Journal of Biomaterials and Nanobiotechnology
Vol.4 No.1(2013), Article ID:26830,11 pages DOI:10.4236/jbnb.2013.41001
Sintering and Mechanical Properties of Magnesium and Fluorine Co-Substituted Hydroxyapatites
![]()
Laboratory of Industrial Chemistry, National School of Engineering, Sfax, Tunisia.
Email: *khaled.bouzouita@ipeim.rnu.tn
Received September 5th, 2012; revised October 17th, 2012; accepted November 10th, 2012
Keywords: Hydroxyapatite; Magnesium, Fluorine; Sintering; Mechanical Properties
ABSTRACT
Biological apatites contain several elements as traces. In this work, magnesium and fluorine co-substituted hydroxyapatites with the general formula Ca9Mg(PO4)6(OH)2-yFy, where y = 0, 0.5, 1, 1.5 and 2 were synthesized by the hydrothermal method. After calcination at 500˚C, the samples were pressureless sintered between 950˚C and 1250˚C. The substitution of F− for OH− had a strong influence on the densification behavior and mechanical properties of the materials. Below 1200˚C, the density steeply decreased for y = 0.5 sample. XRD analysis revealed that compared to hydroxylfluorapatite containing no magnesium, the substituted hydroxyfluorapatites decomposed, and the nature of the decomposition products is tightly dependent on the fluorine content. The hardness, elastic modulus and fracture toughness of these materials were investigated by Vickers’s hardness testing. The highest values were 622 ± 4 GPa, 181 ± 1 GPa and 1.85 ± 0.06 MPa·m1/2, respectively.
1. Introduction
Hydroxyapatite (HA) is widely used in orthopedic and reconstructive surgery thanks to its excellent bioactivity and biocompatibility with the human body [1]. However, the biological apatites contain several species as traces such as F−, Cl−, 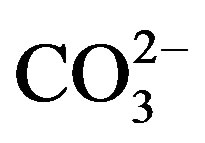 , Na+, Sr2+, Mg2+, etc. Therefore, the incorporation of one or several of these species into the hydroxyapatite structure should improve its biocompatibility and bioactivity [2,3], and affects its physical and chemical properties such as crystallinity, thermal stability, solubility and osteoconductivity [4-6]. Among these latter species, Fluorine is known to delay the caries’ processes [7], improve the bonds between the bones and the implant [8-10], and strengthen the bone structure [11]. Furthermore, this element decreases the solubility and enhances the thermal stability of the hydroxyapatite [12]; therefore, when fluorine is incorporated into bone mineral, its resorption is reduced. On the other hand, Magnesium has a beneficial effect on the mineralization processes [13,14], osteoporosis [15] and might play a role on the osteoblastic and osteoclastic activities [16,17]. In addition, Mg is known to inhibit the crystallization of the hydroxyapatite, increase its dissolution and affect its thermal stability by lowering its decomposition temperature [18-20]. Thus, ceramics designed from hydroxyapatite containing magnesium and fluorine would be more suitable for dental and orthopedic applications.
, Na+, Sr2+, Mg2+, etc. Therefore, the incorporation of one or several of these species into the hydroxyapatite structure should improve its biocompatibility and bioactivity [2,3], and affects its physical and chemical properties such as crystallinity, thermal stability, solubility and osteoconductivity [4-6]. Among these latter species, Fluorine is known to delay the caries’ processes [7], improve the bonds between the bones and the implant [8-10], and strengthen the bone structure [11]. Furthermore, this element decreases the solubility and enhances the thermal stability of the hydroxyapatite [12]; therefore, when fluorine is incorporated into bone mineral, its resorption is reduced. On the other hand, Magnesium has a beneficial effect on the mineralization processes [13,14], osteoporosis [15] and might play a role on the osteoblastic and osteoclastic activities [16,17]. In addition, Mg is known to inhibit the crystallization of the hydroxyapatite, increase its dissolution and affect its thermal stability by lowering its decomposition temperature [18-20]. Thus, ceramics designed from hydroxyapatite containing magnesium and fluorine would be more suitable for dental and orthopedic applications.
Hence, it is thought worthwhile to synthesize and characterize Mg/F-co-substituted hydroxypatites. The present study deals with the sintering of these materials and the investigation of their mechanical properties, all the more as only few studies have dealt with this kind of materials [21,22].
2. Experimental Procedure
2.1. Powder Preparation
Analytical grades Ca(NO3)2∙4H2O, Mg(NO3)2∙6H2O, (NH4)2HPO4 and NH4F were used as starting materials. Appropriate amounts according to the stoichiometric formulas of Ca10(PO4)6(OH)F and Ca9Mg(PO4)6(OH)2-yFy with y = 0, 0.5, 1, 1.5 and 2, were weighed, respectively and dissolved into 5 cm3 of deionised water under vigorous stirring. The pH of the mixed solution was adjusted to 9 by adding a concentrated ammonia solution. After that, the mixed solution was transferred to a Teflon vessel (model 4749 Parr Instrument) and sealed tightly. The autoclave was oven-heated at 180˚C for 6 h, and then cooled to room temperature naturally. The collected precipitates were washed with deionised water and dried at 70˚C overnight. After drying, the powders were calcined under argon flow at 500˚C for 1 h with a heating rate of 10˚C∙min−1.
In the following sections, the compositions Ca10(PO4)6(OH)F, Ca9Mg(PO4)6(OH)2, Ca9Mg(PO4)6(OH)1.5F0.5, Ca9Mg(PO4)6(OH)F, Ca9Mg(PO4)6(OH)0.5F1.5 and Ca9Mg(PO4)6F2 will be named as HFA, MHA, MHF0.5A, MHF1A, MHF1.5A and MFA, respectively.
2.2. Powder Characterization
The (Ca + Mg)/P molar ratios in the as-prepared powders were evaluated by a chemical analysis [23,24]. The fluoride content was measured using a fluoride selective electrode (Ingold, PF4-L).
The XRD patterns of the as-prepared and calcined powders were collected on a Philips X-pert diffractometer operating with Cu-Ka radiation (l = 1.5406 Ǻ) for a 2θ range from 20˚ to 55˚. The scan step was 0.02˚ and the integration time was 1 s per step. The crystalline phases were identified by comparing the experimental XRD patterns to the standards compiled by the Joint Committee on Powder Diffraction and Standards (JCPDS cards).
The 31P magic angle spinning nuclear magnetic resonance (31P MAS NMR) spectra were performed on a Brucker 300 WB spectrometer. The 31P observational frequency was 121.49 MHz with a spin speed 8 kHz. The 31P shift is given in parts per million (ppm) referenced to an aqueous solution of 85 wt% H3PO4.
The specific surface area (SSA) of the as-synthesized and calcined powders was measured with a Belsorp 28 SP apparatus using the BET method, while nitrogen was utilized as an adsorbed gas.
2.3. Sintering
To carry out sintering experiments, the calcined powders were uniaxially pressed under 45 MPa into pellets in a 13 mm diameter steel die, then the pellets were sintered under argon flow in a temperature ranging from 950˚C to 1250˚C with 50˚C in interval for various times.
The sintered samples were characterized based on relative density, X-ray diffraction analysis and microstructural analysis using a scanning electron microscope (PHILIPS XL 30).
Both green and sintered densities (dex) of compacts were determined through dimension and weight, and the relative density was calculated using the formula:
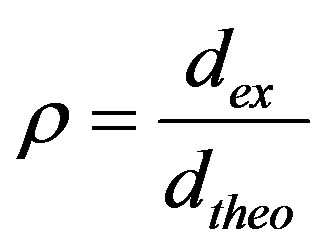 (1)
(1)
The theoretical density for each composition (Ca9Mg(PO4)6(OH)2-yFy) was calculated taking in account its molecular weight (W), the number of units per unit cell (1) and the volume of the unit cell, according to the following equation:
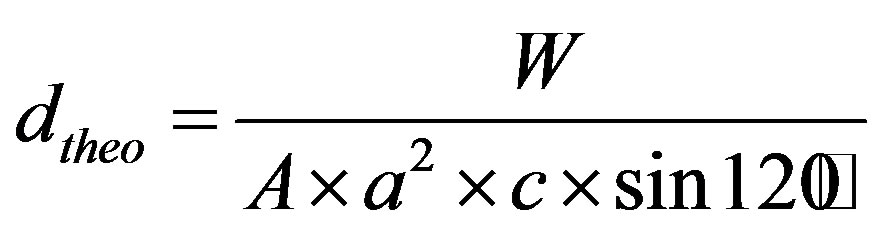 (2)
(2)
where A is Avogadro’s number, and a and c are the lattice parameters.
2.4. Mechanical Characterization
The mechanical properties were investigated on pellets of 13 mm in diameter sintered at different temperatures for 1 h. The sintered samples were polished to mirror finish prior to mechanical investigation using various grade silicon carbide papers (grade 800 - 1200) and a 0.2 mm diamond paste.
The hardness was checked with Vickers’ indentation technique using a Matsuzawa Seiki digital micro-hardness Tester (Japan). Five samples were used for each hardness data point, and for each sample, ten indentations were performed at an applied load of 200 g for 15 s. Thus, the reported hardness Hv is the average of the fifty values calculated according to the equation [25]:
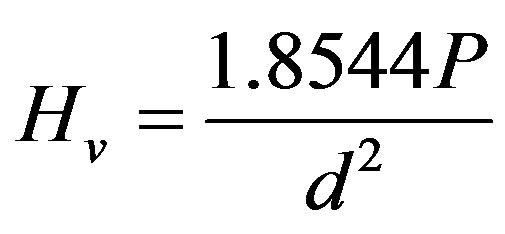 (3)
(3)
where P is the indentation load and d is the length of the diagonal of the indentation.
The Elastic modulus (Young’s modulus) was estimated from an empirical relationship reported by Marshall et al. [26] using Vickers’ microhardness:
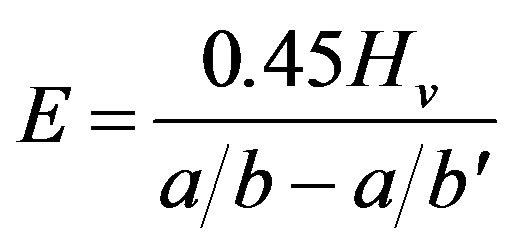 (4)
(4)
where Hv is Vickers’ indentation, a is the length of the shorter diagonal, b is the length of the longer diagonal determined using a Knoop’s indenter and b' is the crack length.
In Equation (4)
 (5)
(5)
with
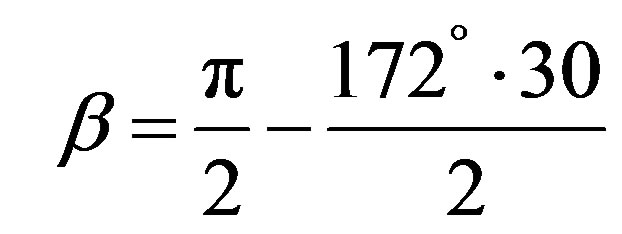 (6)
(6)
The comparison of Vickers and Knoop’ hardness for ceramics material reported by several authors [27,28] showed that the average value of the ratio HK/HV is 1.105.
Under these conditions, the value of a according to the Knoop indentation is determined according to the following equation:
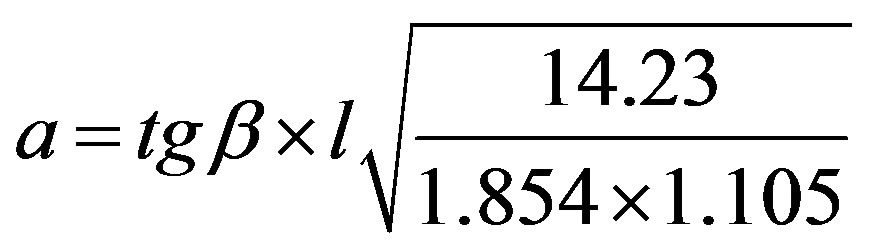 (7)
(7)
where l is the length of Vickers’ indenter diagonal.
