Health
Vol.5 No.1(2013), Article ID:26509,7 pages DOI:10.4236/health.2013.51001
Aged rat heart: Modulation of age-related respiratory defects decreases ischemic-reflow injury
![]()
1Departments of Medicine, Biochemistry and Physiology, McGuire Veterans Affairs Medical Center-Virginia Commonwealth University, Richmond, USA
2Center for Mitochondrial Diseases, Departments of Medicine and Pharmacology, Case Western Reserve University, Cleveland, USA; *Corresponding Author: charles.hoppel@case.edu
Received 1 August 2012; revised 2 September 2012; accepted 9 November 2012
Keywords: Mitochondria; Cytochrome Oxidase; Complex III; Myocardial Infarction; Aging
ABSTRACT
Myocardial injury increases in the elderly heart during ischemia and reperfusion. Mitochondria, the key targets and sources of injury during ischemia and reperfusion, sustain ischemic damage to the electron transport chain that is superimposed upon age-related defects. In the adult heart, interventions to activate endogenous cytoprotective signaling systems meet in mitochondria to decrease cardiac injury. Unfortunately, these systems are largely ineffective in the aged heart. Thus, new treatment concepts are needed to reduce injury in the aged heart. Our group chose a strategy to directly treat the effector of cardiac injury in the aged heart, the mitochondria. We further utilized a novel approach to ask if the reversal of aging defects in cardiac mitochondria before ischemia could decrease ischemia-reperfusion injury in the heart. Three hours following treatment with the small molecule, nutriceutical acetylcarnitine (AcCN), oxidative phosphorylation as well as age-induced defects in electron transport chain complexes III and IV was corrected in the heart. When such hearts were then exposed to ischemia and reperfusion, cardiac injury was markedly reduced. Contraction during reperfusion improved and recovery became similar to that in adult hearts. Cardiac cell death was substantially reduced. Thus, age-related defects in electron transport are a key mechanism of the increased myocardial injury in the elderly heart during ischemia and reperfusion. Modulation of aging-induced defects in mitochondrial metabolism reduces cardiac injury from ischemia and reperfusion, and is a novel strategy to protect myocardium in the elderly patient at risk for an acute myocardial infarction.
1. THE CLINICAL CHALLENGE OF MYOCARDIAL INFARCTION IN THE ELDERLY: INCREASED CARDIAC INJURY CONCOMITANT WITH DECREASED RESPONSIVENESS TO INNATE CYTOPROTECTION
During the course of an acute myocardial infarction, elderly patients experience an increased mortality and sustain greater cardiac damage compared to younger patients despite timely and successful reperfusion [1]. The aged heart suffers greater damage during ischemia and reperfusion in both experimental and clinical settings [1-6]. In a National Institutes on Aging rat model of aging, the Fischer 344 rat (F344), isolated, buffer-perfused hearts from aged rats (24 mo.) exhibit decreased hemodynamic recovery and greater cardiomyocyte death following ischemia and reperfusion compared to hearts from adult (6 mo.) controls [4-7]. Age-enhanced cardiac injury during ischemia and reperfusion has been observed in other rat strains [3] and species [2,8] as well.
Mitochondria are key targets of ischemic damage as well as effectors of cardiac injury during ischemia and reperfusion [2,3,9,10]. Mitochondria-driven effector mechanisms, especially relevant to the aged heart, include oxidative damage [6], calcium accumulation [2], and activation of apoptosis [5]. In the aged heart, ischemic damage to mitochondria is superimposed upon pre-existing age-related alterations in mitochondrial metabolism [10,11]. We posited that intervention to improve aging-induced defects in mitochondrial function would decrease myocardial injury during subsequent ischemia and reperfusion.
Mitochondria-directed strategies to limit cardiac injury during ischemia and reperfusion are highly relevant to the protection of the aged heart and the ultimate improvement of clinical outcomes in high-risk elderly patients. Novel therapies are required because classical approaches widely used in experimental studies of ischemia-reperfusion injury in the adult heart increasingly appear to be of limited efficacy when applied to the aged heart. Interventions that utilize activation of cytoprotective signaling cascades that converge on mitochondria include ischemic preconditioning, where preemptive periods of brief ischemia limit injury from a subsequent longer period of ischemia [7,12-15]; and postconditioning where brief ischemia is applied during early reperfusion [16]. Cytoprotective cascades converge to modulate mitochondrial function via mitochondrial KATP channel via PKC epsilon [17] or to modulate susceptibility to permeability transition via the insulin-signaling related Akt-GSK3beta pathway [18]. These pathways that are highly effective in the adult heart are clearly less effective in the aged heart, as shown by multiple investigators for ischemic preconditioning [19-21] and recently for postconditioning [19,22]. Because ischemic preconditioning and postconditioning share overlapping mechanisms of protection [23] and overlapping extents of cardioprotection [24], it is very likely that observations of the ineffectiveness of ischemic postconditioning in the aged heart will continue to be reinforced as further studies of postconditioning are performed.
