Journal of Biophysical Chemistry
Vol.07 No.03(2016), Article ID:68785,13 pages
10.4236/jbpc.2016.73006
Molecular Modelling of Human Multidrug Resistance Protein 5 (ABCC5)
Natesh Singh
Ranchi College of Pharmacy, Ranchi, India

Copyright © 2016 by author and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 30 April 2016; accepted 18 July 2016; published 21 July 2016
ABSTRACT
Multidrug resistance protein 5 (MRP5/ABCC5) is a 161 kDa member of the super family of ATP- binding cassette (ABC) superfamily of transmembrane transporters that is clinically relevant for its ability to confer multidrug resistance by actively effluxing anticancer drugs. ABCC5 has also been identified as an efflux transporter of cGMP (cyclic guanosine monophosphate). Elevated int- racellular levels of cGMP in cancer cells have been implicated in several clinical studies, that may induce apoptosis, and as a result many different cancer cells seem to overcome this deleterious effect by increased efflux of cGMP through ABCC5. Thus inhibition of ABCC5 may have cytotoxic effects mediated through cGMP and it will also increase the intracellular concentration of other drugs that are aimed for the treatment of cancer which are otherwise exported out of the cells. Considering the functional importance and lack of X-ray crystal structure of ABCC5, present work was undertaken to construct 3D structure of protein using homology modeling protocol of YASARA structure (V. 16.3.28). In this study, five different ABC templates (PDB ID’s: 4F4C, 4Q9H, 4M1M, 4M2T and 4KSD) were used for homology modeling. Five models were constructed on each template and a hybrid model was built using all five templates. All models were refined and ranked as per their overall Z-score. The top ranked ABBC5 model was based on template 4Q9H that had 91.2% of residues in allowed regions as revealed by PROCHECK-NMR and the QMEAN score was 0.54 which indicated a reliable model. The results of the study and the proposed model can be further used for elucidating the structural and functional aspects of ABCC5 and to gain more insights to the molecular basis of ABCC5 inhibition through docking studies.
Keywords:
Multidrug Resistance Protein, ABC Transporter, Homology Modeling, ABCC5

1. Introduction
ABCC5 belongs to the ABC transporters, which are structurally related membrane proteins featuring intracellular motifs that exhibit ATPase activity [1] . This nucleotide binding domain (NBD), which contains the Walker A and Walker B motifs, cleaves ATP’s terminal phosphate to energize the transport of substrate molecules against a concentration gradient. ABC genes are highly conserved within species, indicating that these genes may have been present since the beginning of eukaryotic evolution [1] [2] . The putative structure of ABCC5 consists of 12 transmembrane helices (TMH) and 2 NBDs (Figure 1).
Clinical studies have shown that extracellular cGMP levels are elevated in various types of cancer [3] - [6] . The human ABC transporter ABCC5 transports cGMP out of cells, and the increased cGMP efflux from cancer cells may be caused by an up regulation of ABCC5. The ability of ABCC5 to transport cGMP suggests that it may be playing a biological role in cellular signaling by eliminating the nucleotides from the cells [7] . Increased cGMP efflux by ABCC5 may be one mechanism whereby cancer cells can develop resistance against endogenous growth control, and also against antineoplastic drugs which are substrates for ABCC5. cGMP regulates several apoptosis-associated genes, and therefore intracellular elevation of cGMP concentration by inhibition of ABCC5 efflux may give cytotoxic effects [8] .
No X-ray crystal structure of ABCC5 has been reported, but molecular models of ABCC5 may be constructed by homology using a known 3D crystal structure of an evolutionary related protein as a template. Different models of ABCC5 [9] [10] are previously presented in outward-facing conformations and inward-facing conformations based on the Staphyllococcus aureus ABC transporter Sav1866 [11] , which has been crystallized in an outward-facing ATP-bound state, and on the Escherichia coli MsbA, which has been crystallized in a wide open inward-facing conformation [12] . In this study, multiple homology models of ABCC5 are constructed on the X-ray crystal structure of five different ABC transporters (PDB IDs: 4F4C, 4Q9H, 4M1M, 4M2T and 4KSD) and all models are ranked as per their structural quality. The template resolution of different crystal structures ranged from 3.8 - 4.1 Å and the sequence identity between templates and human ABCC5 are in range 20.7% - 22.8%, clearly implying that the ABCC5 homology models has elements of uncertainty.