So, Equation (4) may be written as follows:
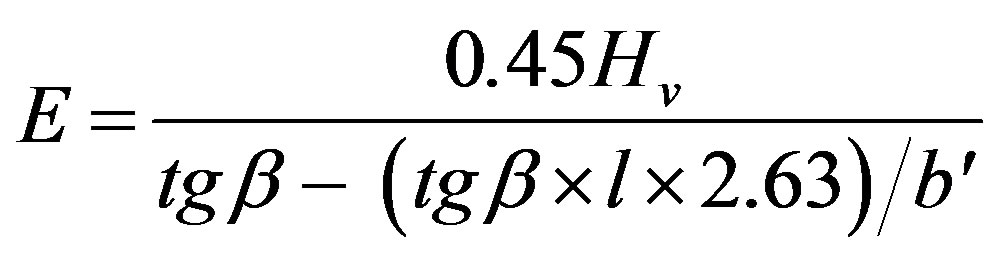 (8)
(8)
The fracture toughness (KIC) was determined using the indentation technique, and following the relationship given below [26]:
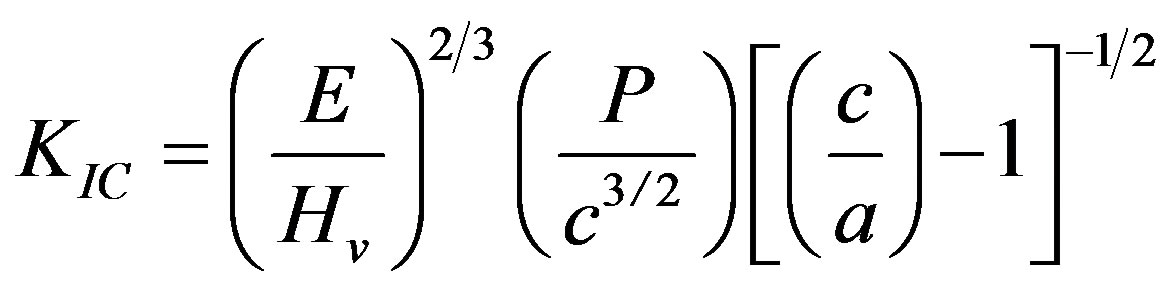 (9)
(9)
where E is Young’s modulus; Hv, Vickers’ hardness; P, the applied load; c, the crack length indentation and a, the length of Vickers’ indenter diagonals.
3. Results and Discussion
3.1. As-Prepared Powders
The quantitative chemical analyses of the samples are listed in Table 1. As has been observed, the amount of Mg in the powders was close to those introduced in the solutions, indicating that all Mg was incorporated in the synthesized materials. The (Ca+Mg)/P molar ratios are very close to the theoretical value of 1.67 for the stoichiometric apatite. On the other hand, fluorine contents were not affected by the presence of Mg, and they are consistent with the nominal composition (Table 1).
The X-ray diffraction patterns of the as-prepared powders are shown in Figure 1. At the available resolution, there was no evidence of any crystalline phase other than the apatite, which is consistent with the JCPDS #01-074-0566 or #00-071-0880 file data for HA and FA, respectively (space group P63/m). As expected, the insertion of Mg into the apatite structure was accompanied by the decrease of both a and c parameters, compared to those of the original HFA (Table 2). This decrease of the lattice parameters, which is consistent with the radius of the Mg2+ ion (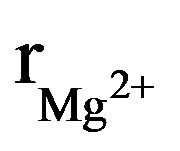 = 0.72 Å), that is smaller than that of Ca2+ (
= 0.72 Å), that is smaller than that of Ca2+ (![]() = 1.00 Ǻ) [29] confirms that the Mg2+ has entered the apatite structure. On the other hand, the substitution of F− for OH− can be also demonstrated by monitoring the variation of the lattice parameters as a function of the incorporated amount of fluoride into the apatite structure. As has been reported in the literature for hydroxyfluorapatite [30-33], with increasing content of F−, a decreased progressively and continuously compared to that of MHA, while c did not vary significantly (Table 2).
= 1.00 Ǻ) [29] confirms that the Mg2+ has entered the apatite structure. On the other hand, the substitution of F− for OH− can be also demonstrated by monitoring the variation of the lattice parameters as a function of the incorporated amount of fluoride into the apatite structure. As has been reported in the literature for hydroxyfluorapatite [30-33], with increasing content of F−, a decreased progressively and continuously compared to that of MHA, while c did not vary significantly (Table 2).
The 31P MAS NMR spectra of the as-prepared samples are shown in Figure 2. All the spectra exhibited a single resonance peak, which is a main feature of phosphorus in an apatite environment. For MHA, the chemical shift is of 2.62 ppm. This value is similar to that reported for this
Table 1. Chemical analysis data of as-prepared powders.
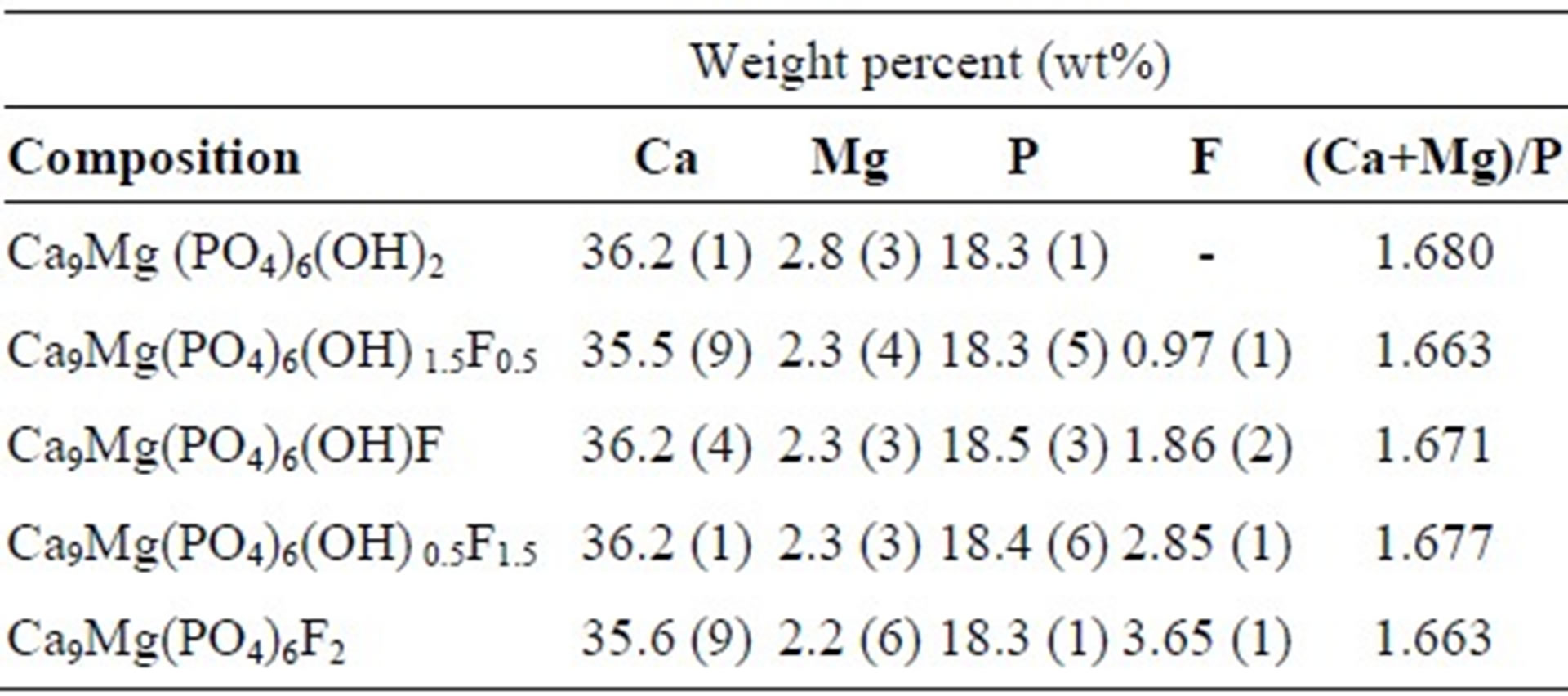
Table 2. Lattice parameters of nonand Mg/F co-substituted hydroxyapatites.
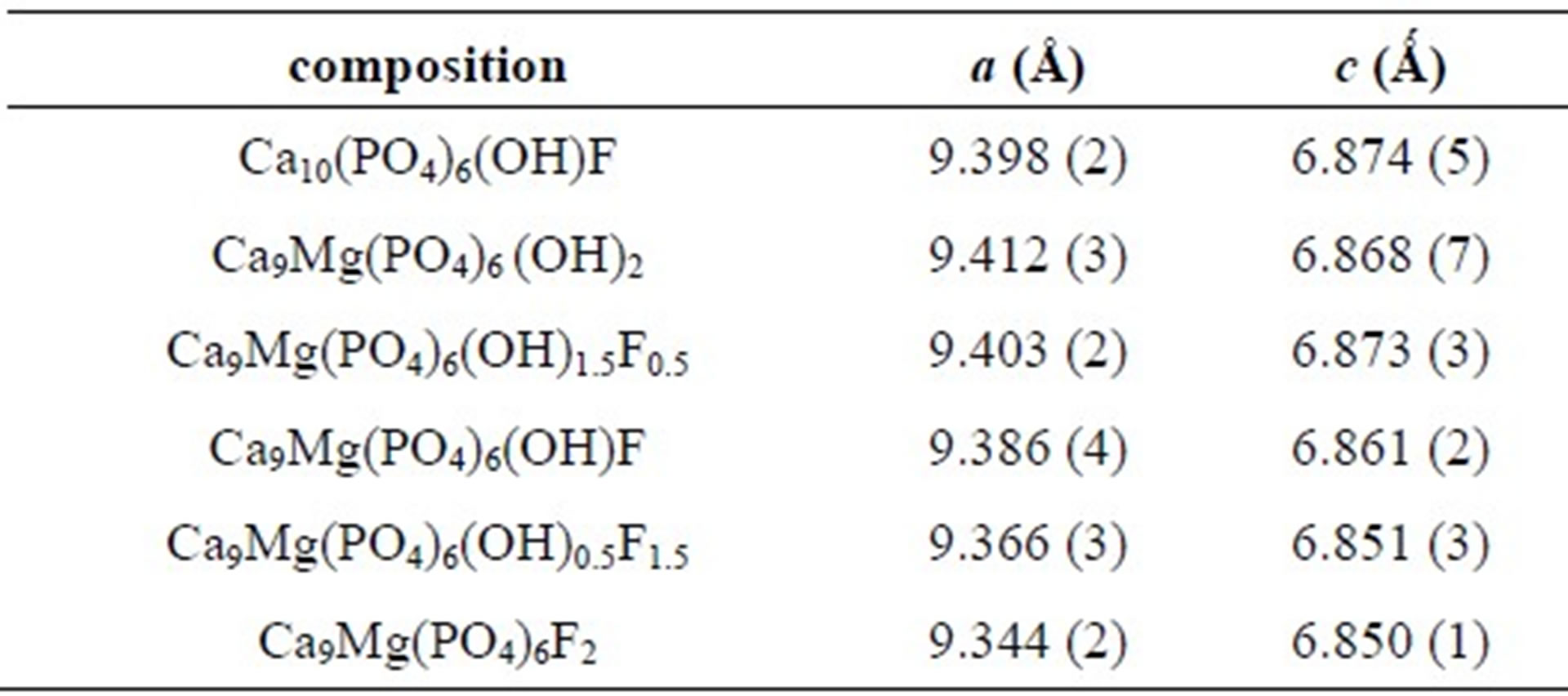
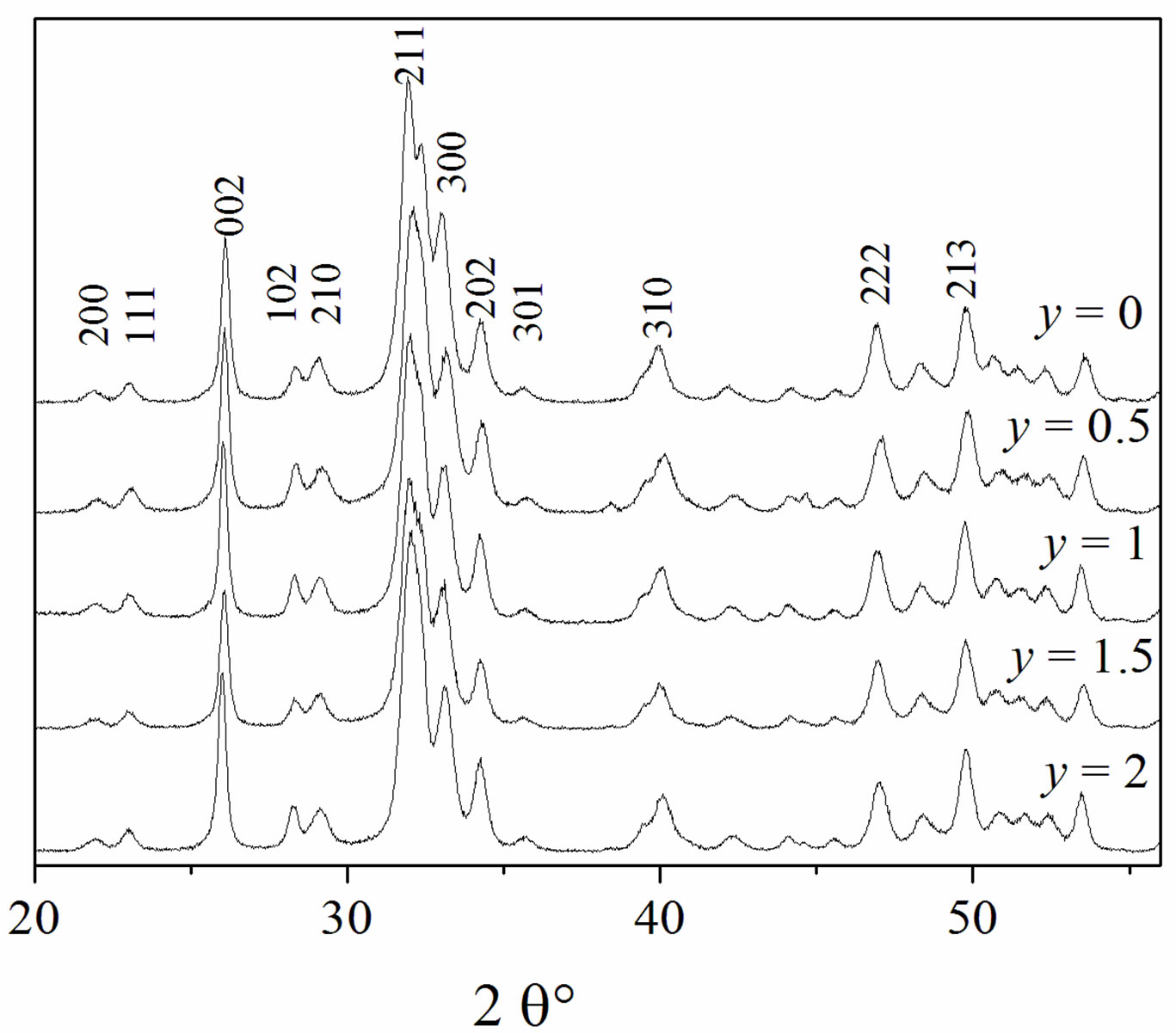
Figure 1. XRD patterns of the as-prepared powders.