It remains unclear if the major mechanism of the ineffectiveness of signaling-based cytoprotection in the aged heart is at the level of the signaling cascades upstream of the mitochondria or the inability of mitochondria in the aged heart to respond to the cytoprotective modulation. Two lines of evidence suggest that mitochondria in the aged heart retain the capacity to respond to cytoprotective modulation. First, the endogenous protective mechanisms can be restored in aging hearts by treatments including caloric restriction [25], exercise [26], and a pharmacologic strategy to inhibit protein phosphatase 2A activity [27]. Second, the attenuation of preconditioning protection observed in the F344 rat experimental model occurs mainly by reduced activation of signaling systems [13] with mitochondria able to respond to direct pharmacologic agonists of the mitochondrial potassium-ATP channel [28]. Thus, the potential for direct mitochondriatargeted intervention remains.
The aged heart exhibits a major clinical challenge for protective therapy. First, not only myocardial injury increases compared to younger hearts for a similar ischemic stress, but the ability to muster endogenous protective pathways is impaired. This challenge in the aged heart makes a strong case for the consideration of novel, direct mitochondrial-centered treatments to mitigate cardiac damage during ischemia and reperfusion. An innovative approach to decrease cardiac ischemia-reperfusion injury by improvement of aging-induced defects in electron transport is the subject of this minireview.
2. REVERSAL OF AGE-RELATED DEFECTS IN MITOCHONDRIAL ELECTRON TRANSPORT TO DECREASE ISCHEMIA-REPERFUSION INJURY IN THE AGED HEART
Age-Induced Defects in Mitochondrial Electron Transport
Cardiac mitochondria exist as two distinct populations. Subsarcolemmal mitochondria (SSM) are located beneath the plasma membrane whereas interfibrillar mitochondria (IFM) are located among the myofibrils [29-31]. Aging impairs oxidative metabolism only in IFM, whereas SSM are unaffected [29]. Age-related defects in electron transport involve complex III [10,11,32] and complex IV [29,33]. Aging also decreases the content of IFM in the 24 and 28 months old F344 rat [29]. Using rat cardiac permeabilized fibers, we observed that Fischer 344 had an approximately 45% decrease in coupled mitochondrial oxidation, similar to that seen using isolated mitochondria [34]. This quasi in situ preparation reinforces the findings of decreased content of IFM with decreased oxidative capacity per mitochondrion observed with isolated mitochondria. In effect, these data are complimentary.
In the Brown Norway rat, which lives longer than the F344, we saw a small (15%) but not significant loss in oxidation in fibers [34]. In contrast, in the hybrid Fischer 344-Brown-Norway, which lives the longest of these strains, there were no oxidative changes in the fibers [34]. Lujbicic and colleagues assessed respiration in an SSMlike population isolated from hearts of the Brown Norway-F344 hybrid, and with complex I substrates noted to decrease approximately 20%, but IFM were not examined [35]. This finding is consistent with the trend toward a minimal decrease in respiration in SSM in the Fischer 344 rat of 18% in SSM in the Fischer 344 rat [29] that reaches statistical significance when a large number of animal are studied [36]. Despite this modest decrease in respiration with NADH-linked substrates, oxidative phosphorylation using substrates that donate electrons to complex III or complex IV, as well as the activities of complexes III and IV, were unaltered with age in SSM [11,29,32].