2. Results and Discussion
The best homology model of ABCC5 was model number 22 (Table 1 lists all models that were built on different templates) and this model was based on crystal structure of P-glycoprotein (PDB ID: 4Q9H) of 3.4 Å (Figure 2). The model was in an inward facing conformation with the NBD’s separated by approximately 26 Å. A large internal cavity open to the cytoplasm was formed by 2 TMD. TMD 1 comprise of TMHs 1, 2, 3, 6, 10, 11 and TMD 2 comprise of TMHs 4, 5, 7, 8, 9, 12 as shown in Figure 3. The secondary structure of the model is depicted in Figure 4.
The Walker A motifs consisted of a coiled loop and a short α-helix (P-loop), and the Walker B motifs were in β-sheet conformation and localized in the NBD’s hydrophobic cores, which consisted of 5 parallel β-sheets and 1 anti-parallel β-sheet. In NBD1 the Walker A has residues from Gly595-Thr602, Q-loop from Gln630-Asn636, C motif from Leu685-Arg692, Walker B from Ile705-Asp709, D-loop from Ser713-Asp716, H-loop from Thr741-Gln743 and in NBD2, the Walker A starts from Gly1227-Ser1234, Q loop from Gln1275-Gly1282,

Figure 1. Topology of ABCC5 showing 2 transmembrane domain (TMD 1 and TMD 2). and 2 NBDs (NBD 1 and NBD 2) with respective Walker A, C Motif and Walker B.

Table 1. Structural analysis of ABCC5 models.
C motif from Phe1330-Gln1336, Walker B from Ile1350-Asp1354, D-loop from Ala1358-Asp1361 and H-loop from Ala1385-Arg1387. The connecting extracellular loop between TMH7 and TMH8 in the model was about 39 amino acids long (Ser872-His910) and this was consistent with the PROSITE [14] predictions of TMH from 848 - 868 and 917 - 937 in ABCC5. NBD1 and TMD2 were connected with 2 α-helices (Thr800-Lys821, Gln832-Gly837) and three loops (Asn783-Glu799, Ala822-Val831 and Gln838-Trp843). The fact that needs to be considered here that this long loop was not present in the template, and that modeling loops of this length is relatively inaccurate and consequently the modeled loop structures must be regarded as ambiguous. The loop is approximately 42 Å from the binding pocket, and accordingly, the inclusion of this loop may not be necessary for the purpose of any future studies like docking on this model.
The Errat option of the Structural Analysis and Verification Server (SAVES)
http://nihserver.mbi.ucla.edu/SAVES/ reported that the overall quality factor of the ABCC5 model was 98.022,

Figure 2. Superimposition of the ABCC5 model (blue) and the template x-ray crystal structure of the p-glycoprotein (red) [13] . Alignment score: 0.123, RMSD: 1.606 Å.

Figure 3. (a) Arrangement and orientation of different TMHs in ABCC5 (The structures are drawn in cartoon: helices as tubes, beta-sheets as arrows, turn and coil as wires. The residues are colored by residue type: non-polar residues in grey, polar-residues in green, basic residues in cyan and acidic residues in red). (b) Molecular surface of ABCC5 model: The residues are colored in blue for the most hydrophilic, to white, and to brown for the most hydrophobic. Horizontal black lines delineate the approximate location of lipid head groups forming bilayer containing 2 TMDs and the curved lines indicate the position of 2 NBDs.
and a value above 90 indicates a good model. According to the Ramachandran plot (Figure 5) provided by the Procheck option, 91.2% of the ABCC5 residues were in the most favored regions, 7.6% were in additional allowed regions, 0.6% were in generously allowed regions, and 0.6% were in disallowed regions. The overall G-factor value of the model was −0.17 indicating that geometry of model corresponds to the probability conformation with 91.2% residue in the core region showing high accuracy of model prediction.

