Advances in Microbiology
Vol.05 No.02(2015), Article ID:53908,9 pages
10.4236/aim.2015.52009
Factors Affecting the Induction of Lignin Peroxidase in Manganese-Deficient Cultures of the White Rot Fungus Phanerochaete chrysosporium
Avi Matityahu1,2, Aya Sitruk1, Yitzhak Hadar3, Paula A. Belinky1*
1Tel Hai Academic College, Kiryat Shmona, Israel
2Faculty of Medicine in the Galilee, Bar-Ilan University, Safed, Israel
3Department of Plant Pathology and Microbiology, The Robert H. Smith Faculty of Agriculture, Food and Environment, The Hebrew University of Jerusalem, Rehovot, Israel
Email: *paudabe@gmail.com
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 19 January 2015; accepted 6 February 2015; published 10 February 2015
ABSTRACT
The lignin peroxidase (LIP) production and regulation, in manganese ions (Mn2+) deficient cultures of the white rot fungus Phanerochaete chrysosporium, is still not clearly understood. Mn2+ deficiency is correlated to low levels of manganese containing superoxide dismutase (MnSOD). In this work, we show that despite the low activity level of MnSOD in Mn2+-deficient cultures, the presence of H2O2 is essential for the expression of the lip-H2 gene, which encodes for the major LIP isoenzyme produced (LIP-H2). Thus, the H2O2 present in Mn2+-deficient cultures is probably produced by other mechanisms rather than dismutation of superoxide ions by MnSOD. Glyoxal oxidase gene (glox) expression was significantly higher than MnSOD (MnSOD1) and cellobiose dehydrogenase (cdh1) expression in Mn2+-deficient cultures, indicating its clear involvement in H2O2 production in those cultures. Glyoxal oxidase may compensate the absence of MnSOD activity in Mn2+-deficient cultures. The high levels of reactive oxygen species (ROS) needed for the enhancement of LIP expression in Mn2+-deficient cultures were not directly correlated to the protein kinase C (PKC) activity involved in signal transduction pathway. High level of oxidative stress was observed in MnSOD silenced mutants, grown in the presence of Mn2+, indicating that oxidative stress in Mn2+-deficient cultures was caused by low levels of MnSOD rather than the deficiency in Mn2+. The results of this work can further contribute to the understanding of LIP regulation in Mn2+-deficient cultures.
Keywords:
ROS, GLOX, MnSOD, PKC, lip-H2 Induction

1. Introduction
Lignin is an amorphous and insoluble polymer lacking stereoregularity, which plays a key role in the carbon cycle as the most abundant aromatic compound and as a protective matrix surrounding the cellulose microfibrils of plant cell walls [1] [2] . Fungi collectively referred to as white rot basidiomycetes are the only microbes capable of efficiently depolymerizing and mineralizing this recalcitrant polymer [3] [4] . The most intensively studied white rot fungus, Phanerochaete chrysosporium, secretes an array of peroxidases that act via the generation of aromatic free radicals, which undergo spontaneous cleavage reactions. Two major families of hydrogen peroxide (H2O2)-requiring extracellular heme-peroxidases designated lignin peroxidase (LIP) and manganese-dependent peroxidase (MNP) were identified [5] . The non-specific nature and exceptional oxidation potential of the LIP have attracted considerable interest in organopollutants degradation and fiber bleaching. The regulation of the gene families encoding extracellular peroxidases is poorly understood, but it is clear that oxidative stress is a key factor [6] - [8] .
LIP expression requires nutrient or carbon starvation, signaled by a rise in intracellular cAMP concentrations [9] and concomitant pulses of pure oxygen gas in the headspace. Starving cultures without excessive oxygen are not enough for triggering LIP expression, indicating that both effects need to be simultaneous [10] - [15] . The effect of such elevated oxygen concentrations on LIP expression can be reproduced by depleting manganese ions (Mn2+) from cultures, in the presence of atmospheric air [7] [8] . Development of a putative oxidative stress is detected in LIP-producing cultures of P. chrysosporium, either oxygenated or Mn2+-deficient. This was evidenced by measuring high levels of reactive oxygen species (ROS) with specific molecular probes, and was further confirmed by the enhancement of the antioxidant defense system and by the high degree of oxidative damage of major macromolecules, lipids, protein and DNA. In oxygenated cultures, increased expression of manganese superoxide dismutase (MnSOD) was the major response of the antioxidant system. In contrast, in Mn2+- deficient cultures, negligible activity of MnSOD was detected [6] [7] .