Complex III activity was decreased in IFM from aged hearts Figure 1 [10,11,32]. Complex III catalyzes electron transfer from ubiquinol to cytochrome c that is coupled to proton translocation [37-39]. Electron transfer within complex III occurs in a bifurcated fashion to cytochromes b and c1 via movement of the iron-sulfur protein during the redox cycle [38-40]. When quinol binds to the oxidation site of cytochrome b (the Qo site), a con-
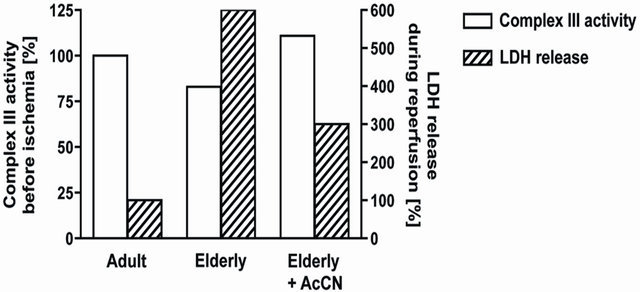
Figure 1. Aging leads to a decrease in the activity of electron transport chain complex III in interfibrillar mitochondria (Open bars; left axis). Treatment three hours before ischemia with acetylcarnitine restored age-related decreases in complex III activity [55]. When elderly hearts underwent ischemia and reperfusion in the presence of restored complex III activity, cardiac injury was decreased following ischemia and reperfusion (Hatched bars; right axis) [55].
certed oxidation of quinol by iron-sulfur protein and cytochrome b occurs [37,40]. Functional studies using partial reactions within complex III localized the site of the age-related defect [11] to the Qo site of cytochrome b in IFM at 24 and 28 months of age in the Fischer 344 rat [11].
The aging phenotype of IFM is a combination of decreased maximal enzyme activity [32], preserved electron paramagnetic resonance signal of the iron-sulfur protein [32], increased myxothiazol-resistant cytochrome b reduction, and preserved inhibition by stigmatellin [11]. Comparison to the functional cytochrome b phenotypes that result from site-directed mutagenesis in the bacterium Rhodobacter sphaeroides [37] suggests that modification of a tyrosine residue may be the mechanism of the complex III phenotype observed in IFM from aged F344 rat hearts [41]. As recently shown by the study of the bacterial cytochrome bc1 complex, this tyrosine residue controls interactions between molecular oxygen and the reduced quinone and attenuates the production of reactive oxygen species by complex III in the baseline state [42].
Complex III is the primary site for the net release of reactive oxygen species from mitochondria [43-46]. The age-related alteration of complex III at the Qo site of cytochrome b in complex III in IFM [11] should augment the net production and release of reactive oxygen species from IFM of elderly hearts, not only in the baseline state, but also during ischemic stress. The aging defect at the Qo site of complex III in IFM increases the production of reactive oxygen species [11]. In contrast, the production of H2O2 was not increased by age in SSM. These findings were supported at the level of intact mitochondria with increased production of reactive oxygen species observed from IFM, but not from SSM [47]. Mitochondria in the aged heart are a source of the increased oxidant production in the baseline resting state and are poised to amplify cell injury during the stress of ischemia and reperfusion. Ischemia decreased the maximal rate of oxidative phosphorylation due to damage to complex I [48], complex III [10], and complex IV [9,30,49]. In contrast to the selectivity of the aging defect to IFM, ischemia-induced damage occurred in both SSM and IFM [10]. The decrease in the rate of respiration due to ischemia was similar in adult and aged hearts [10]. In IFM from the aged heart, ischemia-induced decreases in respiration were additive to the pre-existing aging defects [10].
3. ACETYLCARNITINE THERAPY: REDUCTION OF ISCHEMIA-REPERFUSION INJURY BY THE RESTORATION OF AGE-INDUCED ELECTRON TRANSPORT DEFECTS
If the aging defect in mitochondrial respiration could be diminished or removed, would the aged heart now sustain less injury during ischemia and reperfusion? Acetylcarnitine treatment improves aging-induced decreases in complex IV activity [33,50-54]. Our group used acetylcarnitine treatment to address the question if age-induced decreases in mitochondrial respiration could be improved. Acetylcarnitine treatment improved rates of oxidative phosphorylation and uncoupled respiration in mitochondria from aged hearts to those observed in adult hearts accompanied by a restoration of the activity of complex III in IFM Figure 1 [55]. The content of cytochrome b in IFM from aged hearts was increased [55]. These data suggested that the newly synthesized cytochrome b compensated for the age-damaged cytochrome.