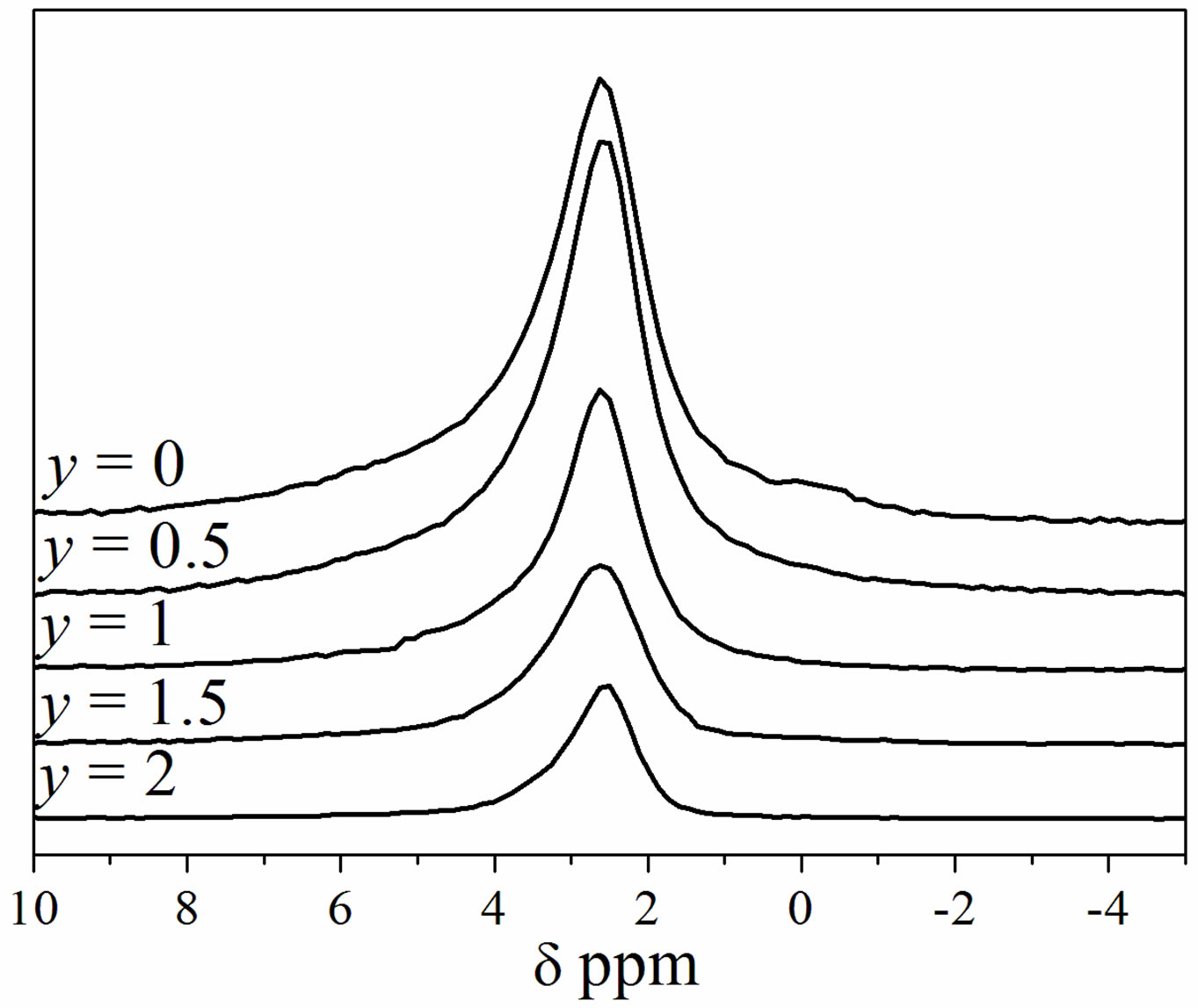
Figure 2. 31P MAS-NMR spectra of Ca9Mg(PO4)6(OH)2-yFy powders.
kind of compounds [34]. For the substituted samples, a slight chemical shift towards higher values occurred as the F− content rose. In agreement with the XRD analysis, Figure 2 confirms that the powders consisted of a single apatite phase.
As can be seen from Table 3, with the increasing of the content of F−, the SSA increased up to y = 0.5 and then decreased. This variation of the SSA as a function of the fluoridation degree is similar to that observed for pure hydroxyfluorapatite [35,36]. The increase of the SSA when fluoride content increased is attributed to the difficulty of the grains to grow due to the interactions between hydroxyl and fluoride ions, which limited the kinetics of the crystallites’ growth [35,37]. It is worth noting that the presence of Mg into the apatite structure, as is well known, inhibits the crystal growth and thus contributes to the increase of the SSA of the substituted powders with respect to the pure powder [38].
3.2. Calcined Powders
After calcination at 500˚C, the XRD patterns of Mg/Fco-substituted hydroxyapatites showed only the apatite reflections, and neither a decomposition sign nor an appearance of a new crystalline phase, resulting from the crystallization of an amorphous phase in the as-prepared powders was detected by XRD (Figure 3). However, the pattern of MHA exhibited reflexions other than those of the apatite. These reflexions whose intensities were very low belong to the b-Mg-substituted tricalcium phosphate (Ca2.81Mg0.19(PO4)2, b-MTCP) (JCPDS #01-070-0682). As the XRD and 31P MAS NMR analyses had shown that
Table 3. Specific surface area of the powders.
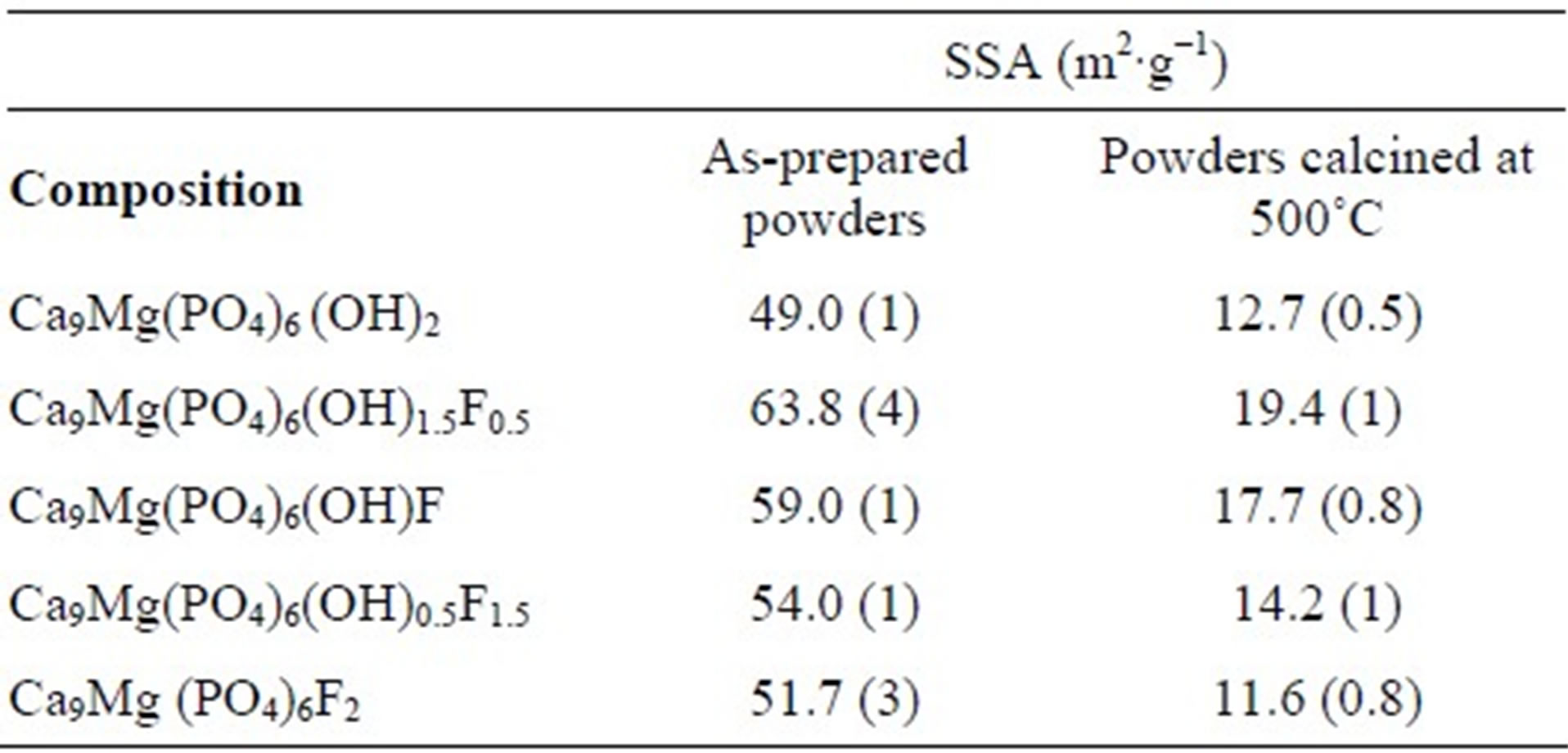

Figure 3. XRD patterns of the powders calcined at 500˚C.
the as-prepared powder was single-phased, b-MTCP would result from the decomposition of MHA. Thus, these findings show the destabilizing effect of Mg on the hydroxyapatite, and the stabilizing role of the fluorine on the Mg-substituted hydroxyapatite.
The values obtained for the SSA of the powders calcined at 500˚C are summarized in Table 3. As expected, the SSA was significantly reduced with respect to that of the as-prepared powders. It decreased from 49 to 12.7 m2∙g−1 and from 51.7 to 11.6 m2∙g−1, for MHA and MFA, respectively. Furthermore, The SSA followed versus the fluorine degree the same evolution as that of the as-prepared powder, the highest SSA value was also observed for y = 0.5.