The existence of oxidative stress in LIP-producing cultures led to the hypothesis that ROS, and hydroxyl radical (OH•), in particular, may act as intracellular messengers, triggering lip-H2 expression (the major LIP enzyme expressed) in liquid cultures of P. chrysosporium, through signal transduction [6] [7] . In a previous work, we found that addition of a OH• scavenger, dimethylsulfoxide (DMSO), to the cultures, completely abolished LIP transcription (both mRNA and heme-protein were undetectable), indicating that these ROS, coupled to high levels of cAMP, were indeed involved in the induction of lip-H2 expression. This effect was confirmed by in-situ generation of OH• via the addition of Fenton reagents (OH• producers), which significantly enhanced lip-H2 expression [6] [7] [16] .
Significantly lower protein kinase C (PKC) activity in oxygenated vs. aerated (grown with free exchange of atmospheric air) low-LIP producing cultures was measured [16] . In oxygenated cultures, inactivation of PKC activity by calphostin C, staurosporin A or H7 (PKC activity inhibitors) caused significant elevation in lip-H2 and MnSOD1 expression. Stimulation of PKC by phorbol 12-myristate 13-acetate (PMA) caused the reverse effect [16] .
Significantly low levels of lip-H2 expression were detected when superoxide anion (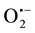 ), H2O2 or OH• was scavenged by 4,5-dihydroxy-1,3-benzene disulfonic acid (tiron), pyruvate or DMSO, respectively, whether the level of PKC was normal, stimulated or inactivated [16] . In addition, in-situ generation of OH•, via addition of Fenton reagents to aerated cultures, reduced pkc expression. In contrast, OH• scavenging stimulated pkc expression [16] . It was suggested that due to high OH• levels, fungal cells activated a complex defensive system which regulated the levels of Fenton components by repressing PKC and stimulating LIP, MnSOD1 and catalase [16] .
), H2O2 or OH• was scavenged by 4,5-dihydroxy-1,3-benzene disulfonic acid (tiron), pyruvate or DMSO, respectively, whether the level of PKC was normal, stimulated or inactivated [16] . In addition, in-situ generation of OH•, via addition of Fenton reagents to aerated cultures, reduced pkc expression. In contrast, OH• scavenging stimulated pkc expression [16] . It was suggested that due to high OH• levels, fungal cells activated a complex defensive system which regulated the levels of Fenton components by repressing PKC and stimulating LIP, MnSOD1 and catalase [16] .
Since the difference between both types of LIP-producing cultures seems to partially relay on inactivation or lack of MnSOD activity, it is possible that ROS production occurs by different mechanisms. MnSOD product, H2O2, might be produced by glyoxal oxidase (GLOX) or cellobiose dehydrogenase (CDH) present in the fungus [17] in Mn2+-deficient cultures. To clarify if deficiency of Mn2+ provokes the generation of high ROS concentrations only via the lack in MnSOD activity, MnSOD silenced mutants (MSC) of P. chrysosporium were previously prepared by using siRNA technique [18] . Significantly lower MnSOD expression, at both the mRNA and protein activity levels, was detected in MSC mutants [18] .
In this work, we try to explain how Mn2+ deficiency in cultures of P. chrysosporium causes enhancement of lip-H2 expression in relation to oxidative stress. The results of this work can contribute to the overall understanding of LIP regulation in white rot fungi.
2. Materials and Methods
2.1. Reagents
Veratryl alcohol, phenylmethanesulfonyl fluoride (PMSF), pyruvate, staurosporin A, calphostin C, H7, leupeptin, aprotinin, ethylenediaminetetraacetic acid (EDTA), N,N,N',N'-tetramethylethylenediamine (TEMED), 4,5-dihy- droxy-1,3-benzene disulfonic acid (Tiron), phosphinothricin (PPT), riboflavin 5,5'-dithiobis-2-nitrobenzoic acid (DTNB), 2',7'-dichlorodihydrofluorescein (DCF) and Tri Reagent were purchased from Sigma (St. Louis, MO). PepTag non-radioactive kit was purchased from Promega (Madison, WI). Reverse-IT™ 1st Strand Synthesis kit and Syber Green were purchased from Abgene (Epsom, UK).