From a mitochondrial perspective, the age-related defect in complex III thus was effaced, accompanied by normalization of oxidative phosphorylation and integrated mitochondrial function. To determine if the mitochondrial improvement is relevant to the heart in toto, we looked at myocardial injury in treated hearts during ischemia and reperfusion. Adult and aged rats were treated with acetylcarnitine followed by removal of the heart three hours later for the study of ischemia and reperfusion in vitro without any further treatment [55]. In the aged heart, acetylcarnitine enhanced the recovery of contractile function following ischemia and reperfusion, whereas recovery in adult hearts was unchanged by treatment [55]. In hearts from acetylcarnitine-treated aged rats, recovery was now similar to the recovery occurring in treated or untreated adults. Acetylcarnitine treatment substantially decreased the release of lactate dehydrogenase from the aged heart during reperfusion, indicating less myocyte necrosis Figure 1 [55], without any protection observed in the adult heart. Acetylcarnitine did not alter mechanisms of ischemia-reperfusion damage common to both adult and aged hearts. These data from acetylcarnitine provide convincing and compelling evidence that the age-related defects in mitochondrial oxidative metabolism contribute to enhanced cardiac injury that follows ischemia and reperfusion in the elderly heart. Thus, these studies provide strong support for the concept developed herein.
Acetylcarnitine has been postulated to improve aging defects in electron transport by several possible mechanisms. First, acetylcarnitine was proposed to increase the content of cardiolipin [50-54,56] a phospholipid present in mitochondria that is required for optimal function of the electron transport chain [56]. Although aging leads to a functional membrane defect [29,41], aging does not decrease the content or composition of cardiolipin in SSM or in IFM in the aged F344 rat heart [57], rendering cardiolipin an unlikely target of acetylcarnitine therapy in the F344 model. Second, and of much greater interest, treatment with acetylcarnitine results in increased transcription of mitochondrial DNA in the aged F344 heart with increased mtRNA synthesis [58,59]. The optimal response to acetylcarnitine occurs at 3 hours at a dose of 300 mg/kg, the treatment regimen used in our ischemia-reperfusion study [55]. The increased transcription is in turn linked to a greater content of mitochondrial-encoded electron transport subunits [58]. The increase in cytochrome content observed in F344 heart mitochondria following acetylcarnitine treatment is in line with this observation.
Thus, stimulation of mitochondrial protein synthesis may contribute to acetylcarnitine-mediated improvement in respiration in the aged heart and the concomitant decrease in susceptibility to ischemia-reperfusion injury [60]. Potential mechanisms of increased transcriptional responses may occur via acetylation of mitochondrial proteins with subsequent activation of mitochondrial sirtuins [61,62]. Additionally, acetylcarnitine, via increases in intracellular acetyl-CoA, may impact nuclear transcription via acetylation of nuclear proteins or an increase in mitochondrial biogenesis [60].
Despite successful and sustained reperfusion of the infarct zone, the mortality of acute myocardial infarction in elderly patients remains markedly elevated [1]. Although pharmacologic treatment at the onset of reperfusion can reduce myocardial injury [63,64] the option of utilizing chronic pretreatment of high risk elders in a preemptive fashion before ischemia represents a novel cardio protective strategy for the aged heart. The modulation of aging-induced defects in mitochondrial metabolism in order to reduce cardiac injury from ischemia and reperfusion is an attractive potential strategy to protect myocardium in the high-risk elderly patient suffering an acute myocardial infarction. We are suggesting that elderly individuals take this compound on a regular basis in order to lessen the effect of a heart attack, in the event that such a happenstance occurs. Furthermore, a problem with other cardio protective strategies is that efficacy is observed in young adult models of experimental ischemia [22], but not in the presence of aging [16,19-21] or other comorbid diseases [65] that impact the elderly who suffer acute coronary syndromes. In contrast, acetylcarnitine therapy exerts a therapeutic specificity for the elderly heart [55]. The approach to rejuvenate mitochondrial function in order to directly address age-enhanced ischemic injury [55] remains an attractive and potentially clinically useful strategy [66].
4. ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (2PO1AG15885) and by the Office of Research and Development, Medical Research Service, Department of Veterans Affairs. The editorial assistance of Dr. Bernard Tandler is greatly appreciated.
![]()
![]()
REFERENCES
- Lesnefsky, E.J., Lundergan, C.F., Hodgson, J.M., et al. (1996) Increased left ventricular dysfunction in elderly patients despite successful thrombolysis: The GUSTO-I angiographic experience. Journal of the American College of Cardiology, 28, 331-337. doi:10.1016/0735-1097(96)00148-9
- Ataka, K., Chen, D., Levitsky, S., et al. (1992) Effect of aging on intracellular Ca2+, pHi, and contractility during ischemia and reperfusion. Circulation, 86, 371-376.