Figure 4. Secondary structure of ABCC5 model (Helix is represented as blue cylinders, β-strand as red arrows, coils as cyan and, turns as green connecting lines).

Figure 5. Ramachandran plot of ABCC5 model.
The summary of the what check option reported that the ABCC5 model was found to be satisfactory. ProSA was used to check the three dimensional model of ABCC5 for potential errors [15] . The program displays 2 characteristics of the input structure: its Z-score and a plot of its residue energies. The Z-score of −11.61 indi
cates the overall model quality of ABCC5 (Figure 6) and it also measures the deviation of total energy of the structure with respect to an energy distribution derived from random conformations. The score is indicating a highly reliable structure that is well within the range of scores typically found for proteins of similar size. The energy plot shows the local model quality by plotting knowledge-based energies as a function of amino acid sequence position.
QMEAN analysis was also used to evaluate and validate the model [16] . The QMEAN score of the model obtained was 0.54 and the Z-score was −2.44. The QMEAN analysis reflects the good quality of the model because the estimated reliability of the model was expected to be in between 0 and 1 and this can be inferred from the density plot for QMEAN scores of the reference set (Figure 7). Comparison with non-redundant set of PDB structures in the plot between normalized QMEAN score and protein size revealed different set of Z-values for different parameters such as C-beta interactions (−2.37), interactions between all atoms (−1.40), solvation (−2.25), torsion (−3.69), SSE agreement (0.37) and ACC agreement (−1.55) which can be clearly observed (Figure 8). Some local error was also obtained for the ABCC5 model which was higher somewhere in between the residue from 1250 and 1300. The PROCHECK and QMEAN analysis of other best ranked ABCC5 models are summarized in Table 2.
All 26 models were validated using DOPE [17] which computes the energy of the tertiary structure of a protein as the sum of the energy of pairs of atoms in the protein. The detailed structural analysis of all the models is provided in Table 1. The top ranked ABCC5 model (Model no. 22) has normalized dope score of −0.43, suggesting that 60% of its C-α atoms are within 3.5 Å of their correct positions implying a reliable model. The overall Z-score of this model as determined by structural validation tools of YASARA was −1.433 and this model was ranked 1 in terms of stability and stereo chemical aspects.
The presence of a well-defined binding site, shown in Figure 9, was probed through the MOE Site Finde program. Table 3 reports the binding site score for the ABCC5 model and lists the residues composing the site. Predicted active site residues like ASN 441, GLN 1138 were consistent with ABCC5 model [9] .
Other ABCC5 models that were ranked best among the five models built on each template are shown in Figure 10. The PDB files of the ABCC5 models are available upon request.
3. Methodology
Homology modelling of ABCC5 was performed using YASARA Structure (V. 16.3.28) that fully automatically takes all the steps from an amino acid sequence to a refined high-resolution model using a CASP approved protocol [18] . The target sequence (Uniprot ID: O15440) was PSI-BLASTed [19] against Uniprot to build a
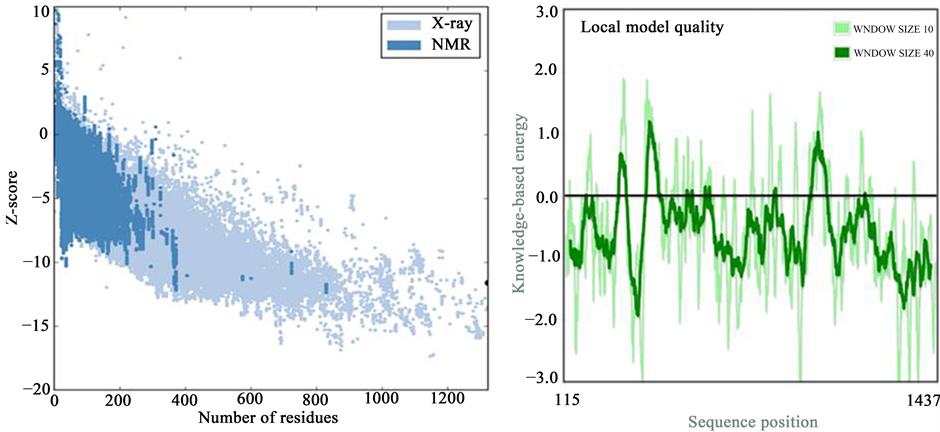
Figure 6. ProSA web service analysis of ABCC5 model. ProSA-web zscore of all protein chains in PDB determined by x-ray crystallography (light blue) or NMR-spectroscopy (dark blue) with respect to their length. The z-score of modeled ABCC5 is highlighted as large dot and the right graph is showing energy plot of model.