3.3. Sintering of Powders
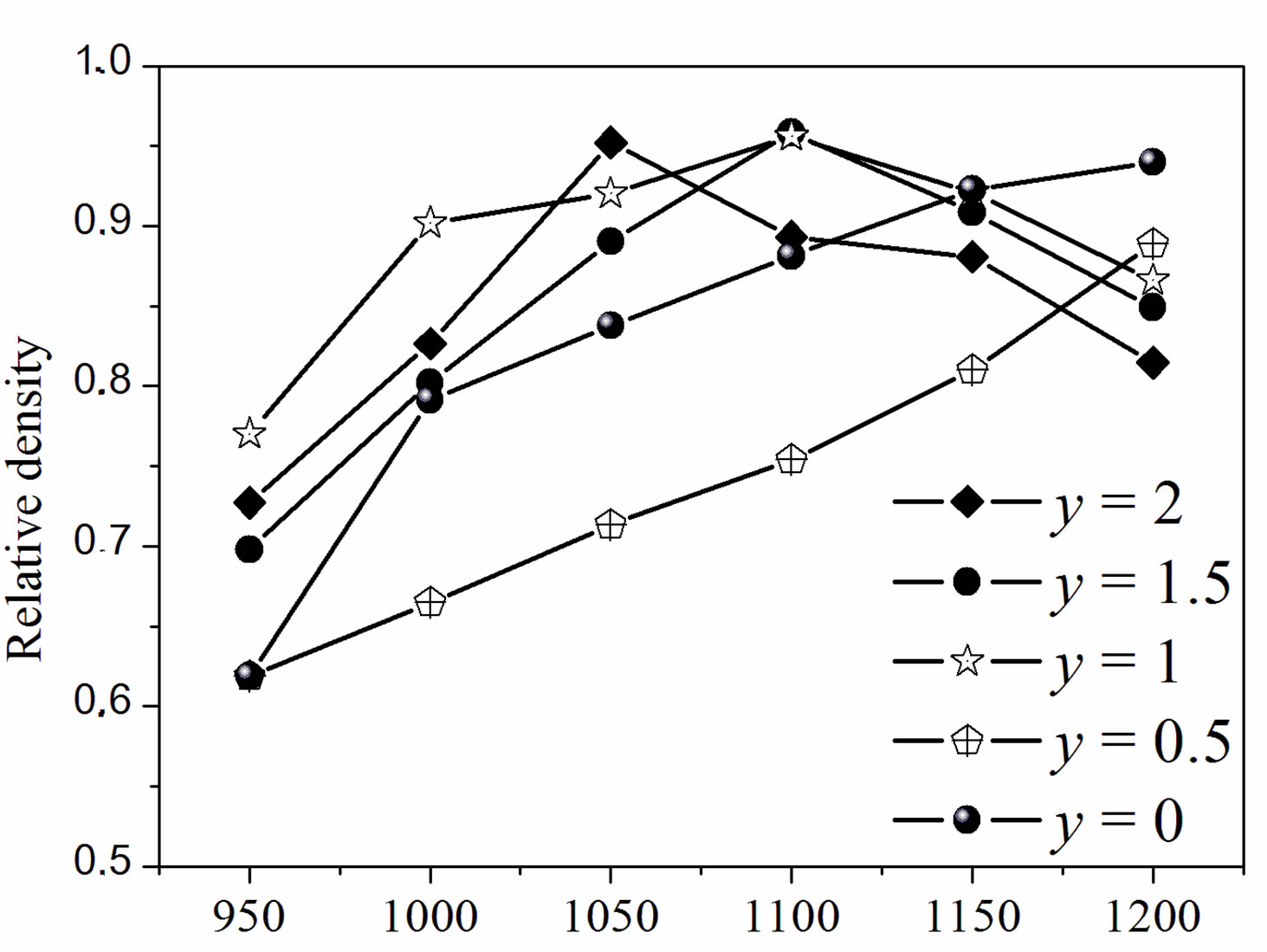
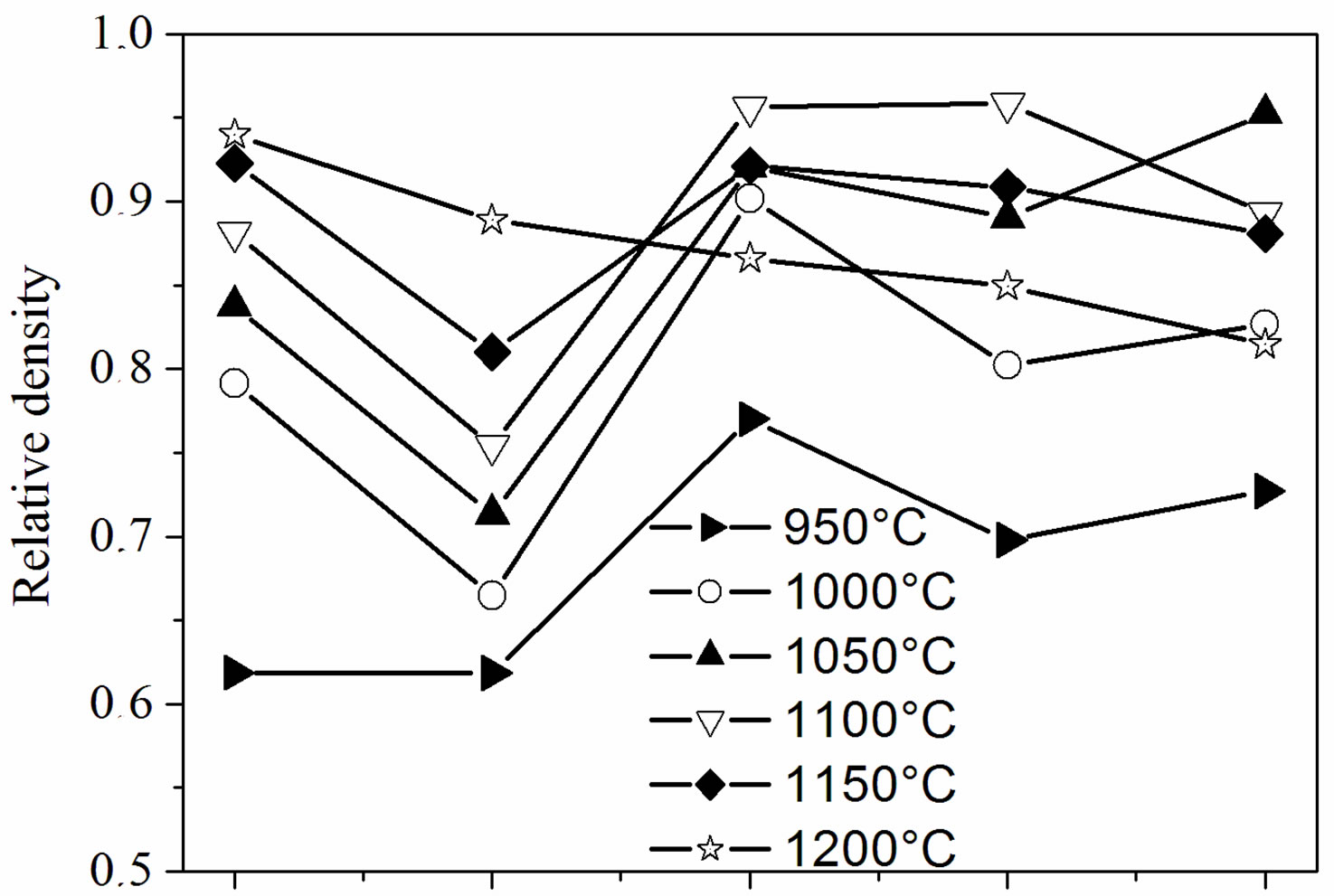
Figure 4 illustrates the relative density of the samples with different fluoridation degrees sintered at various temperatures. As shown in Figure 4(a), two composition ranges can be distinguished. For MHA and MHF0.5A, the relative density continuously increased as the sintering temperature increased. However, the density of MHF0.5A between 1000˚C and 1200˚C was much lower than that of MHA. For MHF1A, MHF1.5A and MFA, the relative density rose with the increase in temperature, reached a
T ˚C
(a)
0.0 0.5 1.0 1.5 2.0 y(F)
(b)
Figure 4. Relative density of the sintered samples as a function of: (a) sintering temperatures and (b) fluorine contents.
maximum of about 96% at around 1050˚C - 1100˚C, and then decreased. For MFA, the maximum density was observed at 1050˚C, while for the two other samples it was attained at 1100˚C, indicating that MFA densified better than the partially fluoridate samples, and obviously hydroxyapatite. Above 1200˚C, the sintered samples of the latter group were distorted, making impossible the determination of their density. The difference in the curve shape of the densities suggests that the mechanisms responsible for the densification are different for the two groups of composition.
According to Figure 4(b), showing the relative density against the fluoride content, two kinds of curves can be also distinguished. At 1200˚C, the relative density, whose highest value (0.94) was observed for MHA, decreased continuously and uniformly with the fluoridation degree to 0.82 for MFA. Below 1200˚C, and apart from a temperature reaching 950˚C, all the curves have roughly the same shape with a large decrease in density for y = 0.5. Such a shape of the curves has been previously observed for the sintering of the hydroxyfluorapatites [37]. However, Senamaud observed the decrease in density for y = 1 [35], while for Gross [37], the decrease occurred for y = 1.2. This low sinterability was attributed to the interactions between OH− and F−, which reduce the species mobility [35,37]. If, for the samples with y = 0 and 0.5, the relative density increased with the increasing of temperature, it did not follow the temperatures’ increase for y > 0.5. For example, the density of the y =1 sample was higher at 1100˚C than at 1150˚C. Similarly, for the y = 2 sample, the density was higher at 1050˚C than at 1100˚C or 1150˚C.
The effect of time on the relative density of the sintered samples was examined by fixing the temperature at 1050˚C (Figure 5). This figure highlights the low sinterability of MHF0.5A, and the high densification of the other compositions. Indeed, the samples with y = 1, 1.5 and 2 sintered to a relative density of ~0.97 after a heat treatment of 0.5 h. This density was slightly higher than that of MHA. We note that, for the latter compositions, the effect of the fluorine substitution on the densification of MHA was not noticeable at 1050˚C since there was no significant difference between the sintered densities of the fluoridate compositions. However, the y = 0.5 sample sintered only to 66.6%, and 75.4% of the theoretical density after 0.5 and 1 h, respectively. After that, the density decreased for 2 h and increased again to reach its maximum (81%) for 4 h.