- Frolkis, V.V., Frolkis, R.A., Mkhitarian, L.S., et al. (1991) Age-dependent effects of ischemia and reperfusion on cardiac function and Ca2+ transport in myocardium. Gerontology, 37, 233-239. doi:10.1159/000213266
- Lesnefsky, E.J., Gallo, D.S., Ye, J., et al. (1994) Aging increases ischemia-reperfusion injury in the isolated, buffer-perfused heart. Journal of Laboratory and Clinical Medicine, 124, 843-851.
- Liu, L., Azhar, G., Gao, W., et al. (1998) Bcl-2 and Bax expression in adult rat hearts after coronary occlusion: Age-associated differences. American Journal of Physiology, 275, R315-R322.
- Lucas, D.T. and Szweda, L.I. (1998) Cardiac reperfusion injury: Aging, lipid peroxidation, and mitochondrial dysfunction. Proceedings of the National Academy of Sciences, 95, 510-514. doi:10.1073/pnas.95.2.510
- Tani, M., Suganuma, Y., Hasegawa, H., et al. (1997) Decrease in ischemic tolerance with aging in isolated perfused Fischer 344 rat hearts: Relation to increases in intracellular Na+ after ischemia. Journal of Molecular and Cellular Cardiology, 29, 3081-3089. doi:10.1006/jmcc.1997.0533
- Azhar, G., Gao, W., Liu, L., et al. (1999) Ischemiareperfusion in the adult mouse heart influence of age. Experimental Gerontology, 34, 699-714. doi:10.1016/S0531-5565(99)00031-5
- Lesnefsky, E.J., Moghaddas, S., Tandler, B., et al. (2001) Mitochondrial dysfunction in cardiac disease: Ischemiareperfusion, aging, and heart failure. Journal of Molecular and Cellular Cardiology, 33, 1065-1089. doi:10.1006/jmcc.2001.1378
- Lesnefsky, E.J., Gudz, T.I., Migita, C.T., et al. (2001) Ischemic injury to mitochondrial electron transport in the aging heart: Damage to the iron-sulfur protein subunit of electron transport complex III. Archives of Biochemistry and Biophysics, 385, 117-128. doi:10.1006/abbi.2000.2066
- Moghaddas, S., Hoppel, C.L. and Lesnefsky, E.J. (2003) Aging defect at the Qo site of complex III augments oxyradical production in rat heart interfibrillar mitochondria. Archives of Biochemistry and Biophysics, 414, 59- 66. doi:10.1016/S0003-9861(03)00166-8
- Abete, P., Testa, G., Ferrara, N., et al. (2002) Cardioprotective effect of ischemic preconditioning is preserved in food-restricted senescent rats. American Journal of Physiology—Heart and Circulatory Physiology, 282, H1978- H1987.
- Tani, M., Honma, Y., Hasegawa, H., et al. (2001) Direct activation of mitochondrial KATP channels mimics preconditioning but protein kinase C activation is less effective in middle-aged rat hearts. Cardiovascular Research, 49, 56-68. doi:10.1016/S0008-6363(00)00240-6
- Ishihara, M., Sato, H., Tateishi, H., et al. (2000) Beneficial effect of prodromal angina pectoris is lost in elderly patients with acute myocardial infarction. American Heart Journal, 139, 881-888. doi:10.1016/S0002-8703(00)90021-8
- Lee, T.M., Su, S.F., Chou, T.F., et al. (2002) Loss of preconditioning by attenuated activation of myocardial ATPsensitive potassium channels in elderly patients undergoing coronary angioplasty. Circulation, 105, 334-340. doi:10.1161/hc0302.102572
- Przyklenk, K., Maynard, M., Darling, C.E., et al. (2008) Aging mouse hearts are refractory to infarct size reduction with post-conditioning. Journal of the American College of Cardiology, 51, 1393-1398. doi:10.1016/j.jacc.2007.11.070
- Quinlan, C.L., Costa, A.D., Costa, C.L., et al. (2008) Conditioning the heart induces formation of signalosomes that interact with mitochondria to open mitoKATP channels. American Journal of Physiology—Heart and Circulatory Physiology, 295, H953-H961. doi:10.1152/ajpheart.00520.2008
- Juhaszova, M., Zorov, D.B., Yaniv, Y., et al. (2009) Role of glycogen synthase kinase-3beta in cardioprotection. Circulation Research, 104, 1240-1252. doi:10.1161/CIRCRESAHA.109.197996
- Vessey, D.A., Kelley, M., Li, L., et al. (2009) Sphingosine protects aging hearts from ischemia/reperfusion injury: Superiority to sphingosine 1-phosphate and ischemic preand post-conditioning. Oxidative Medicine and Cellular Longevity, 2, 146-151. doi:10.4161/oxim.2.3.8622
- Boengler, K., Konietzka, I., Buechert, A., et al. (2007) Loss of ischemic preconditioning’s cardioprotection in aged mouse hearts is associated with reduced gap junctional and mitochondrial levels of connexin 43. American Journal of Physiology—Heart and Circulatory Physiology, 292, H1764-H1769. doi:10.1152/ajpheart.01071.2006
- Schulman, D., Latchman, D.S. and Yellon, D.M. (2001) Effect of aging on the ability of preconditioning to protect rat hearts from ischemia-reperfusion injury. American Journal of Physiology—Heart and Circulatory Physiology, 281, H1630-H1636.
