, but normoxic environments. Several of the up-regulated genes expressed in the hypoxic cells are associated with pluriand multipotent stem cells including LnX2, SOX21, Nestin, NFATc1, Krt15, POU5F1 (Oct4), SOX2 and Nanog. In addition to gene upregulation, stem cell markers were secreted, including Nestin, Oct4, Brachyury, and Nodal [17]. These results were independently confirmed and reported by Page et al. [18].
We have simulated the embryonic environment of hypoxia and suspension culture, so that the bioengineer could produce soluble and insoluble embryonic proteins for use in the study of modulating the tumor microenvironment. We have previously demonstrated that human ECM and CCM secreted by cells grown in a hypoxic bioreactor are able to support the rapid growth of human embryonic stem cells and mesenchymal stem cells, while killing a variety of malignant cells in a dose dependant manner [17]. In addition to our initial work with human cancer cell lines, PC3 human prostate cancer inoculated onto the chorioallentoic membrane (CAM) of chicken embryos showed a statistically significant reduction (p < 0.0001) in brain metastasis when treated with the hECM and CCM [19]. Given these results, we have studied the effect of hECM and CCM on the growth of basal cell carcinoma (BCC), squamous cell carcinoma (SCC) and melanoma cell lines using in vitro and in vivo models.
2. Experimental Methods and Procedure
2.1. hECM and CCM Production
Cryogenically preserved human neonatal dermal fibroblasts stored in cell banks were recovered and grown through multiple sub-cultivations to generate inoculums for bioreactors. The fibroblast cells are native and not engineered to posses specialized characteristics. This initial cell expansion step was performed using roller bottles. Roller bottles were prepared, inoculated and grown according to predetermined criteria using commercially available culture medium. At conclusion of each passage, cells were removed from roller bottles by enzymatic disassociation and used to seed subsequent passages. Upon completing cell expansion, harvested cells were prepared to specific conditions and processed into bioreactor inoculums.
Microcarrier type bioreactors were used to produce test material. The 10 L liter working volume reactor was prepared for cultivation through several steps including hydration of dextran microcarrier beads, sterilization, calibration of instruments and preconditioning beads for seeding. A specific bead loading and cell concentration were used to initiate reactor cultivation. The cells were fed predetermined quantities of growth medium at specific intervals following a fed batch method. At each feeding interval, spent cell conditioned medium (CCM) was replaced with fresh medium. When cells reached working density they were adapted to a serum free production medium. CCM was only collected from serum free medium cultivation for processing of test material. CCM was collected during the production step and retained for further processing.
CCM collected from a bioreactor was tested for specific growth factors by ELISA tests and certain physical attributes and concentrated approximately 10 fold using ultrafiltration methods. The filtrate was separated from concentrate using a 10,000 MWCO membrane and was retained for processing CCM. After initial molecular separation, the filtrate was again separated by ultrafiltration using a 3 MWCO membrane, with the filtrate from this step retained. Approximately 90% - 95% of the starting material is recovered from ultrafiltration.
The hECM production was started after final CCM removal. The cells on the beads were removed through lysis with sterile water and the beads were pooled, pelleted, then frozen overnight. The pellet was thawed then treated with 10,000 u/ml dextranase in PBS at 37˚C. The remaining material was pelleted at high speed and served as the raw insoluble hECM. Routine Western Blot analysis was performed on the hECM to characterize the predominant matrix proteins.
2.2. Cell Lines and Culture Media Used
SCC-25 (human squamous cell carcinoma), TE354.T (human basal cell carcinoma), and B16 (mouse melanoma) cell lines were purchased from ATCC (American Type Cell Culture, Baltimore). The human NRAS mutant melanoma line MM485 and primary human melanocytes (FOM) were used as a courtesy of Dr. Susana Ortiz-Urda. Dulbecco’s Modified Eagle Medium (DMEM) and RPMI 1640 + 10% fetal bovine serum + 1% penicillin-streptomycin (Life Technologies, Carlsbad, CA) were used to carry cell lines and as controls where appropriate. Primary human melanocytes were grown in supplemented M254 medium (Gibco, Life Technologies).