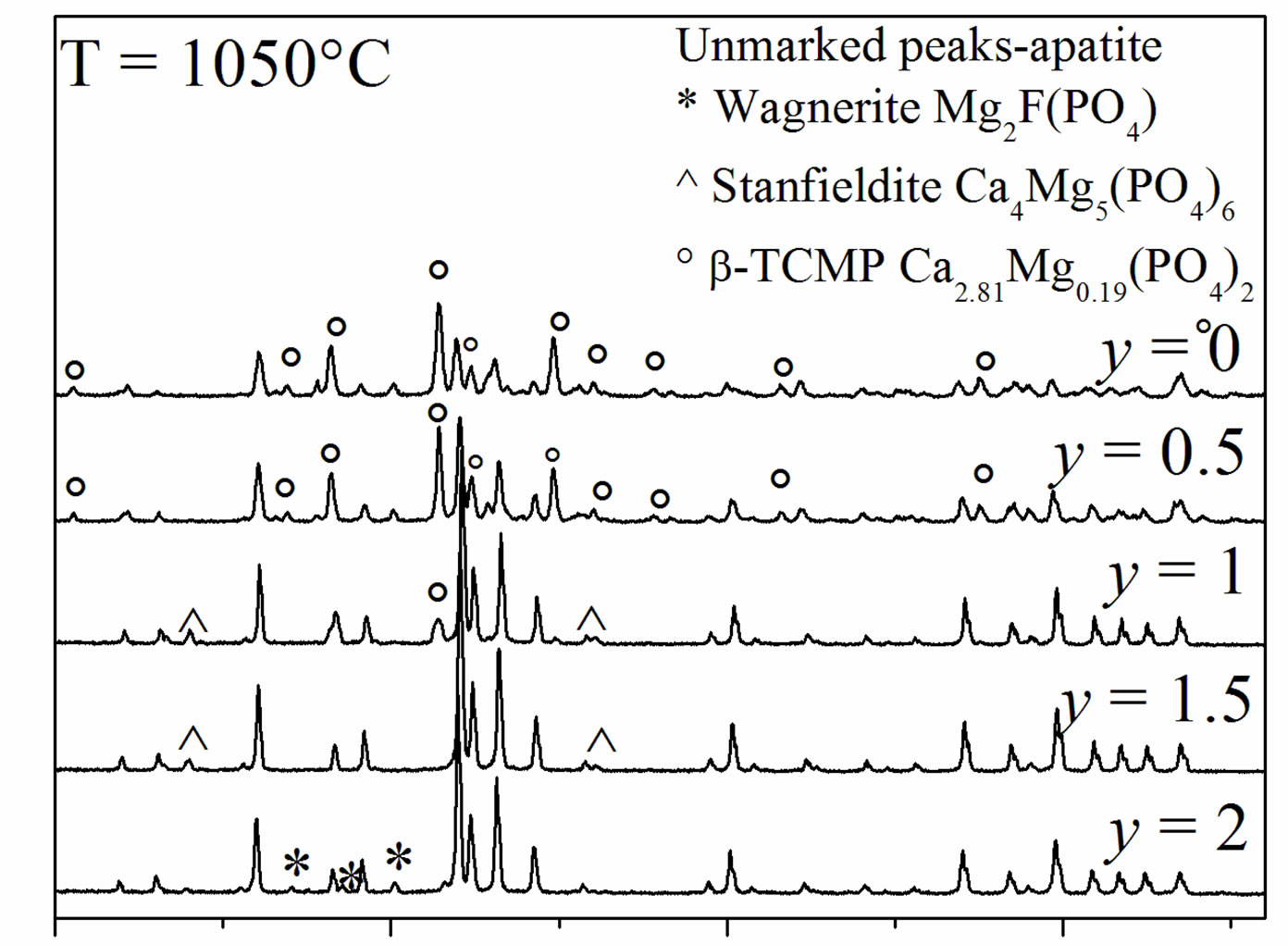
Figure 6 shows the XRD patterns of the samples sintered at 1050˚C. The patterns of MHA and MHF0.5A showed the presence of b-MTCP, indicating the decomposition of MHA, as has been reported in the literature [19,20,39,40]. This phase remained stable up to 1300˚C [18]. Indeed, it is well-known that the Mg substitution
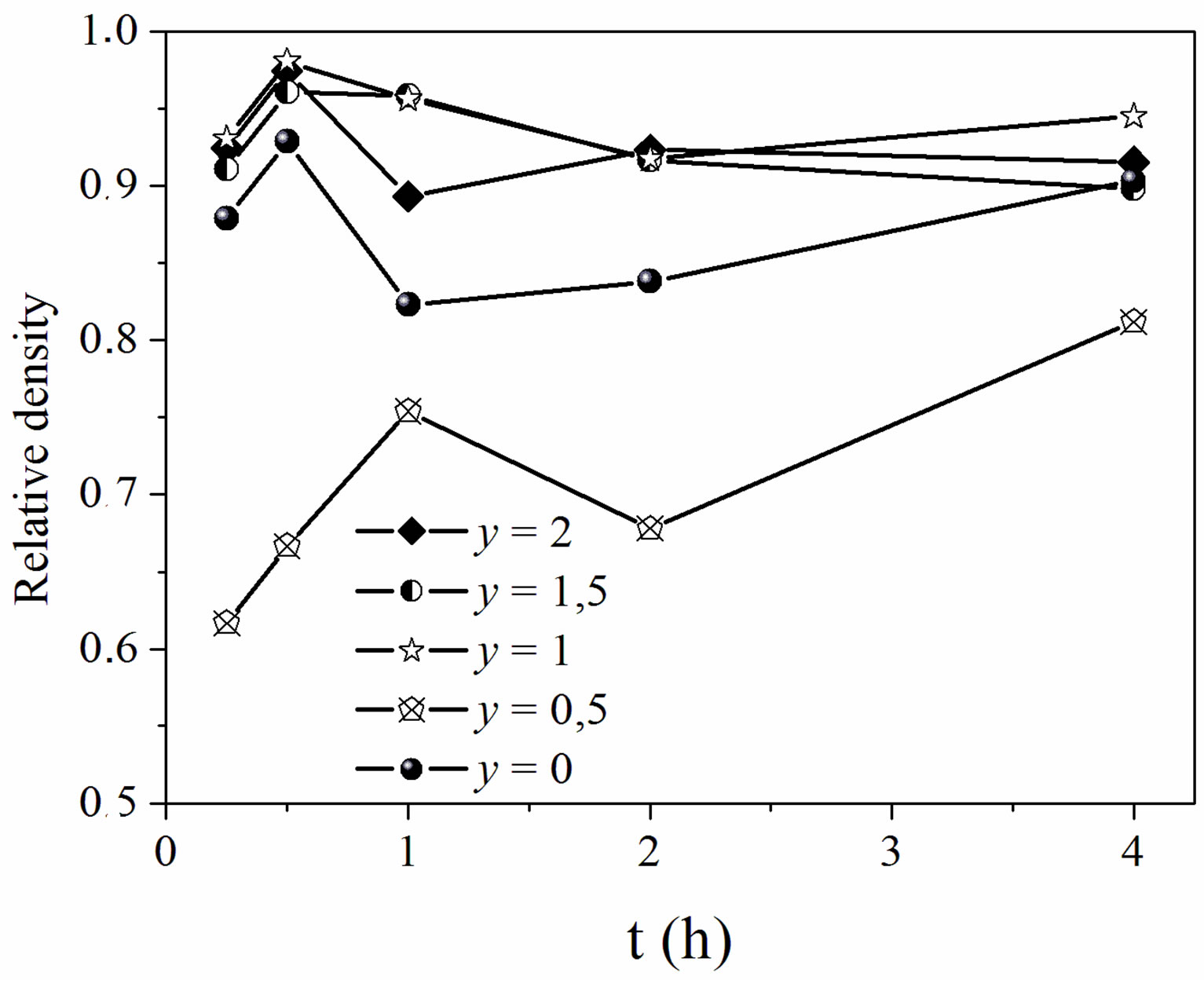
Figure 5. Relative density of the samples sintered at 1050˚C versus holding times.
20 30 40 50 2θ˚
Figure 6. XRD patterns of the samples sintered at 1050˚C for 1 h.
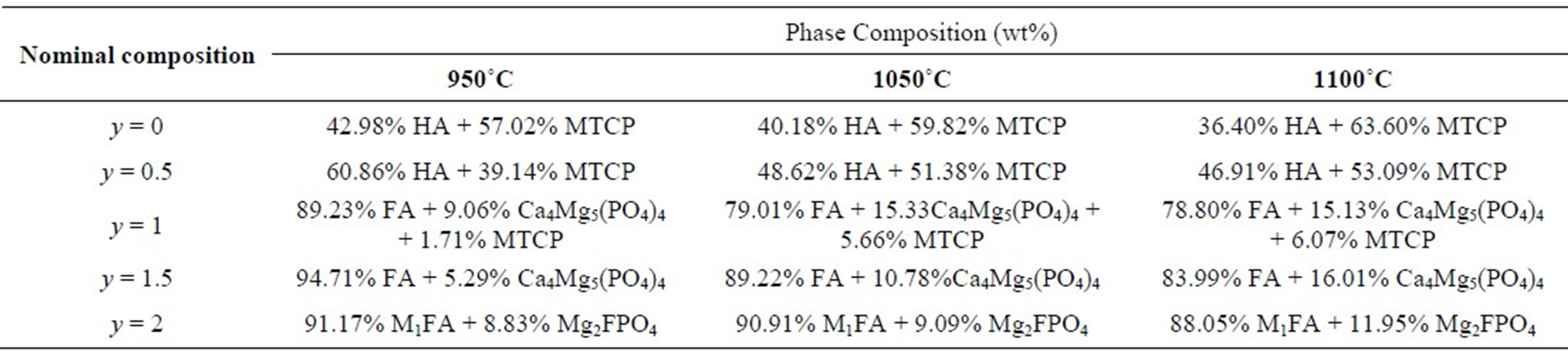
stabilizes the b-TCP phase at temperatures above 1300˚C [18]. For MHF1A, the XRD analysis revealed in addition to b-MTCP the stanfieldite (Ca4Mg5(PO4)6) (JCPDS #11-0231), while for MHF1.5A, this latter phase was the only secondary phase, which was detected. In the case of MFA, the secondary phase was the wagnerite, Mg2FPO4 (JCPDS #00-074-1236). With the increasing of the sintering temperature, there was no variation in the nature of the formed secondary phases. The phase quantification performed by XRD Rietveld analysis using the Fullprof program [41] showed that the amount of the secondary phases increased with the ascent of the sintering temperature (Table 4). On the other hand, at a given temperature, as expected, the decomposition degree decreased as the samples became rich in fluorine. One will keep in mind that this decomposition would affect the calculated relative densities, which were determined taking into account the theoretical densities of MHFyA.
Figure 7 shows the SEM micrographs of the fractured surfaces of the samples sintered at 1000˚C, 1100˚C and 1250˚C as the composition becomes rich in fluorine. According to the density curves, the microstructures of MHA and MHF0.5A samples were different from those of
Table 4. Phase composition of sintered samples (XRD data).
 (a)
(a)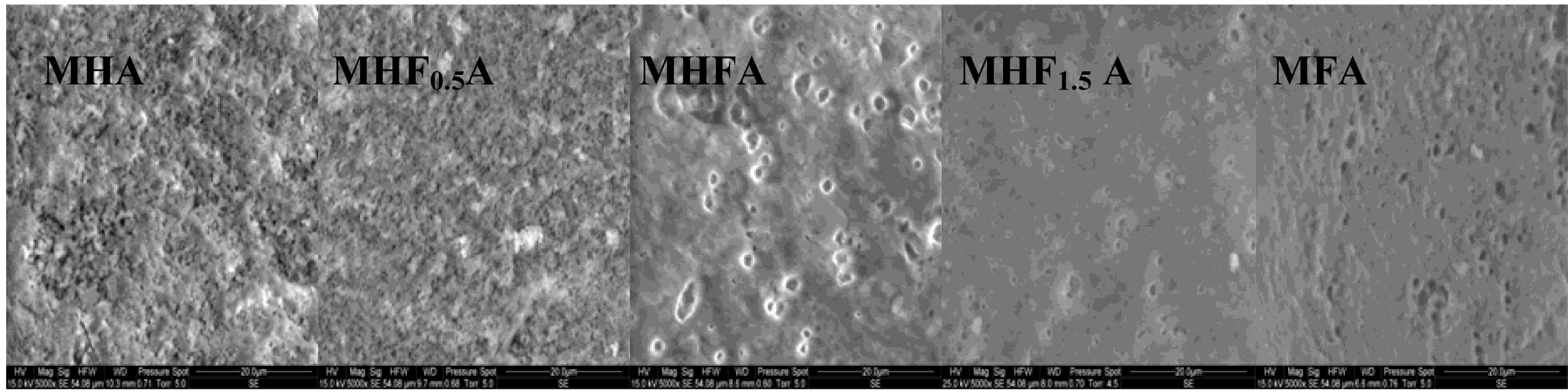 (b)
(b)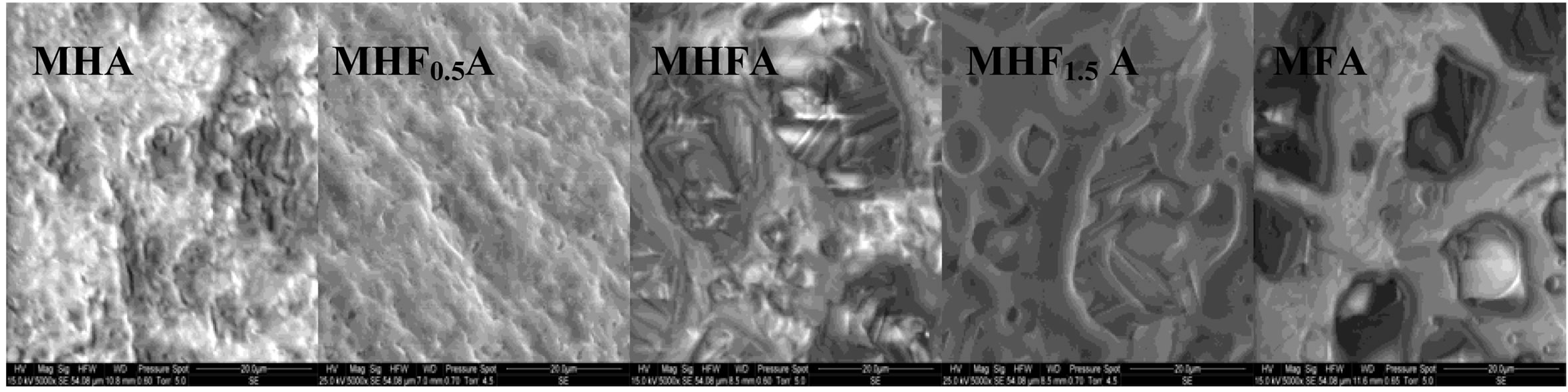 (c)
(c)
Figure 7. SEM micrographs of the fracture surfaces of the MHFyA (0 ≤ y ≤ 2) samples sintered for 1 h at: (a) 1000˚C; (b) 1100˚C and (c) 1250˚C.