- Downey, J.M. and Cohen, M.V. (2009) Why do we still not have cardioprotective drugs? Circulation Journal, 73, 1171-1177. doi:10.1253/circj.CJ-09-0338
- Hausenloy, D.J., Ong, S.B. and Yellon, D.M. (2009) The mitochondrial permeability transition pore as a target for preconditioning and postconditioning. Basic Research in Cardiology, 104, 189-202. doi:10.1007/s00395-009-0010-x
- Vinten-Johansen, J., Zhao, Z.Q., Jiang, R., et al. (2007) Preconditioning and postconditioning: Innate cardioprotection from ischemia-reperfusion injury. Journal of Applied Physiology, 103, 1441-1448. doi:10.1152/japplphysiol.00642.2007
- Long, P., Nguyen, Q., Thurow, C., et al. (2002) Caloric restriction restores the cardioprotective effect of preconditioning in the rat heart. Mechanisms of Ageing and Development, 123, 1411-1413. doi:10.1016/S0047-6374(02)00068-4
- Abete, P., Testa, G., Galizia, G., et al. (2005) Tandem action of exercise training and food restriction completely preserves ischemic preconditioning in the aging heart. Experimental Gerontology, 40, 43-50. doi:10.1016/j.exger.2004.10.005
- Fenton, R.A., Dickson, E.W. and Dobson Jr., J.G. (2005) Inhibition of phosphatase activity enhances preconditioning and limits cell death in the ischemic/reperfused aged rat heart. Life Sciences, 77, 3375-3388. doi:10.1016/j.lfs.2005.05.047
- Jahangir, A., Ozcan, C., Holmuhamedov, E.L., et al. (2001) Increased calcium vulnerability of senescent cardiac mitochondria: Protective role for a mitochondrial potassium channel opener. Mechanisms of Ageing and Development, 122, 1073-1086. doi:10.1016/S0047-6374(01)00242-1
- Fannin, S.W., Lesnefsky, E.J., Slabe, T.J., et al. (1999) Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Archives of Biochemistry and Biophysics, 372, 399-407. doi:10.1006/abbi.1999.1508
- Lesnefsky, E.J., Tandler, B., Ye, J., et al. (1997) Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. American Journal of Physiology, 273, H1544-H1554.
- Palmer, J.W., Tandler, B. and Hoppel, C.L. (1977) Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. Journal of Biological Chemistry, 252, 8731-8739.