Figure 7. Density plot for QMEAN showing the value of Z-score and QMEAN score.
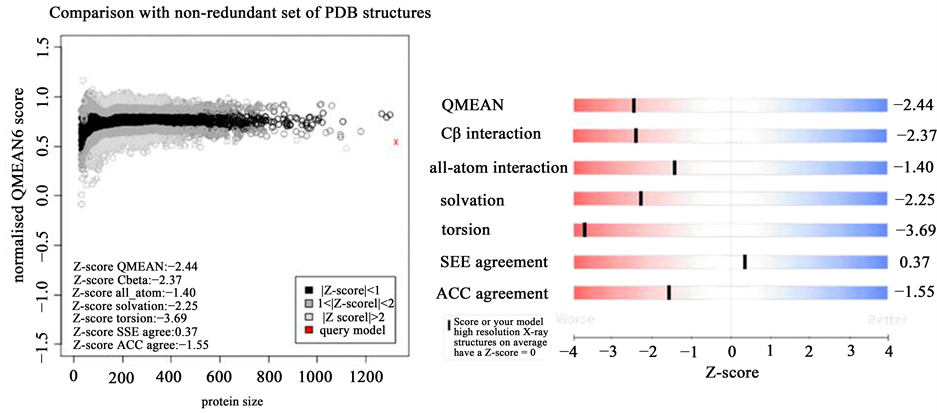
Figure 8. Plot showing the QMEAN value as well as Z-score.
position-specific scoring matrix (PSSM) from related sequences, then this profile was used to search the PDB for potential modelling templates. Common protein purification tags were excluded to avoid false positives. The templates were then ranked based on the alignment score and the structural quality according to WHATCHECK [20] obtained from the PDB Finder 2 database [21] . Usually models built using high-resolution X-ray templates are more accurate than those created from lower resolution X-ray or NMR templates, even if the latter share a higher percentage sequence identity. Models were finally built for the top scoring five templates in this study (Table 4).

Figure 9. Molecular surface of the binding site and filling dummy atoms in the ABCC5 model.
Table 2. PROCHECK and QMEAN analysis of other ABCC5 models.
Table 3. Binding site features of ABCC5 model (Size indicates the number of alpha spheres comprising the site. PLB is the propensity for ligand binding score for the contact residues. Hyd indicates the number of hydrophobic contact atoms in the receptor. Side indicates the number of sidechain contact atoms in the receptor).
To aid alignment correction and loop modelling, a secondary structure prediction for the target sequence was obtained by running PSI-BLAST to create a target sequence profile and feeding it to the PSI-Pred secondary structure prediction algorithm [25] . For each available template, the alignment with the target sequence was performed after taking into account additional information like sequence-based profiles of target and template calculated from related Uniprot sequences, optionally augmented with structure-based profiles from related template structures. The alignment of the ABCC5 sequence with all five templates is depicted in Figure 11.
The alignment also considered the structural information contained in the template (avoiding gaps in second- dary structure elements, keeping polar residues exposed etc.), as well as the predicted target secondary structure [27] . This structure-based alignment correction is partly based on SSALN scoring matrices [28] . If the alignment is not certain, alternative high-scoring alignments were created using a stochastic approach [29] , and models were built for all of them. If templates exist in oligomeric states (according to the PQS database), models may

Figure 10. ABCC5 models (a) model number 4 based on template 4F4C; (b) model number 8 based on template 4KSD; (c) model number 13 based on template 4M1M; (d) model number 16 based on template 4M2T and (e) model number 26, a hybrid model based on all five templates).