2.3. Proliferation Assay
Cells were bound to wells of 24-well plates (Nunc, Denmark) for 60 minutes then treated with dilutions of CCM. Plates were incubated for 3 days at 37˚C in a 5% CO2 incubator (Forma, Thermo, Pittsburg, PA). After incubation the medium was replaced with 1 mL of 1 mg/mL MTT (Sigma, St. Louis) and incubated for one hour. The MTT solution was removed and 0.25 - 0.5 mL of isopropyl alcohol was added to extract the dye. 200 uL of alcohol/dye solution was placed in a 96-well plate and read at 540 nm by a Tecan Sunrise Microplate Reader using Magellan software (Tecan Systems, San Jose, CA) Data were processed using Microsoft Excel and the percentage reduction in cell number was calculated with untreated cells representing 100% viability.
2.4. Xenograft Tumor Model on the Chorioallantoic Membrane (CAM) of the Fertilized Chicken Egg
A burr hole was made at the air sac end of the egg and another hole was drilled through the shell at the equator. Vacuum was applied at the first hole and the air in the sac was displaced to create a blister between the shell and chorioallantoic membrane. Using a cutting wheel a square hole was cut through the shell, allowing access to the CAM. Skin cancer cells and treatments (CCM, hECM, or no treatment) were applied through the square access hole. Final tumor load assessment was performed at 7 days.
2.5. Subcutaneous Xenograft Mouse Tumor Model
Athymic mice were maintained in a barrier facility on high efficiency particulate air (HEPA)-filtered racks. The animals were fed with autoclaved laboratory rodent diet (Teckland LM-485; Western Research Products, Orange, CA). Nude mice were anaesthetized using inhalatory Isoflurane. While unconscious, the mice were injected subcutaneously with cells and treatments (CCM or hECM). Mice were followed over time and tumor mass was tracked. Final tumor removal and growth assessment was performed after 14 days.
2.6. Statistical Analysis
Student’s t-test was performed to generate p-values comparing growth curves of treated and untreated cell lines in vitro as well as tumor size in both the CAM and the orthotopic mouse model.
2.7. Caspase 9 Immunostaining
Cells were cultured on glass chamber slides (Nunc, Denmark) and were incubated for three days in dilutions of CCM. The slides were then fixed for 30 minutes in 4% paraformaldehyde in PBS, washed, then blocked with 4% bovine serum albumin for 30 minutes at room temp. The “blocker” was removed and the primary antibody, rabbit anti-human/mouse caspsase 9 (Santa Cruz Biotech., Santa Cruz), was added to the chambers and incubated overnight at 4˚C. Wells were washed three times with PBS then goat anti-rabbit IgG, 1 μg/ml in blocker, was added and incubated at room temperature for one hour. Wells were washed with PBS then visualized using OV-100 flourescent microscope (Olympus, Japan). In addition to this assay, flow cytometry was performed to measure the positivity of annexin V and propidum iodide, markers of apoptotic and dead cells, respectively.
2.8. Immunoblotting
Cell lines were plated in 6-well plates 24 hours prior to treatment. After 6 hours of incubation with the compound, cells were washed with 1x PBS and lysed using radioimmunoprecipitation (RIPA) buffer [150 mM NaCl, 1% (vol/vol) Nonidet P-40, 0.5% (wt/vol) sodium deoxycholate, 0.1% (wt/vol) SDS] in 50 mM Tris-HCl (pH 8.0) supplemented with protease and phosphatase inhibitors (78442; Pierce). Protein concentrations were determined using the BCA Protein Assay kit (23235; Pierce). Total protein in 1x Laemmli buffer with 10% 2-mercaptoethanol was separated by SDS-PAGE, transferred after 1 hour to a PVDF membrane (IPVH00010; Millipore) by electro-blotting with 20% (vol/vol) methanol and blocked for 1 hour in 5% (wt/vol) dry milk (or BSA)/TBS/0.1% (vol/vol) Tween-20. Membranes were incubated overnight at 4˚C with primary antiserum following incubation with horseradish peroxidase-conjugated secondary antiserum for 1 hour, and developed using enhanced chemiluminescence (32105; Pierce or 64201BP; Millipore). Cyclin D1 (2926), phospho-ERK (4370), phospho-S6 (4857), cleaved caspase 3 (9664), beta actin (3700), BIM (2933), phospho-MEK (9154) and phospho-AKT (4060) antibodies were purchased from Cell Signaling Technologies.