- Lesnefsky, E.J., Gudz, T.I., Moghaddas, S., et al. (2001) Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. Journal of Molecular and Cellular Cardiology, 33, 37-47. doi:10.1006/jmcc.2000.1273
- Paradies, G., Ruggiero, F.M., Dinoi, P., et al. (1993) Age-dependent decrease in the cytochrome c oxidase activity and changes in phopsholiids in rat-heart mitochondria. Archives of Gerontology and Geriatrics, 16, 262- 272. doi:10.1016/0167-4943(93)90037-I
- Lemieux, H., Vazquez, E.J., Fujioka, H., et al. (2010) Decrease in mitochondrial function in rat cardiac permeabilized fibers correlates with the aging phenotype. Journals of Gerontology Series A: Biological Sciences and Medical Sciences, 65, 1157-1164. doi:10.1093/gerona/glq141
- Ljubicic, V., Menzies, K.J. and Hood, D.A. (2010) Mitochondrial dysfunction is associated with a pro-apoptotic cellular environment in senescent cardiac muscle. Mechanisms of Ageing and Development, 131, 79-88. doi:10.1016/j.mad.2009.12.004
- Kypriotakis, G., Vazquez, E., Lesnefsky, E.J., et al. (2007) Oxidative phosphorylation provides a valuable tool for identifying the aging mitochondrial phenotype. 12th Congress of the International Association of Biomedical Gerontology Molecular Mechanisms and Models of Aging, Spetses Island, 20-24 May 2007, Abstratct, p. 31.
- Crofts, A.R., Barquera, B., Gennis, R.B., et al. (1999) Mechanism of ubiquinol oxidation by the bc(1) complex: Different domains of the quinol binding pocket and their role in the mechanism and binding of inhibitors. Biochemistry, 38, 15807-15826. doi:10.1021/bi990962m
- Crofts, A.R., Guergova, K.M., Huang, L., et al. (1999) Mechanism of ubiquinol oxidation by the bc(1) complex: Role of the iron sulfur protein and its mobility. Biochemistry, 38, 15791-15806. doi:10.1021/bi990961u
- Trumpower, B.L. (1990) The protonmotive Q cycle. Energy transduction by coupling of proton translocation to electron transfer by the cytochrome bc1 complex. Journal of Biological Chemistry, 265, 11409-11412.
- Trumpower, B.L. (2002) A concerted, alternating sites mechanism of ubiquinol oxidation by the dimeric cytochrome bc(1) complex. Biochimica et Biophysica Acta, 1555, 166-173. doi:10.1016/S0005-2728(02)00273-6
- Lesnefsky, E.J. and Hoppel, C.L. (2003) Ischemiareperfusion injury in the aged heart: Role of mitochondria. Archives of Biochemistry and Biophysics, 420, 287-297. doi:10.1016/j.abb.2003.09.046
- Lee, D.W., Selamoglu, N., Lanciano, P., et al. (2011) Loss of a conserved tyrosine residue of cytochrome b induces reactive oxygen speices production by cytochrome bc1. Journal of Biological Chemistry, 286, 18139-18148. doi:10.1074/jbc.M110.214460
- Chen, Q., Vazquez, E.J., Moghaddas, S., et al. (2003) Production of reactive oxygen species by mitochondria: Central role of complex III. Journal of Biological Chemistry, 278, 36027-36031. doi:10.1074/jbc.M304854200
- Gille, L. and Nohl, H. (2001) The ubiquinol/bc1 redox couple regulates mitochondrial oxygen radical formation. Archives of Biochemistry and Biophysics, 388, 34-38. doi:10.1006/abbi.2000.2257
- Han, D., Antunes, F., Canali, R., et al. (2003) Voltagedependent anion channels control the release of the superoxide anion from mitochondria to cytosol. Journal of Biological Chemistry, 278, 5557-5563. doi:10.1074/jbc.M210269200
- St-Pierre, J., Buckingham, J.A., Roebuck, S.J., et al. (2002) Topology of superoxide production from different sites in the mitochondrial electron transport chain. Journal of Biological Chemistry, 277, 44784-44790. doi:10.1074/jbc.M207217200
- Suh, J.H., Heath, S.H. and Hagen, T.M. (2003) Two subpopulations of mitochondria in the aging rat heart display heterogenous levels of oxidative stress. Free Radical Biology & Medicine, 35, 1064-1072. doi:10.1016/S0891-5849(03)00468-4
- Chen, Q., Moghaddas, S., Hoppel, C.L., et al. (2008) Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. American Journal of Physiology— Cell Physiology, 294, C460-466. doi:10.1152/ajpcell.00211.2007
- Lesnefsky, E.J., Slabe, T.J., Stoll, M.S., et al. (2001) Myocardial ischemia selectively depletes cardiolipin in rabbit heart subsarcolemmal mitochondria. American Journal of Physiology, 280, H2770-H2778.