Table 4. Templates used for homology modelling.

Figure 11. Amino acid sequence alignment of human ABCC5 and 5 templates (4F4C, 4Q9H, 4M1M, 4M2T and 4KSD). The alignment was visualized by using Jalview [26] . The aligned residues are colored based on their type using the clustlx color scheme.
be built in the same state, so that interactions between side-chains across the interface can be considered. This
includes all kinds of hetero-oligomers, e.g. a homo-dimer of two hetero-dimers. In case of insertions and deletions, an indexed version of the PDB is used to determine the optimal loop anchor points and collect possible loop conformations. If templates contain ligands, these molecules were parameterized and fully considered in the homology modelling procedure, including hydrogen bonding and other interactions with the peptide chain. After initial model building the loops were optimized by trying hundreds of different conformations and re-optimizing the side-chains for all of them. Side-chain rotamers were fine-tuned considering electrostatic and knowledge-based packing interactions as well as solvation effects. After the side-chains optimization the newly modelled parts were subjected to a combined steepest descent and simulated annealing minimization (i.e. the backbone atoms of aligned residues were kept fixed to avoid potential damage). The model’s hydrogen bonding network was optimized, including pH-dependence and ligands. An unrestrained high-resolution refinement with explicit solvent molecules was run, using the latest knowledge-based force fields. The result was validated to ensure that the refinement did not move the model in the wrong direction. The tasks above were performed for all combinations of templates and alignments, per-residue quality indicators for the resulting models were determined. A hybrid model (model no. 26) was also built in which bad regions in the top scoring model were iteratively replaced with corresponding fragments from the other models. The binding site of the model was identified through the MOE Site finder program, which uses a geometric approach to calculate putative binding sites in a protein, starting from its tridimensional structure. This method is not based on energy models, but only on alpha spheres, which are a generalization of convex hulls [30] .
4. Conclusion
In silico studies in general and molecular modeling techniques have been of great help in understanding the structure, function and mechanism of the action of proteins, particularly the membrane proteins. The present investigation was carried out with major objective to model the ABCC5 protein using different templates and subjecting models so obtained for structural validation using different analysis tools like PROCHECK, ProSA and QMEAN. ABCC5 model based on 4Q9H was found to be reliable in terms of stereochemistry with G-factor value of −0.17 indicating that the geometry of model correlated to the probability conformation with 91.2% residue in the core region of Ramachandran plot showing very high accuracy of model prediction. Z score of −11.61 predicted by ProSA represented the good quality of the model. The predicted binding site had the residues as enumerated in previously reported ABCC5 model. The predicted 3D structure will provide more insight in understanding the structure and function of the protein. Moreover, this structure can be used for drug designing or understanding the interactions of ligand with the active site residues.
Cite this paper
Natesh Singh, (2016) Molecular Modelling of Human Multidrug Resistance Protein 5 (ABCC5). Journal of Biophysical Chemistry,07,61-73. doi: 10.4236/jbpc.2016.73006
References
- 1. Peracchi, M., Bamonti-Catena, F., Lombardi, L., Toschi, V., Bareggi, B., Cortelezzi, A., Maiolo, A.T. and Polli, E.E. (1985) Plasma Cyclic Nucleotide Levels in Monitoring Acute Leukemia Patients. Cancer Detection and Prevention, 8, 291-295.
- 2. Turner, G.A., Greggi, S., Guthrie, D., Benedetti Panici, P., Ellis, R.D., Scambia, G. and Mancuso, S. (1990) Monitoring Ovarian Cancer Using Urine Cyclic GMP. A Two-Centre Study. European Journal of Gynaecological Oncology, 11, 421-427.