3. Results
Western blot analysis performed on the hECM confirmed the presence of decorin, which has been associated with growth suppression of mammary carcinoma cells [9]. Large amounts of SPARC (osteonectin) was also seen, along with collagens I, III, IV, and V, fibrillin, lumican, fibromodulin and several laminins (Figure 1). The CCM contains follistatin, which has been shown to suppress organ metastasis in small cell lung cancer cells [20]. In addition to other growth factors and morphogens, the CCM contains soluble hyaluronic acid and other soluble matrix proteins.
A dose-dependent reduction of viability in skin cancer cells (SCC-25, TE354.T, B16, MM485) was seen in vitro with the CCM as well as with the hECM, as measured by MTT analysis. Figure 2 shows 70% inhibition of growth of the SCC-25 cells at a concentration of 2 mg/ml. The basal cell carcinoma cell line TE354.T was inhibited by 40% (p < 0.05) at a concentration of 7 mg/ml and the melanoma cell lines B16 and MM485 by over 60% (p = 0.01) at a concentration of 0.5 mg/ml with CCM treatment.
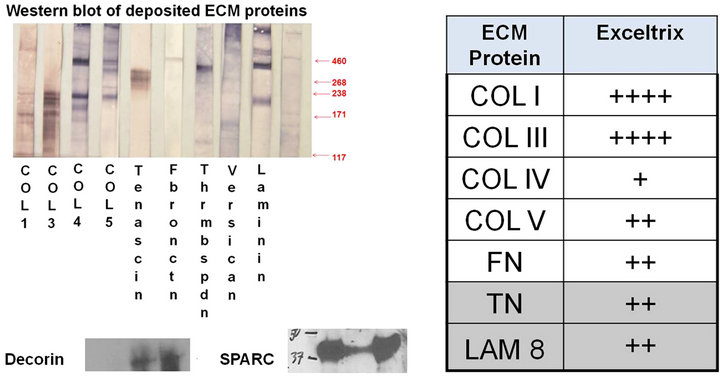
Figure 1. Western blot analysis of hECM showed the presence of a diverse mixture of matrix proteins.

Figure 2. MTT studies showed a dose dependent reduction in SCC cells treated with the CCM.
In the CAM assay, melanoma tumor load was reduced by up to 80% with the addition of lyophilized reconstituted hECM compared to vehicle-treated controls (p < 0.05). The reduction in tumor load was dose dependent (Figure 3). Similar inhibition was seen with the squamous cell carcinoma cell line SCC-25.
In the xenograft model of subcutaneous melanoma, tumor growth was inhibited by 70% - 90% in the treatment group using either CCM or hECM (Figure 4).