the samples with y > 0.5. At 1000˚C, MHA and MHF0.5A were not much sintered. The microstructures were constituted by small particles with an apparent diameter of 0.9 ± 0.09 and 0.7 ± 0.04 mm, respectively (Figure 7(a)). As fluorine content increased (y > 0.5), the samples were better densified, but there were still many pores. The densification was accompanied by a coarsening of the grains. For MHF1A and MHF1.5A, the grain’s size was of about 2 ± 0.09 and 1.7 ± 1 µm, respectively (Figure 7(a)). At 1100˚C (Figure 7(b)), the microstructures of MHA and MHF0.5A showed little change compared to 1000˚C, a slight coarsening occurred but not enough to induce the densification. However, a strong change was observed with the samples richer in fluorine (Figure 7(b)). The pores in the samples were subsequently reduced. This is consistent with the measured values for the densities. At 1250˚C (Figure 7(c)), the difference in the microstructure is more conspicuous: the microstructure of MHF1A, MHF1.5A and MFA samples, which suggested that a liquid phase was formed during sintering showed many large pores in which there are needle-like crystals. The shrinkage curves of these samples (not shown here) exhibited an expansion at about 1100˚C. This expansion was due to the formation of a liquid phase resulting from an eutectic between apatite and fluorite, which was probably present in the as-synthesized powder as impurity [42-44]. As fluorine content was increased, the amount of the liquid phase seemed to increase (Figure 7(c), MFA), causing the distortion of the samples. As shown from Figure 7(c), the microstructure of MHF0.5A sample appears well-densified, only few pores appear at the triple grain boundaries. With respect to MHF0.5A, the MHA sample was less densified, its microstructure was crossed by numerous cracks and presented more pores (Figure 7(c)).
According to the above findings, Mg destabilized hydroxyapatite. However, fluorine content seems to play an important role in the densification of the samples. For MHA and MHF0.5A, as the microstructural observations did not reveal the formation of a liquid phase, the sintering was governed by a solid state diffusion mechanism. This explains the high sintering temperatures needed to achieve a high density. The decomposition of the apatite is also another reason to delay the densification. Nevertheless, at a given temperature, the decomposition became less important with the increase of the fluorine content. Compared to MHA, MHF0.5A exhibited a lower sinterability in spite of a larger SSA of the powder (Table 3) and a lower decomposition (Table 4). A high SSA will increase the driving force for densification during sintering and lead to a high sinterability of the materials. Furthermore, the mobility of fluoride ions, higher than that of hydroxide ions, should improve the densification. But in the present case, the low sinterability of MHF0.5A should be attributed to the interactions between these two latter species, which limited, especially at moderate temperatures, their mobility. This is corroborated by the grain’s size, which was smaller than that of MHA sintered at a temperature below 1200˚C. The absence of the drop in density at 1200˚C for the y = 0.5 sample can be related to the influence of the temperature, which became much larger than the effect of the interactions between OH− and F− on the diffusion of these latter species.
For the samples richer in fluorine, two mechanisms are responsible for the sintering. Before the formation of the liquid phase, i.e., below 1100˚C, the densification was due to the volume and/or grain boundary diffusions. Compared to MHA and MHF0.5A, the densification of the y > 0.5 samples was larger. It seems, therefore, that the effect of the interactions between OH− and F− occurred only for y = 0.5. The reason for this remains unclear. Indeed, for MHF1A and MHF1.5A, these interactions should exist, and their maximum effect should be observed for MHF1A [35]. As these interactions are no longer a limiting step, the improvement of the densification for the samples with y > 0.5 can be explained, in the absence of the liquid phase, by the great mobility of F− following its substitution for OH−, leading to a slight grain coarsening and a better densification of the samples. Notice that the maximum interaction at y = 0.5 between OH− and F− could be related to the presence of Mg, especially as this atom substitutes preferentially the calcium lining the tunnel containing the OH− and F− species [20].
After the formation of the liquid phase, the densification would be governed by the dissolution-diffusionreprecipitation process. Usually, a liquid phase formed by using appropriate sintering additives, is employed to promote densification, especially in covalent materials. In the present study, the liquid phase was probably due, as indicated above, to the eutectic between the apatite and fluorite, this latter was not detected by XRD because of its low amount. The magnesium might also promote the liquid phase formation. At a moderate temperature (1100˚C) and an average content of fluorine, the amount of liquid phase was low. It is just enough to improve the densification (Figure 4(a)). In contrast, at high temperatures and high fluorine contents, the quantity of liquid phase became important, and then a process of dissolution-diffusion-reprecipitation took place (Figure 7(c)). After the dissolution of the apatite up to saturation, facilitated probably by its high solubility, there was a crystallization of the needle-like crystals, which hindered the densification. Such a process was observed for magnesium-substituted fluorapatite sintered with additives [44].
3.4. Mechanical Properties of Sintered Samples
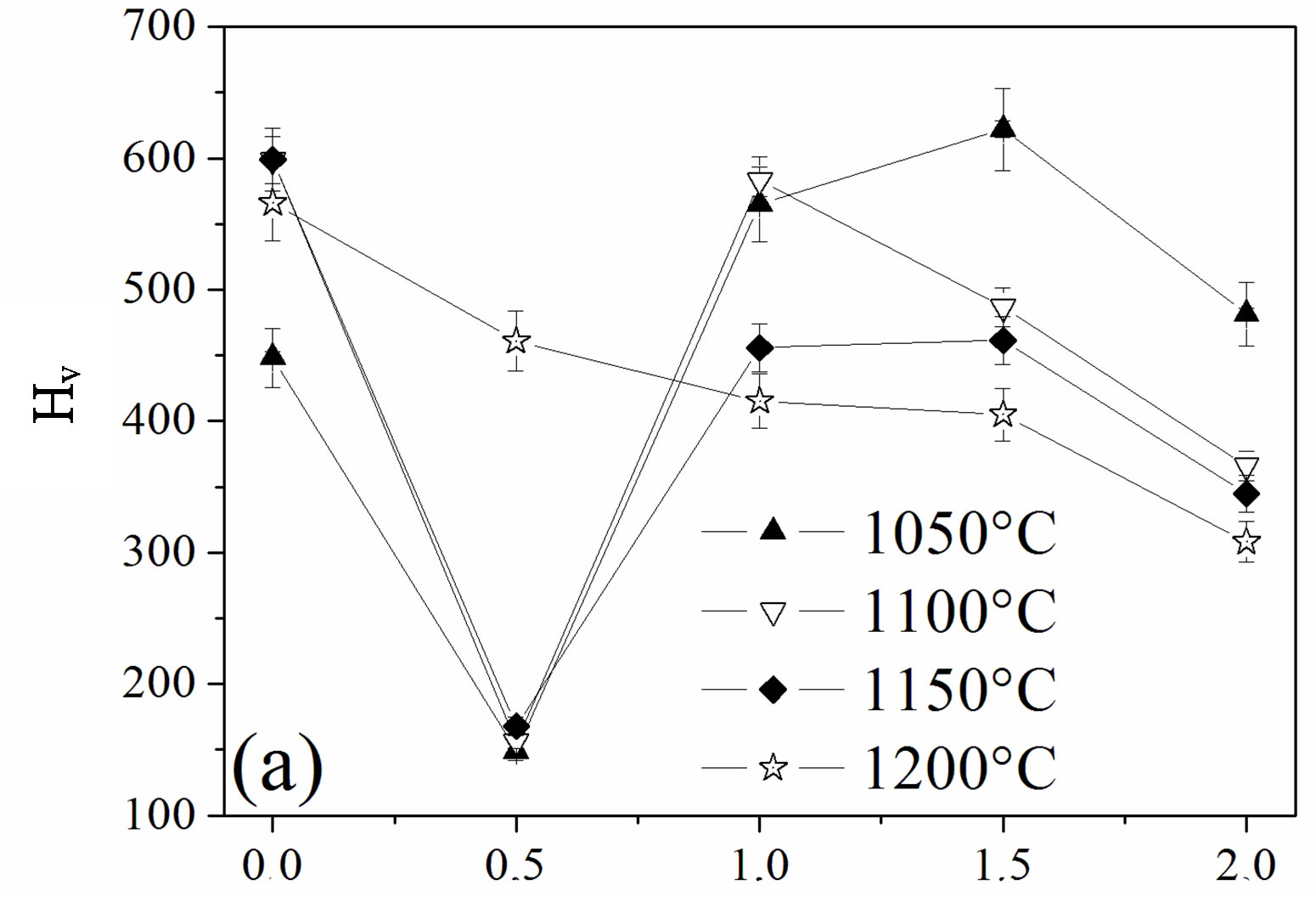