2.2. Fungal strain andculture conditions
The filamentous fungus P. chrysosporium Burds BKM-F-1767 (ATCC 24725) was used for this study. The fungus was maintained at 4˚C on 2% (wt/vol) malt extract agar slants and inoculated by the method of Tien and Kirk [19] . The growth medium was prepared as previously described [8] [20] with initial concentrations of glucose, diammonium tartrate and MnSO4・H2O of 56, 2.4 and 0.225 mM , respectively. Two different cultures of the fungus were studied: aerated cultures: The fungus was grown in submerged liquid cultures (90 ml) at 175 rpm at 37˚C in 250-ml flasks with free exchange of atmospheric air (flasks sealed with dense-paper plugs). Mn2+-deficient cultures: The fungus was grown at the same culture conditions as above, but with depletion of Mn2+. Unless otherwise specified, the cultures were incubated for 5 days.
MSC mutants preparation was described in a previous work using siRNA silencing technique [18] . The MSC mutants were grown in oxygenated cultures, containing 0.225 mM MnSO4・H2O. The MSC mutants were grown in submerged liquid cultures (90 ml) at 175 rpm at 37˚C in 250-ml flasks sealed with rubber stoppers, and the headspace was flushed twice a day with O2 for 2 min at a flow rate of 1 l/min. Oxygen gas was of medical-grade purity.
2.3. Measurement of enzyme activities
LIP activity was measured in the extracellular medium of the fungus culture. PKC and catalase activities were measured in protein extracts. For preparation of protein extracts, fungal pellets, harvested after 120 h, were separated from extracellular fluid by filtration through cheesecloth, and the biomass was frozen at −80˚C. Biomass samples were ground with a mortar and pestle in the presence of liquid nitrogen. The powder was then suspended in 0.5 ml of cold extraction buffer, 25 mM Tris-HCl (pH 7.4), 0.5 mM EDTA, 0.5 mM EGTA, 0.05% Triton X-100, 10 mM β-mercaptoethanol, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 0.5 mM PMSF and 200 mM NaCl. The lysates were centrifuged at 14,000 ´ g for 5 min at 4˚C.
2.4. LIP Activity
LIP activity was measured according to the method of Tien and Kirk [19] . The oxidation of veratryl alcohol to veratryl aldehyde was recorded at 310 nm for 40 s, with a unit being defined as 1 µmol of veratryl alcohol oxidized to veratryl aldehyde per minute.
2.5. PKC Activity
Samples of protein extracts (5 μg) from the different experimental conditions tested were assayed for PKC activity using the PepTag non-radioactive kit (Promega) according to the manufacturer’s directions. This kit uses the brightly colored fluorescent peptide substrate PLSRTLSVAAK. The hot pink color is imparted by a dye molecule conjugated to the substrate. The reaction was performed for 30 min at 30˚C. The non-phosphorylated peptide substrate was then separated from the phosphorylated substrate by electrophoresis on a 0.8% agarose gel, according to their migration to the anode and cathode, respectively. The gels were photographed on a transilluminator. PKC activity was estimated by the fluorescence intensity of the band corresponding to the non-phos- phorylated substrate. More intensive band was corresponding to low PKC activity. The densities of the areas of activity were measured and compared by using TINA program software (Raytest Isotopenmessgeräte GmbH).
2.6. Catalase activity
Catalase activity was assayed by measuring the degradation of H2O2. The rate of disappearance of H2O2 was monitored at an absorbance of 240 nm [21] . One unit of catalase decomposed 1 mol of H2O2 in 1 min (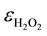 = 39.4 M−1 cm−1).
= 39.4 M−1 cm−1).
2.7. Influence of H2O2 on theinduction of lip-H2 expression
To determine the effect of H2O2, on lip-H2 expression, the fungus was grown under the conditions described above, with or without the addition of a H2O2 scavenger, pyruvate, at different concentrations (0.1 - 5 mM ), at 72 h, and at 96 h. lip-H2 expression from cultures incubated with or without pyruvate was measured as described in Section 2.7. H2O2 levels in 120 h liquid cultures of P. chrysosporium were detected by adding 2',7'-dichlorodihydrofluorescein (a fluorescent indicator of peroxide). This compound was added to the cultures at a final concentration of 30 μM and incubated for 30 min at 37˚C and 250 rpm. The mycelia were then harvested, washed with double-distilled water, and treated with 2 ml of 5% 5-sulfosalicylic acid (vol/vol) for 20 min at 4˚C and a cellular extract was obtained. The treated mycelia were centrifuged at 20,000 ´ g for 15 min at 4˚C. The production of 2',7'-dichlorofluorescein was measured in the extracellular medium and in the cellular extract with a spectrofluorimeter (Victor3, PerkingElmer, USA). To detect 2',7'-dichlorofluorescein, the extinction and emission wavelengths were 501 and 521 nm.