The inhibitory effect is pronounced in malignant cells and preferably targets rapidly dividing cancer cells through the upregulation of caspase 9, and cleaved caspase 3 leading to apoptosis (Figure 5). B16 cells cultured with varying concentrations of CCM or hECM, then immunostained with antibody against caspase 9, expressed the apoptotic enzyme in a dose dependant fashion. More than 90% of cells expressed caspase 9 when treated with 4 mg/ml of the CCM while less than 10% of cells expressed the enzyme when treated with 0.5 mg/ml CCM. Immunoblot analysis of MM485 cells incubated with indicated concentrations of CCM show upregulation of pERK, slight induction of BIM and dramatic increase of cleaved caspase-3. Furthermore levels of p-AKT and pS6 are reduced (Figure 6). These results support the hypothesis that the CCM and hECM kill skin cancer cells
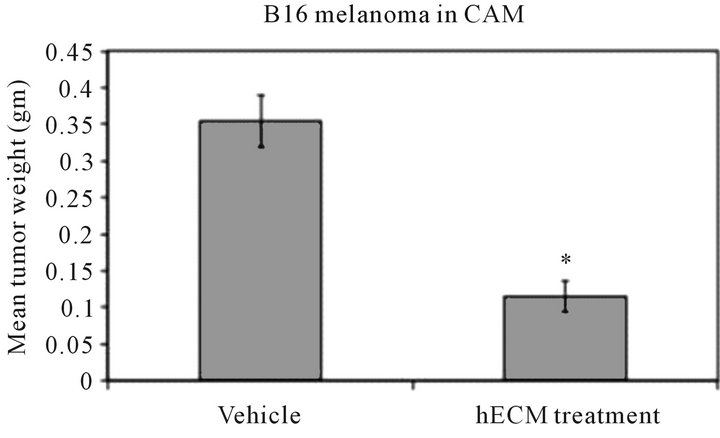
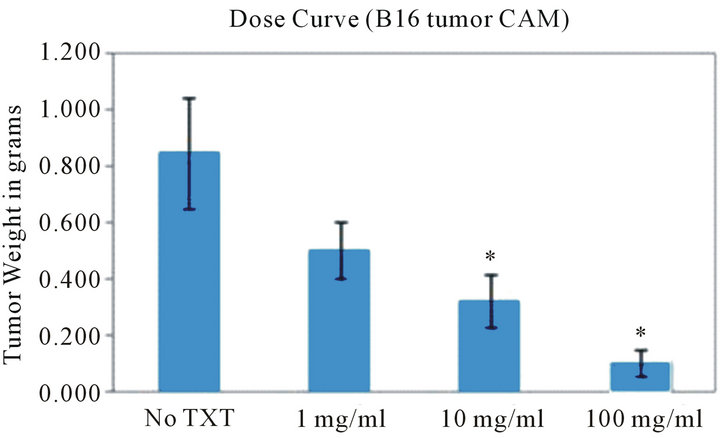
Figure 3. hECM inhibition of B16 melanoma in the CAM model. 2 × 106 B16 cells were treated with 670 µg of hECM at the time of cell seeding. Data represents excised tumor weight p < 0.05 (left graph). B16 melanoma cells treated with CCM showed a dose dependent response in cell death.
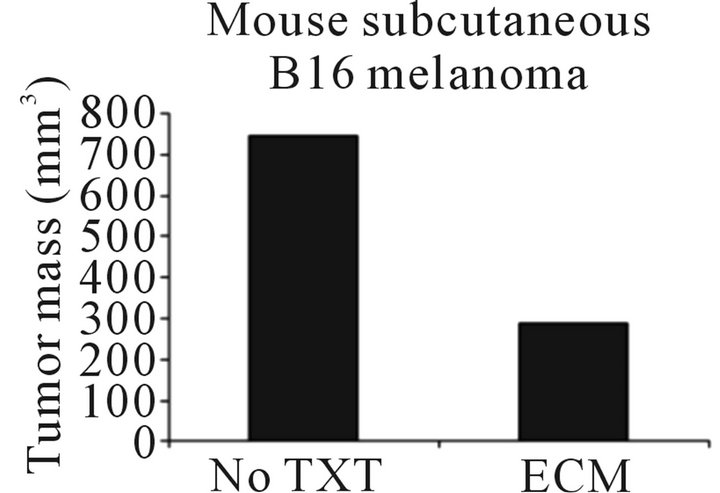
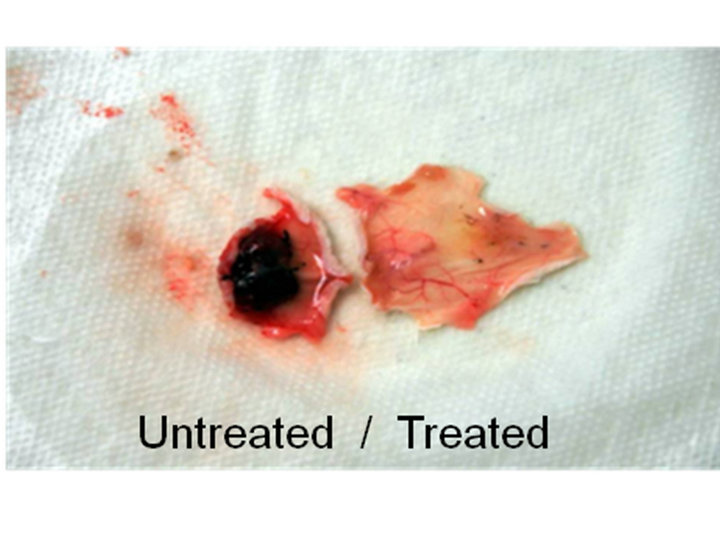
Figure 4. B16 melanoma tumor in NOD/SCID mice with and without hECM co-injection (left photo). hECM resulted in a 2.5 fold decrease in tumor mass.