- Paradies, G., Petrosillo, G., Gadaleta, M.N., et al. (1999) The effect of aging and acetyl-L-carnitine on the pyruvate transport and oxidation in rat heart mitochondria. FEBS Letters, 454, 207-209. doi:10.1016/S0014-5793(99)00809-1
- Paradies, G., Ruggiero, F.M., Petrosillo, G., et al. (1992) The effect of aging and acetyl-L-carnitine on the acitivity of the phophate carrier and on the phospholipid composition in rat heart mitochondria. Biochimica et Biophysica Acta, 406, 136-138.
- Paradies, G., Ruggiero, F.M., Petrosillo, G., et al. (1994) The effect of aging and acetyl-L-carnitine on the function and on the lipid composition of rat heart mitochondria. Annals of the New York Academy of Sciences, 717, 233- 243. doi:10.1111/j.1749-6632.1994.tb12093.x
- Paradies, G., Ruggiero, F.M., Petrosillo, G., et al. (1994) Effect of aging and acetyl-L-carnitine on the activity of cytochrome oxidase and adenine nucleotide translocase in rat heart mitochondria. FEBS Letters, 350, 213-215. doi:10.1016/0014-5793(94)00763-2
- Paradies, G., Ruggiero, F.M., Petrosillo, G., et al. (1996) Age-dependent impairment of mitochondrial function in rat heart tissue. Effect of pharmacological agents. Annals of the New York Academy of Sciences, 786, 252-263. doi:10.1111/j.1749-6632.1996.tb39068.x
- Lesnefsky, E.J., He, D., Moghaddas, S., et al. (2006) Reversal of mitochondrial defects before ischemia protects the aged heart. FASEB Journal, 20, 1543-1545. doi:10.1096/fj.05-4535fje
- Sparagna, G.C. and Lesnefsky, E.J. (2009) Cardiolipin remodeling in the heart. Journal of Cardiovascular Pharmacology, 53, 290-301. doi:10.1097/FJC.0b013e31819b5461
- Moghaddas, S., Stoll, M.S., Minkler, P.E., et al. (2002) Preservation of cardiolipin content during aging in rat heart interfibrillar mitochondria. Journals of Gerontology Series A: Biological Sciences and Medical Sciences, 57, B22-B28. doi:10.1093/gerona/57.1.B22
- Gadaleta, M.N., Petruzzella, V., Daddabbo, L., et al. (1994) Mitochondrial DNA transcription and translation in aged rat. Effect of acetyl-L-carnitine. Annals of the New York Academy of Sciences, 717, 150-160. doi:10.1111/j.1749-6632.1994.tb12082.x
- Gadaleta, M.N., Petruzzella, V., Fracasso, F., et al. (1990) Acetyl-L-carnitine increases cytochrome oxidase subunit I mRNA content in hypothyroid rat liver. FEBS Letters, 277, 191-193. doi:10.1016/0014-5793(90)80841-6
- [61] Rosca, M.G., Lemieux, H. and Hoppel, C.L. (2009) Mitochondria in the elderly: Is acetylcarnitine a rejuvenator? Advanced Drug Delivery Reviews, 61, 1332-1342. doi:10.1016/j.addr.2009.06.009
- [62] Ahn, B.H., Kim, H.S., Song, S., et al. (2008) A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proceedings of the National Academy of Sciences, 105, 14447-14452. doi:10.1073/pnas.0803790105
- [63] Lombard, D.B., Alt, F.W., Cheng, H.L., et al. (2007) Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Molecular and Cellular Biology, 27, 8807-8814. doi:10.1128/MCB.01636-07
- [64] Carrea, F.P., Lesnefsky, E.J., Repine, J.E., et al. (1991) Reduction of canine myocardial infarct size by a diffusible reactive oxygen metabolite scavenger. Efficacy of dimethylthiourea given at the onset of reperfusion. Circulation Research, 68, 1652-1659. doi:10.1161/01.RES.68.6.1652
- [65] Stewart, S., Lesnefsky, E.J. and Chen, Q. (2009) Reversible blockade of electron transport with amobarbital at the onset of reperfusion attenuates cardiac injury. Translational Research, 153, 224-231.
- [66] Przyklenk, K., Maynard, M., Greiner, D.L., et al. (2011) Cardioprotection with postconditioning: Loss of efficacy in murine models of type-2 and type-1 diabetes. Antioxidants & Redox Signaling, 14, 781-790.
- [67] Chen, Q., Camara, A.K., Stowe, D.F., et al. (2007) Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. American Journal of Physiology— Cell Physiology, 292, C137-C147. doi:10.1152/ajpcell.00270.2006