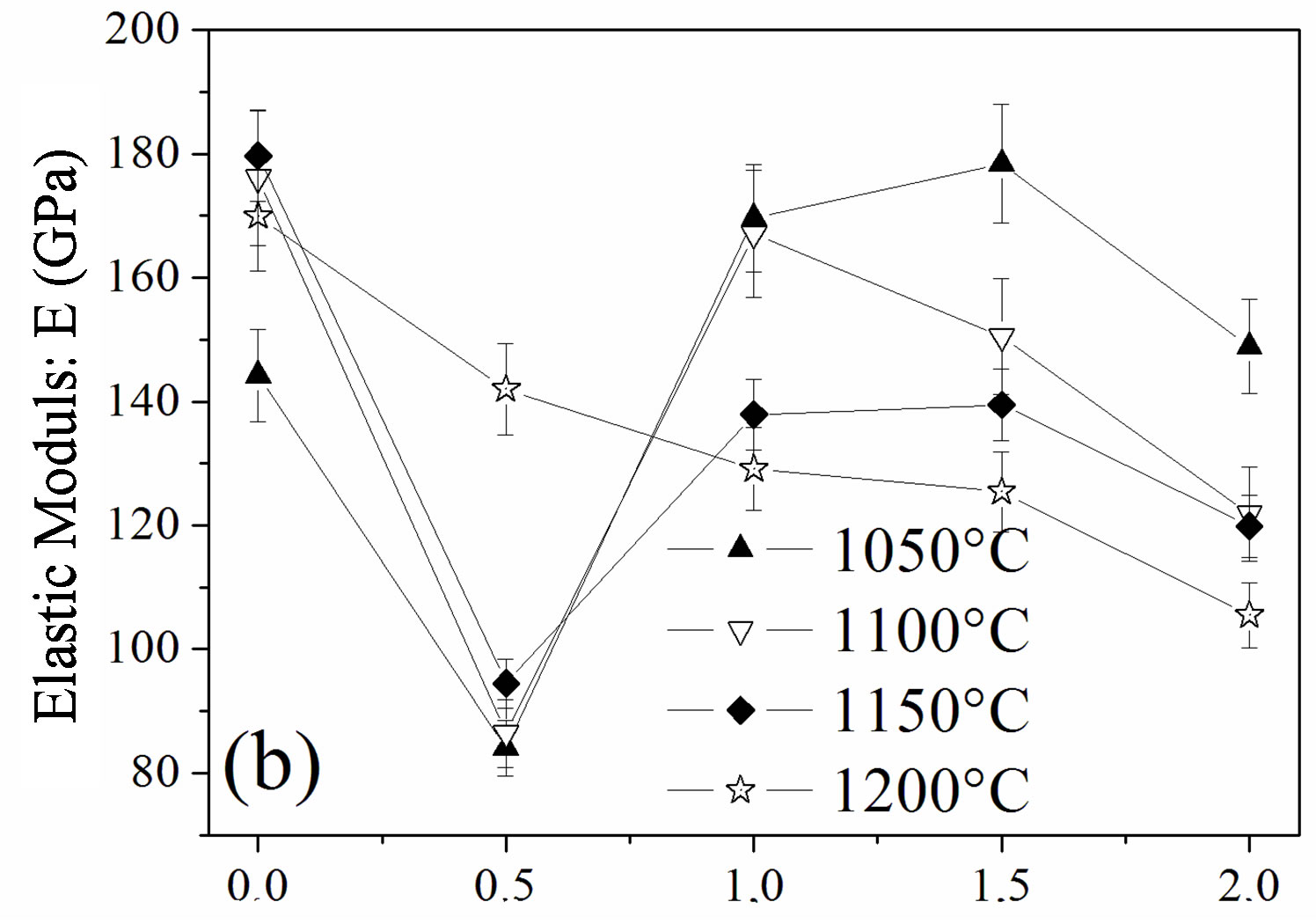
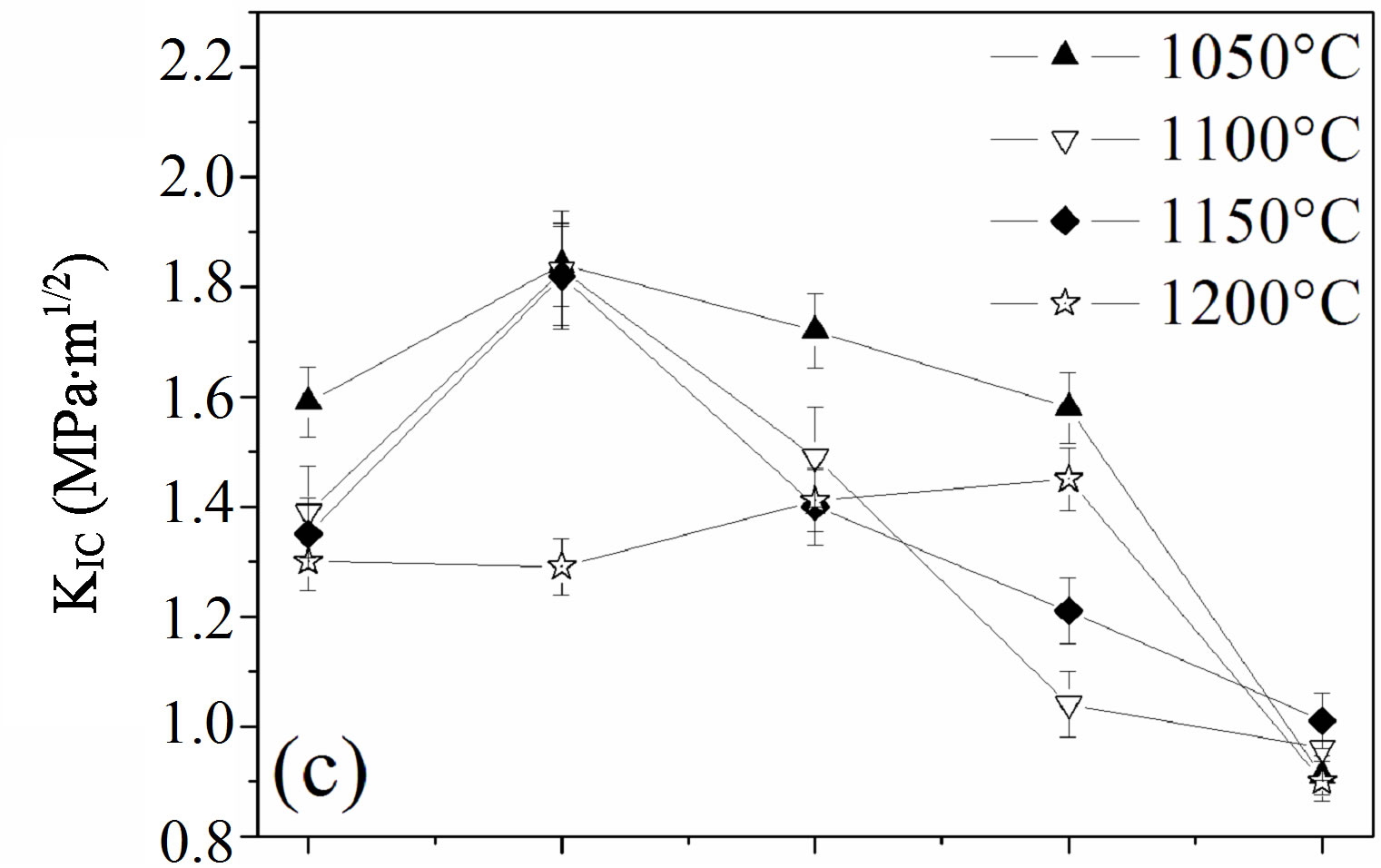
The influence of the fluorine substitution on the mechanical properties of the magnesium-substituted hydroxyapatites sintered in the temperature range 1050˚C - 1200˚C was determined by measuring the hardness, elastic modulus and fracture toughness (Figure 8). As shown from Figures 8(a) and (b), the hardness and elastic modulus’ curves presented the same shape. For the MHA sample, both properties increased with the temperature up to 1150˚C to attain their maximum values of 599 ± 9 Hv and 179.7 ± 1 GPa, respectively. If the value of the hardness is comparable with previous works, the value of the elastic modulus is much higher [45-47]. Usually, the hardness of ceramic materials is sensitive to the porosity and grain size [48,49]. Therefore, the increase with the temperature up 1150˚C is consecutive to the reduction of the porosity, and the decrease at 1200˚C although the density was higher would be related to the grain size, which was much higher than that of samples sintered at 1150˚C. As expected, between 1050˚C and 1150˚C, both properties were severely deteriorated for a fluorine content of y = 0.5, in good agreement with the density and SEM results, which indicated that, for this composition, the samples were poorly densified. The achieved hardness and elastic modulus were only of ~166 ± 4, and ~94.5 ± 3 GPa, respectively. In the case of the y > 0.5 samples, the highest values for both properties were observed for the y = 1.5 sample sintered at 1050˚C, 622 ± 4 and 181 ± 1 GPa, respectively, while when this sample was sintered at 1100˚C, the hardness and elastic modulus values were much lower, 486 ± 6 and 150.5 ± 1 GPa, respectively. The values of the microhardness and elastic modulus higher at 1050˚C than at 1100˚C despite the better densification of the samples at this latter temperature can be deemed as the consequence of the microstructure, which is fine and homogeneous while the lower values at 1100˚C reflected, in contradiction with a higher density of the sample, the change of the microstructure, which was constituted by large grain sizes induced by the high temperature and the liquid phase. Furthermore, it can be seen from Figures 8(a) and (b) that
y(F)
y(F)
0.0 0.5 1.0 1.5 2.0 y(F)
Figure 8. Mechanical properties of the samples sintered at different temperatures: (a) hardness; (b) elastic modulus and (c) fracture toughness.
compared to all the other compositions, MFA displayed lower values for both mechanical properties for any sintering temperature, except for 1050˚C, for which the density was the highest. At this temperature, there was no formation of the liquid phase.
The effect of the fluorine substitution on the fracture toughness, KIC, of MHFyA solid solutions sintered between 1050˚C and 1200˚C, is presented in Figure 8(c). In the range 1050˚C - 1150˚C, in contrast to the hardness and elastic modulus, i.e., to the density, the fracture toughness increased to a maximum value for y = 0.5, then it decreased. At 1200˚C, the fracture toughness rose slightly and linearly as a function of fluorine content up to 1.5 and then decreased. The maximum value is of 1.45 ± 0.08 MPa·m1/2. It was much lower than 1.85 ± 0.06 MPa·m1/2 exhibited by the y = 0.5 sample sintered at 1050˚C: the more the temperature was low, the more the fracture toughness value was higher. Such a behavior was previously reported for the hydroxyfluorapatite solid solutions [40]. The increase of the facture toughness value with porosity can be explained, as it was suggested by this latter author, by the role of the pores, which acted as stoppers of the cracks. The increase of fluorine content caused the decrease fracture toughness whatever the sintering temperature was. Above 1050˚C, the decrement was due to the change of the microstructure following the formation of the liquid phase. There was a crystallization of the needle-like crystals and a coarsening of the enlarged grains. This induced a change of the fracture mechanism, which became transgranular (Figure 7). Moreover, these results show that the mechanical properties are more sensitive to the presence of the liquid phase than the decomposition of the apatite. After formation, the liquid phase induced the crystallization of new grains with abnormal sizes having a negative effect on the mechanical properties.
4. Conclusions
The aim of this work is to investigate the sintering and the mechanical properties of magnesium and fluorine co-substituted hydroxyapatites (Ca9Mg(PO4)6(OH)2−yFy, where y = 0, 0.5, 1, 1.5 and 2) prepared by the hydrothermal method. The sinterability, microstructure and mechanical properties depended tightly on fluorine content and temperature. In the temperature range 950˚C - 1250˚C, the sintering of the y = 0 and 0.5 samples, was governed by a solid state diffusion mechanism, while for the other samples, at T ³ 1100˚C a liquid phase was formed, whose amount increased with the increase of the sintering temperature and fluorine content. The liquid phase induced the crystallization of needle-like crystals, which hindered the densification.
A positive correlation was roughly observed between density, microhardness and elastic modulus, in contrast to the fracture toughness, which was inversely correlated to the density. The maximum values for the hardness and elastic modulus, 624 ± 9 and 192 ± 4 GPa, respectively were obtained for the y = 1.5 sample sintered at 1050˚C. However, the maximum fracture toughness value was obtained for the y = 0.5 sample whose density was the lowest.
5. Acknowledgements
The authors would like to thank T. Ben Amer for his useful discussions of the mechanical properties, and A. Hadroug for his help with English.