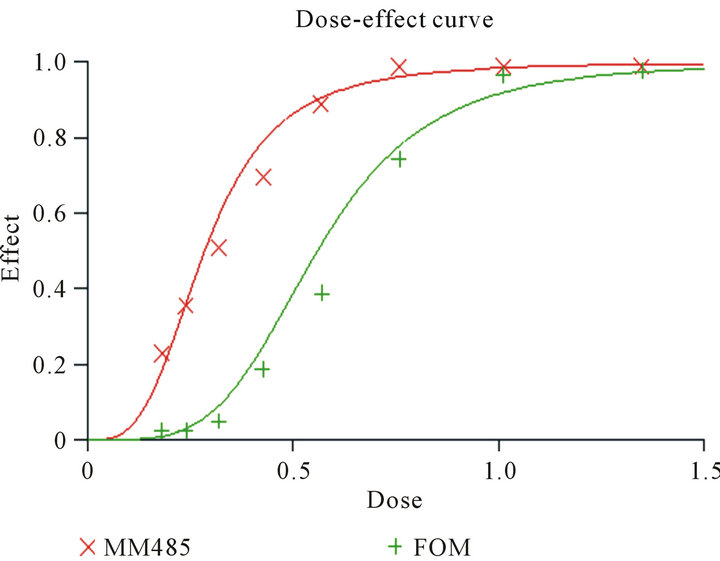
Figure 5. CCM and hECM show specificity in targeting malignant cells. Primary human melanocytes (FOM) and MM485 melanoma cells were incubated for 72 h with different concentrations of the compound. The dose effect curve shows that malignant cells (red line) are affected by the CCM and hECM at lower concentrations compared to primary melanocytes.
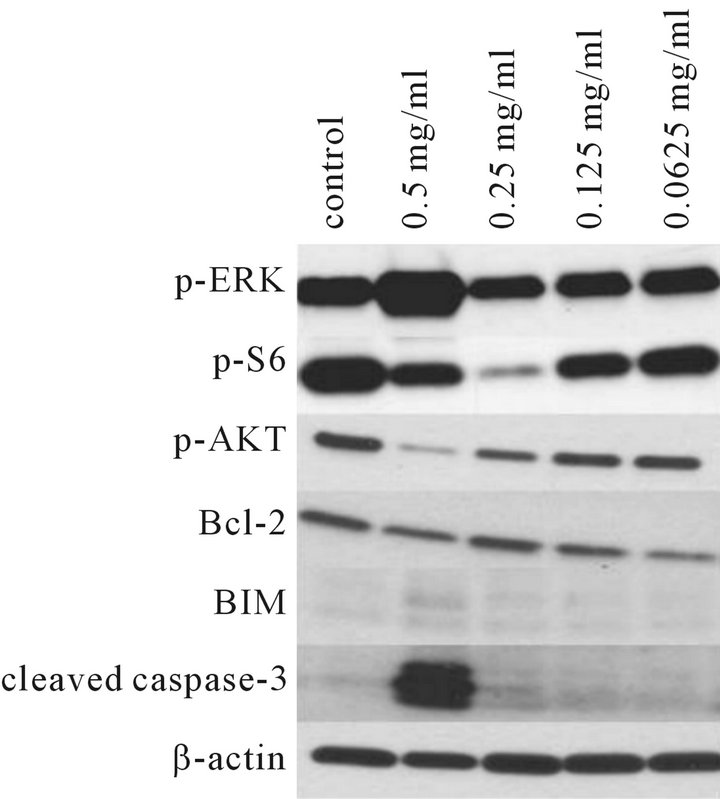
Figure 6. Immunoblot analysis of human MM485 melanoma cells incubated with CCM. Significant upregulation of p-ERK and dramatic induction of cleaved caspase-3 at a concentration of 0.5 mg/ml of CCM was noted. There was a slight increase of BIM expression. Reduction of levels of p-AKT and p-S6 was also seen at 0.5 mg/ml and 0.25 mg/ml of CCM.