- 3. Jedlitschky, G., Burchell, B. and Keppler, D. (2000) The Multidrug Resistance Protein 5 Functions as an ATP-Dependent Export Pump for Cyclic Nucleotides. The Journal of Biological Chemistry, 275, 30069-30074.
http://dx.doi.org/10.1074/jbc.M005463200 - 4. Pilz, R.B. and Broderick, K.E. (2005) Role of Cyclic GMP in Gene Regulation. Frontiers in Bioscience, 10, 1239-1268.
http://dx.doi.org/10.2741/1616 - 5. Sager, G., Orvoll, E.O., Lysaa, R.A., Kufareva, I., Abagyan, R. and Ravna, A.W. (2012) Novel cGMP Efflux Inhibitors—Identified by Virtual Ligand Screening (VLS) and Confirmed by Experimental Studies. Journal of Medicinal Chemistry, 55, 3049-3057.
http://dx.doi.org/10.1021/jm2014666 - 6. Ravna, A.W., Sylte, I. and Sager, G. (2008) A Molecular Model of a Putative substrate Releasing Conformation of Multidrug Resistance Protein 5 (MRP5). European Journal of Medicinal Chemistry, 43, 2557-2567.
http://dx.doi.org/10.1016/j.ejmech.2008.01.015 - 7. Dawson, R.J.P. and Locher, K.P. (2006) Structure of a Bacterial Multidrug ABC Transporter. Nature, 443, 180-185.
http://dx.doi.org/10.1038/nature05155 - 8. Ward, A., Reyes, C.L., Yu, J., Roth, C.B. and Chang, G. (2007) Flexibility in the ABC Transporter MsbA: Alternating Access with a Twist. Proceedings of the National Academy of Sciences of the United States of America, 104, 19005- 19010.
http://dx.doi.org/10.1073/pnas.0709388104 - 9. Szewczyk, P., Tao, H., McGrath, A.P., Villaluz, M., Rees, S.D., Lee, S.C., Doshi, R., Urbatsch, I.L., Zhang, Q. and Chang, G. (2015) Snapshots of Ligand Entry, Malleable Binding and Induced Helical Movement in P-Glycoprotein. Acta Crystallographica Section D Biological Crystallography, 71, 732-741.
- 10. Sigrist, C.J.A., Cerutti, L., Hulo, N., Gattiker, A., Falquet, L., Pagni, M., Bairoch, A. and Bucher, P. (2002) PROSITE: A Documented Database Using Patterns and Profiles as Motif Descriptors. Briefings in Bioinformatics, 3, 265-274.
http://dx.doi.org/10.1093/bib/3.3.265 - 11. Wiedersteinand, M. and Sippl, M.J. (2007) ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Research, 35, W407-W410.
- 12. Benkert, P., Tosatto, S.C.E. and Schomburg, D. (2008) QMEAN: A Comprehensive Scoring Function for Model Quality Assessment. Proteins: Structure, Function, and Bioinformatics, 71, 261-277.
http://dx.doi.org/10.1002/prot.21715 - 13. Shen, M.-Y. and Sali, A. (2006) Statistical Potential for Assessment and Prediction of Protein Structures. Protein Science, 15, 2507-2524.
http://dx.doi.org/10.1110/ps.062416606 - 14. Krieger, E., Joo, K., Lee, J., Lee, J., Raman, S., Thompson, J., Tyka, M., Baker, D. and Karplus, K. (2009) Improving Physical Realism, Stereochemistry, and Side-Chain Accuracy in Homology Modeling: Four Approaches that Performed Well in CASP8. Proteins: Structure, Function, and Bioinformatics, 77, 114-122.
http://dx.doi.org/10.1002/prot.22570 - 15. Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W. and Lipman, D.J. (1997) Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Research, 25, 3389- 3402.
http://dx.doi.org/10.1093/nar/25.17.3389 - 16. Hooft, R.W., Vriend, G., Sander, C. and Abola, E.E. (1996) Errors in Protein Structures. Nature, 381, 272.