REFERENCES
- S. V. Dorozhkin, “Bioceramics of Calcium Orthophosphates,” Biomaterials, Vol. 31, No. 7, 2010, pp. 1465- 1485. doi:10.1016/j.biomaterials.2009.11.050
- E. Landi, S. Sprio, M. Sandri, G. Celotti and A. Tampieri, “Development of Sr and CO3 Co-Substituted Hydroxyapatites for Biomedical Applications,” Acta Biomaterialia, Vol. 4, No. 3, 2007, pp. 656-663. doi:10.1016/j.actbio.2007.10.010
- M. E. Fleet, X. Liu and X. Wang, “Magnesium Carbonate-Phosphate Glass,” Journal of Non-Crystalline Solids, Vol. 355, No. 52-54, 2009, pp. 2604-2609. doi:10.1016/j.jnoncrysol.2009.09.006
- R. Z. LeGeros, “Apatites in Biological Systems,” Progress in Cryatal Growth Characterization of Materials, Vol. 4, No. 1-2, 1981, pp. 1-45. doi:10.1016/0146-3535(81)90046-0
- C. Capuccini, P. Torricelli, F. Sima, E. Boanini, C. Ristoscu, B. Bracci, G. Socol, M. Fini, I. N. Mihailescu and A. Bigi, “Strontium-Substituted Hydroxyapatite Coatings Synthesized by Pulsed-Laser Deposition: In Vitro Osteoblast and Osteoclast Response,” Acta Biomaterialia, Vol. 4, No. 6, 2008, pp. 1885-1893. doi:10.1016/j.actbio.2008.05.005
- B. Bracci, P. Torricelli, S. Panzavolta, E. Boanini, R. Giardino and A. Bigi, “Effect of Mg2+, Sr2+, and Mn2+ on the Chemico-Physical and in Vitro Biological Properties of Calcium Phosphate Biomimetic Coatings,” Journal of Inorganic Biochemistry, Vol. 103, No. 12, 2009, pp. 1666-1674. doi:10.1016/j.jinorgbio.2009.09.009
- J. F. McClendon, W. C. Foster and N. V. Ludwick, “Prevention of Dental Caries by Brushing the Teeth with Powders Containing Fluorapatite,” Journal of Dental Research, Vol. 26, No. 3, 1947, pp. 233-239. doi:10.1177/00220345470260030601
- M. Sundfeldt, M. Widmark, A. Wennerberg, J. Kärrholm, C. B. Johansson and L. V. Carlsson, “Does Sodium Fluoride in Bone Cement Affect Implant Fixation? Part I: Bone Tissue Response, Implant Fixation and Histology in Nine Rabbits,” Journal of Materials Science: Materials in Medicine, Vol. 13, No. 11, 2002, pp. 1037-1043. doi:10.1023/A:1020336404407
- M. Sundfeldt, J. Persson, J. Swanpalmer, A. Wennerberg, J. Kärrholm, C. B. Johansson and L. V. Carlsson, “Does Sodium Fluoride in Bone Cement Affect Implant Fixation Part II: Evaluation of the Effect of Sodium Fluoride Additions to Acrylic Bone Cement and the Fixation of Titanium Implants in Ovariectomized Rabbits,” Journal of Materials Science: Materials in Medicine, Vol. 13, No. 11, 2002, pp. 1045-1050. doi:10.1023/A:1020340521246
- H. Qu and M. Wei, “The Effect of Fluoride Contents in Fluoridated Hydroxyapatite on Osteoblast Behavior,” Acta Biomaterialia, Vol. 2, No. 1, 2006, pp. 113-119. doi:10.1016/j.actbio.2005.09.003
- K. A. Bhadang, C. A. Holding, H. Thissen, K. M. McLean, J. S. Forsythe and D. R. Haynes, “Biological Responses of Human Osteoblasts and Osteoclasts to FlameSprayed Coatings of Hydroxyapatite and Fluorapatite Blends,” Acta Biomaterialia, Vol. 6, No. 4, 2010, pp. 1575-1583. doi:10.1016/j.actbio.2009.10.029
- S. M. Barinov, S. V. Tumanov, I. V. Fadeeva and V. Yu. Bibikov, “Environment Effect on the Strength of Hydroxyand Fluorohydroxyapatite Ceramics,” Journal of Inorganic Materials, Vol. 39, No. 8, 2003, pp. 877-880. doi:10.1023/A:1025041817017
- P. Spencer, C. Barnes, J. Martini, R. Garcia, C. Elliott and R. Doremus, “Magnesium Distribution in Human Bone,” Archives of Oral Biology, Vol. 34, No. 10, 1989, pp. 767-771. doi:10.1016/0003-9969(89)90026-5
- L. N. Y. Wu, B. R. Genge and R. E. Wuthier, “Differential Effects of Zinc and Magnesium Ions on Mineralization Activity of Phosphatidylserine Calcium Phosphate Complexes,” Journal of Inorganic Biochemistry, Vol. 103, No. 7, 2009, pp. 948-962. doi:10.1016/j.jinorgbio.2009.04.004
- R. K. Rude, “Cause of Heterogenous Disease in Humans,” Journal of Bone and Mineral Research, Vol. 13, No. 4, 1998, pp. 749-758. doi:10.1359/jbmr.1998.13.4.749
- E. Landi, A. Tampieri, M. Mattioli-Belmonte, G. Celotti, M. Sandri, A. Gigante, P. Fava and G. Biagini, “Biomimetic Mgand Mg,CO3-Substituted Hydroxyapatites: Synthesis Characterization and in Vitro Behaviour,” Journal of European Ceramic Society, Vol. 26, No. 13, 2006, pp. 2593-2601. doi:10.1016/j.jeurceramsoc.2005.06.040
- G. Qi, S. Zhang, K. A. Khor, S. W. Lye, X. Zeng, W. Weng, C. Liu, S. S. Venkatraman and L. L. Ma, “Osteoblastic Cell Response on Magnesium-Incorporated Apatite Coatings,” Applied Surface Science, Vol. 255, No. 2, 2008, pp. 304-307. doi:10.1016/j.apsusc.2008.06.106
- S. Kannan, I. A. F. Lemos, J. H. G. Rocha and J. M. F. Ferreira, “Synthesis and Characterization of Magnesium Substituted Biphasic Mixtures of Controlled Hydroxyapatite/β-Tricalcium Phosphate Ratios,” Journal of Solid State Chemistry, Vol. 178, No. 10, 2005, pp. 3190-3196. doi:10.1016/j.jssc.2005.08.003
- W. L. Suchanek, K. Byrappa, P. Shuk, R. E. Riman, V. F. Janas and K. S. TenHuisen, “Preparation of MagnesiumSubstituted Hydroxyapatite Powders by the Mechanochemical-Hydrothermal Method,” Biomaterials, Vol. 25, No. 19, 2004, pp. 4647-4657. doi:10.1016/j.biomaterials.2003.12.008
- A. Bigi, G. Falini, E. Foresti, A. Ripamonti, M. Gazzano and N. Roveri, “Magnesium Influence on Hydroxyapatite Crystallization,” Journal of Inorganic Biochemistry, Vol. 49, No. 1, 1993, pp. 69-78. doi:10.1016/0162-0134(93)80049-F
- M. Okazaki, “Crystallographic Properties of Heterogeneous Mg-Containing Fluoridated Apatites Synthesized with a Two-Step Suphly System,” Biomaterials, Vol. 16, No. 9, 1995, pp. 703-707. doi:10.1016/0142-9612(95)99698-L
- Y. Cai, S. Zhang, X. Zeng, Y. Wang, M. Qian and W. Weng, “Improvement of Bioactivity with Magnesium and Fluorine Ions Incorporated Hydroxyapatite Coatings via Sol-Gel Deposition on Ti6Al4V Alloys,” Thin Solid Films, Vol. 517, No. 17, 2009, pp. 5347-5351. doi:10.1016/j.tsf.2009.03.071
- A. Gee and V. R. Deitz, “Determination of Phosphates by Differential Spectrometric,” Annales de Chemie, Vol. 25, No. 9, 1953, pp. 1320-1324. doi:10.1021/ac60081a006
- G. Charlot, “Méthodes de la Chimie Analytique: Analyse Quantitative Minérale,” Masson, Paris, 1966, p. 551.
- ASTM, “E384-99: A Standard Test Method for Microindentation Hardness of Materials,” ASTM, Philadelphia, 1984.
- D. B. Marshall, T. Noma and A. G. Evans, “A Simple Method for Determining Elastic-Modulus to Hardness Ratios Using Knoop Indentation Methods,” Journal of the American Ceramic Society, Vol. 65, No. 10, 1982, pp. C175-C176. doi:10.1111/j.1151-2916.1982.tb10357.x
- Z. Li, A. Ghosh, A. S. Kobayashi and R. C. Bradt, “Indentation Fracture Toughness of Sintered Silicon Carbide in the Palmqvist Crack Regimes,” Journal of the American Ceramic Society, Vol. 72, No. 6, 1989, pp. 904-911. doi:10.1111/j.1151-2916.1989.tb06242.x
- J. Gong, J. Wang and Z. Guan, “A Comparison between Knoop and Vickers Hardness of Silicon Nitride Ceramics,” Material Letter, Vol. 56, No. 6, 2002, pp. 941-944. doi:10.1016/S0167-577X(02)00641-9
- R. D. Shannon, “Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides,” Acta Crystallographica, Vol. A32, 1976, pp. 751-767.
- M. J. Larsen and S. J. Jensen, “Solubility, Unit Cell Dimensions and Crystallinity of Fluoridated Human Dental Enamel,” Archives of Oral Biology, Vol. 34, No. 12, 1989, pp. 969-973. doi:10.1016/0003-9969(89)90054-X
- M. Okazaki, “Fluoridated Hydroxyapatites Synthesized with Organic Phosphate Ester,” Biomaterials, Vol. 12, No. 1, 1991, pp. 46-49. doi:10.1016/0142-9612(91)90131-S
- L. M. Rodríguez-Lorenzo, J. N. Hart and K. A. Gross, “Influence of Fluorine in the Synthesis of Apatites. Synthesis of Solid Solutions of Hydroxy-Fluorapatite,” Biomaterials, Vol. 24, No. 21, 2003, pp. 3777-3785. doi:10.1016/S0142-9612(03)00259-X
- Y. Chen and X. Miao, “Thermal and Chemical Stability of Fluorohydroxyapatite Ceramics with Different Fluorine Contents,” Biomaterials, Vol. 26, No. 11, 2005, pp. 1205-1210. doi:10.1016/j.biomaterials.2004.04.027
- P. Hartmann, C. Jäger, St. Barth, J. Vogel and K. Meyer, “Solid State NMR, X-Ray Diffraction, and Infrared Characterization of Local Structure in Heat-Treated Oxyhydroxyapatite Microcrystals: An Analog of the Thermal Decomposition of Hydroxyapatite during Plasma-Spray Procedure,” Journal of Solid State Chemistry, Vol. 160, No. 2, 2001, pp. 460-468. doi:10.1006/jssc.2001.9274
- N. Senamaud, D. Bemache-Assollant, E. Champion, M. Heughebaert and C. Rey, “Calcination and Sintering of Hydroxyfluorapatite Powders,” Solid State Ionics, Vol. 101-103, 1997, pp. 1357-1362. doi:10.1016/S0167-2738(97)00242-7
- L. M. Rodríguez-Lorenzo, J. N. Hart and K. A. Gross, “Influence of Fluorine in the Synthesis of Apatites. Synthesis of Solid Solutions of Hydroxy-Fluorapatite,” Biomaterials, Vol. 24, No. 21, 2003, pp. 3777-3785. doi:10.1016/S0142-9612(03)00259-X
- K. A. Gross and L. M. Rodríguez-Lorenzo, “Sintered Hydroxyfluorapatites. Part I: Sintering Ability of Precipitated Solid Solution Powders,” Biomaterials, Vol. 25, No. 7-8, 2004, pp. 1375-1384. doi:10.1016/S0142-9612(03)00565-9
- M. Hidouri, K. Bouzouita, F. Kooli and I. Khattech, “Thermal Behaviour of Magnesium-Containing Fluorapatite,” Materials Chemistry and Physics, Vol. 80, No. 2, 2003, pp. 496-505. doi:10.1016/S0254-0584(02)00553-9
- J. Marchi, A. C. S. Dantas, P. Greil, J. C. Bressiani, A. H. A. Bressiani and F. A. Müller, “Influence of Mg-Substitution on the Physicochemical Properties of Calcium Phosphate Powders,” Materials Research Bulletin, Vol. 42, No. 6, 2007, pp. 1040-1050. doi:10.1016/j.materresbull.2006.09.015
- I. Cacciotti, A. Bianco, M. Lombardi and L. Montanaro, “Mg-Substituted Hydroxyapatite Nanopowders: Synthesis, Thermal Stability and Sintering Behavior,” Journal of European Ceramic Society, Vol. 29, No. 14, 2009, pp. 2969-2978. doi:10.1016/j.jeurceramsoc.2009.04.038
- J. Rodríguez-Carvajal, “A Program for Rietveld Refinement and Pattern Matching Analysis,” Physica, Vol. B192, No. 1-2, 1993, pp. 55-69. doi:10.1016/0921-4526(93)90108-I
- E. D. Franz, “Fluorapatit Ca5F(PO4)3: Ein Modell Zur Synthese der Zahnhartsubstanz im System CaF2-Ca3(PO4)2,” Z Naturforschung, Vol. 38b, 1983, pp. 1037-1040.
- F. Ben Ayed, J. Bouaziz and K. Bouzouita, “Pressureless Sintering of Fuorapatite under Oxygen Atmosphere,” Journal of European Ceramic Society, Vol. 20, No. 8, 2000, pp. 1069-1076. doi:10.1016/S0955-2219(99)00272-1
- M. Hidouri, K. Bouzouita and N. Fattah, “Effect of Additives on the Densification and the Microstructure of Magnesium-Containing Fluorapatite,” Annales de Chimie: Science de Matériaux, Vol. 30, 2005, pp. 133-138. doi:10.3166/acsm.30.133-148
- K. A. Gross and K. A. Bhadang, “Sintered Hydroxyfluorapatites. Part II: Mechanical Properties of Solid Solutions Determined by Microindentation,” Biomaterials, Vol. 25, No. 7-8, 2004, pp. 1385-1394. doi:10.1016/S0142-9612(03)00636-7
- M. Hidouri, K. Boughzala, J. P. Lecompte and K. Bouzouita, “Sintering and Mechanical Properties of Magnesium-Containing Fluorapatite,” Comptes Rendus Physique, Vol. 10, No. 2-3, 2009, pp. 242-248. doi:10.1016/j.crhy.2009.04.001
- H. Eslami, M. Solati-Hashjin and M. Tahriri, “The Comparison of Powder Characteristics and Physicochemical, Mechanical and Biological Properties between Nanostructure Ceramics of Hydroxyapatite and Fluoridated Hydroxyapatite,” Materials Science and Engineering, Vol. Vol. C29, No. 4, 2009, pp. 1387-1398. doi:10.1016/j.msec.2008.10.033
- D. J. Clinton and R. Morrell, “Hardness Testing of Ceramic Materials,” Materiel Chemistry and Physics, Vol. 17, No. 5, 1987, pp. 461-473. doi:10.1016/0254-0584(87)90096-4
- R. W. Rice, C. C. Wu and F. Borchelt, “HardnessGrain-Size Relations in Ceramics,” Journal of the American Ceramic Society, Vol. 77, No. 10, 1994, pp. 2539- 2553. doi:10.1111/j.1151-2916.1994.tb04641.x
NOTES
*Corresponding author.