through upregulation of caspase enzymes and the induction of apoptosis.
4. Conclusions
These data show that bioengineered soluble and insoluble proteins produced by induced multipotent human stem cells significantly reduce viability of skin cancer cell lines in vitro and in two different in vivo models. This work substantiates research by other investigators demonstrating the ability of matrix proteins from ESCs to selectively target the growth of cancer cells in vitro. We have used three different methods to elucidate the mechanism of action of these proteins. Initial indications in our research point to the activation of the apoptotic enzymes leading to programmed cancer cell death.
There are a number of applications where CCM and hECM materials could be used to augment cancer therapies. We hypothesize that the insoluble ECM could be utilized as a post resectional adjuvant in cutaneous or metatstatic skin cancers including melanoma, BCC or SCC, where improved wound healing and the prevention of growth of remnant cancer cells might be achieved.
5. Acknowledgements
The authors wish to thank Kayler Brintle and Eileen Brandt for their technical assistance.
REFERENCES
- C. T. Mierke, “The Biomechanical Properties of 3D Extracellular Matrices and Embedded Cells Regulate the Invasiveness of Cancer Cells,” Cell Biochemistry and Biophysics, 2011.
- P. M. Kulesa, J. C. Kasemeier-Kulesa, J. M. Teddy, N. V. Margaryan, E. A. Seftor, R. E. B. Seftor and M. J. C. Hendrix, “Reprogramming Metastatic Melanoma Cells to Assume a Neural Crest Cell-Like Phenotype in an Embryonic Microenvironment,” PNAS, Vol. 103, No. 10, 2006, pp. 3752-3753. doi:10.1073/pnas.0506977103
- M. J. C. Hendrix, E. A. Seftor, A. R. Hess and R. E. B. Seftor, “Molecular Plasticity of Human Melanoma Cells,” 2003, pp. 3070-3075.
- T. Mizuno, H. Fukamachi and Y. S. Kim, “Gland Formation of Colon Cancer Cells Combined with Foetal Rat Mesenchymal in Organ Culture: An Ultrastructural Study,” Journal of Cell Science, Vol. 87, No. P5, 1987, pp. 615- 621.
- N. Hayashi and G. Cunha, “Mesenchymal-Induced Changes in the Neoplastic Characteristics of the Dunning Prostatic Adenocarcinoma,” Cancer Research, Vol. 51, No. 18, 1991, pp. 4924-4930.
- M. Cooper and H. Pinkus, “Intrauterine Transplantation of Rat Basal Cell Carcinoma as Model for Reconversion of Malignant to Benign Growth,” Cancer Research, Vol. 37, No. 8, 1977, pp. 2544-2552.
- L.-M. Posavit, E. Seftor, R. Seftor and M. Hendrix, “Influence of Microenvironment on Melanoma Cell Fate Determination and Cell Fate,” Cancer Research, Vol. 66, No. 16, 2006, pp. 7833-7836. doi:10.1158/0008-5472.CAN-06-0731
- S. Goldoni, D. G. Seidler, J. Heath, M. Fasson, R. Baffa, M. L. Thakur, R. T. Owens, D. J. McQuilan and R. V. Iozza, “An Antimetastatic Role for Decorin in Breast Cancer,” American Journal of Pathology, Vol. 173, No. 3, 2008, pp. 844-855. doi:10.2353/ajpath.2008.080275
- M. Santra, I. Eichstetter and R. V. Iozza, “An Anti-Oncogenic Role for Decorin: Downregulation of ErbB2 Leads to Growth Suppression and Cytodifferentiation of Mammary Carcinoma Cells, JBC,” 2000.