http://dx.doi.org/10.1038/381272a0 - 17. Hooft, R.W., Sander, C., Scharf, M. and Vriend, G. (1996) The PDBFINDER Database: A Summary of PDB, DSSP and HSSP Information with Added Value. Computer Applications in the Biosciences, 12, 525-529.
http://dx.doi.org/10.1093/bioinformatics/12.6.525 - 18. Jin, M.S., Oldham, M.L., Zhang, Q. and Chen, J. (2012) Crystal Structure of the Multidrug Transporter P-Glycoprotein from Caenorhabditis elegans. Nature, 490, 566-569.
http://dx.doi.org/10.1038/nature11448 - 19. Li, J., Jaimes, K.F. and Aller, S.G. (2014) Refined Structures of Mouse P-Glycoprotein. Protein Science, 23, 34-46.
http://dx.doi.org/10.1002/pro.2387 - 20. Ward, A.B., Szewczyk, P., Grimard, V., Lee, C.-W., Martinez, L., Doshi, R., Caya, A., Villaluz, M., Pardon, E., Cregger, C., Swartz, D.J., Falson, P.G., Urbatsch, I.L., Govaerts, C., Steyaert, J. and Chang, G. (2013) Structures of P- Glycoprotein Reveal Its Conformational Flexibility and an Epitope on the Nucleotide-Binding Domain. Proceedings of the National Academy of Sciences of the United States of America, 110, 13386-13391.
http://dx.doi.org/10.1073/pnas.1309275110 - 21. Jones, D.T. (1999) Protein Secondary Structure Prediction Based on Position-Specific Scoring Matrices. Journal of Molecular Biology, 292, 195-202.
http://dx.doi.org/10.1006/jmbi.1999.3091 - 22. Clamp, M., Cuff, J., Searle, S.M. and Barton, G.J. (2004) The Jalview Java Alignment Editor. Bioinformatics, 20, 426- 427.
http://dx.doi.org/10.1093/bioinformatics/btg430 - 23. King, R.D. and Sternberg, M.J. (1996) Identification and Application of the Concepts Important for Accurate and Reliable Protein Secondary Structure Prediction. Protein Science, 5, 2298-2310.
http://dx.doi.org/10.1002/pro.5560051116 - 24. Qiu, J. and Elber, R. (2006) SSALN: An Alignment Algorithm Using Structure-Dependent Substitution Matrices and Gap Penalties Learned from Structurally Aligned Protein Pairs. Proteins, 62, 881-891.
http://dx.doi.org/10.1002/prot.20854 - 25. Mückstein, U., Hofacker, I.L. and Stadler, P.F. (2002) Stochastic Pairwise Alignments. Bioinformatics, 18, S153-S160.
http://dx.doi.org/10.1093/bioinformatics/18.suppl_2.S153 - 26. Edelsbrunner, H., Facello, M. and Liang, J. (1996) On the Definition and the Construction of Pockets in Macromolecules. Pacific Symposium on Biocomputing, the Big Island of Hawaii, 3-6 January 1996, 272-287.
- 27. Orbo, A., Jaeger, R. and Sager, G. (1998) Urinary Levels of Cyclic Guanosine Monophosphate (cGMP) in Patients with Cancer of the Uterine Cervix: A Valuable Prognostic Factor of Clinical Outcome? European Journal of Cancer, 34, 1460-1462.
- 28. Luesley, D.M., Blackledge, G.R., Chan, K.K. and Newton, J.R. (1986) Random Urinary Cyclic 3’,5’Guanosine Monophosphate in Epithelial Ovarian Cancer: Relation to Other Prognostic Variables and to Survival. British Journal of Obstetrics and Gynaecology, 93, 380-385.
- 29. Dean, M., Rzhetsky, A. and Allikmets, R. (2001) The Human ATP-Binding Cassette (ABC) Transporter Superfamily. Genome Research, 11, 1156-1166.
http://dx.doi.org/10.1101/gr.GR-1649R - 30. Saier, M.H., Tran, C.V. and Barabote, R.D. (2006) TCDB: The Transporter Classification Database for Membrane Transport Protein Analyses and Information. Nucleic Acids Research, 34, D181-D186.