- J. Muschler, D. Levy and R. Boudreau, “The Role for Dystroglycan in Epithelial Polarization: Loss of Unction in Breast Tumor Cells,” Cancer Research, Vol. 62, No. 23, 2002, pp. 7102-7109
- M. Henry, M. Cohen and K. Campbell, “Reduced Expression of Dystroglycan in Breast and Prostate Cancer,” Human Pathology, Vol. 32, No. 8, 2001, pp. 791-795. doi:10.1053/hupa.2001.26468
- A. Sudhakar, H. Sugimoto, C. Yang, J. Lively, M. Zeisberg and R. Kalluri, “Human Tumstatin and Human Endostatin Exhibit Distinct Antiangiogenic Activities Mediated by Alpha v Beta 3 and Alpha 5 Beta 1 Integrins,” Proceedings of the National Academy of Sciences of the USA, Vol. 100, No. 8, 2003, pp. 4766-4771. doi:10.1073/pnas.0730882100
- N. S. Adzick and H. P. Lorenz, “Cells, Matrix, Growth Factors, and the Surgeon. The Biology of Scarless Fetal Wound Repair,” Annals of Surgery, Vol. 220, No. 1, 1994, pp. 10-18. doi:10.1097/00000658-199407000-00003
- M. V. Chakravarthy, E. E. Spangenburg and F. W. Booth, “Culture in Low Levels of Oxygen Enhances in Vitro Proliferation Potential of Satellite Cells from Old Skeletal Muscles,” Cellular and Molecular Life Sciences CMLS, Vol. 58, No. 8, 2001, pp. 1150-1158. doi:10.1007/PL00000929
- K. L. Covello, J. Kehler, H. Yu, J. D. Gordan, A. M. Arsham, C. J. Hu, P. A. Labosky, M. C. Simon and B. Keith, “HIF-2 Alpha Regulates Oct-4: Effects of Hypoxia on Stem Cell Function, Embryonic Development, and Tumor Growth,” Genes Development, Vol. 20, No. 5, 2006, pp. 557-570. doi:10.1101/gad.1399906
- W. L. Grayson, F. Zhao, R. Izadpanah, B. Bunnell and T. Ma, “Effects of Hypoxia on Human Mesenchymal Stem Cell Expansion and Plasticity in 3D Constructs,” Journal of Cellular Physiology, Vol. 207, No. 2, 2006, pp. 331- 339. doi:10.1002/jcp.20571
- E. Pinney, M. Zimber, A. Schenone, M. Montes-Camacho, F. Zeigler and G. K. Naughton, “Human EmbryonicLike E CM (hECM) Stimulates Proliferation and Differentiation in Stem Cells While Killing Cancer Cells,” International Journal of Stem Cells, Vol. 4, No. 1, 2011, pp. 70-75.
- R. L. Page, S. Ambady, W. F. Holmes, L. Vilner, D. Kole, O. Kashpur, V. Huntress, I. Vojtic, H. Whitton and T. Dominko, “Induction of Stem Cell Gene Expression in Adult Human Fibroblasts without Transgenes,” Cloning and Stem Cells, Vol. 11, No. 3, 2009, pp. 417-426. doi:10.1089/clo.2009.0015
- R. S. Menen, E. Pinney, et al., “Inhibition of Metastasis of Circulating Human Prostate Cancer Cells in the Chick Embryo by an Extracellular Matrix Produced by Foreskin Fibroblasts in Culture,” Anticancer Research, Vol. 32, No. 5, 2012, pp. 1573-1578.
- H. Ogino, S. Yano, S. Kakiuchi, H. Mugguruma, K. Ikuta, M. Hanibuchi, H. Uehara, K. Tsuchida, H. Sugino and S. Sone, “Follistatin Suppresses the Production of Experimental Multiple-Organ Metastasis by Small Cell Lung Cancer Cells in Natural Killer Cell-Depleted SCID Mice,” Clinical Cancer Research, Vol. 14, No. 3, 2008, pp. 660- 667. doi:10.1158/1078-0432.CCR-07-1221