2.8. Influence of PKC on the induction of lip-H2 expression
To determine the effect of PKC on lip-H2 expression, the fungus was grown for 120 h under the conditions described above, then PKC inhibitors were added, or not, for 2 h. The PKC inhibitors staurosporin A, calphostin C or H7 were added at concentrations of 40 ng/ml, 122 ng/ml or 5 µg/ml, respectively. lip-H2 expression from cultures incubated with or without PKC inhibitors were measured as described below.
2.9. Measurement of glox, MnSOD1, cdh1 and lip-H2 Expression by real-Time PCR
Frozen biomass was ground with a mortar and pestle in the presence of liquid nitrogen. Total RNA was extracted from each sample using Tri Reagent (Sigma). Total cDNA was generated by reverse transcription using the Reverse-IT™ 1st Strand Synthesis kit (ABgene, Epsom, UK). The amount of each gene in relation to 18S rRNA transcript was determined by real-time PCR, based on the high-affinity double-stranded DNA-binding dye Syber Green (Absolute™ QPCR SYBER® Green ROX mix, ABgene, Epsom, UK). Each reaction was performed in triplicate in a spectrofluorometric thermal cycler (Rotor-Gene™ 3000, Corbett Research, Sydney, Australia), using the primers AM42, AM43 for 18S rRNA, AM63, AM64 for MnSOD1, AM44, AM45 for lip-H2, AM83, AM84 for glox and AS114, AS115 for cdh1 (Table 1). The real-time PCR program included a 15-min polymerase activation step at 95˚C followed by up to 45 cycles of 15 s at 95˚C, 20 s at the optimal annealing temperature (60˚C) and 25 s at 72˚C. Assay specificity was confirmed by subjecting the PCR products to SYBER Green I melting curves. The efficiency of real-time amplification was determined by running a standard curve with serial dilution of cDNA and defined as E = [10(?1/m)] ? 1 (m = slope of reaction). Optimal melting points for 18S rRNA, MnSOD1, lip-H2, glox and cdh1 were 84˚C, 93˚C, 91˚C, 89˚C, 90.5˚C and 84˚C, respectively. The reaction efficiencies for 18S rRNA, MnSOD1, lip-H2, glox and cdh1 were 93% (R2 = 0.999), 80% (R2 = 0.994), 85% (R2 = 0.997), 97% (R2 = 0.998), 99% (R2 = 0.999) and 84% (R2 = 0.991) respectively.
2.10. Determination of oxidative damage to Macromolecules
2.10.1. Measurements of oxidative damage
Oxidative damage was determined by detecting the levels of oxidized lipids with LPO-586 kit (Oxis Research)

Table 1. Primers used in this study.
following the manufacturer’s directions. This kit detects a chromogen that absorbs at 586 nm and is formed by the reaction between malondialdehyde (MDA), an end product of the peroxidation of polyunsaturated fatty acids, and N-methyl-2-phenylindole.
2.10.2. Reduced glutathione:oxidized Glutathione (GSH:GSSG) Ratio
The GSH/GSSG molar ratio was determined with a Glutathione assay kit (Sigma) according to the manufacturer’s directions, with slight modifications. This method employs 5,5'-dithiobis-2-nitrobenzoic acid (DTNB), which reacts with GSH to form 2-nitro-5-thiobenzoate anion (TNB2−), a product which can be detected spectrophotometrically at 412 nm with an extinction coefficient of 14,150 M−1 cm−1. The concentration of GSH was calculated from a calibration curve (0 - 3 M GSH). The assay was conducted on two parallel sets of samples. One set, preincubated with glutathione reductase, gave total glutathione (reduced and oxidized) as GSH. The second set, used for GSSG determination, was first treated with the thiol-scavenging reagent 1-methyl-2-vinyl- pyridinium trifluoromethanesufonate to scavenge GSH. The remaining GSSG was then reduced to GSH with NADPH catalyzed by glutathione reductase and the formed GSH was measured. GSSG at concentrations from 0 to 1.5 M was used as the standard to calibrate the curves. The GSH/GSSG ratio was calculated as follows: GSH/GSSG = [GSH − (2GSSG)]/GSSG.
3. Results
3.1. Thelow level of MnSOD, rather than the Deficiency in Mn2+, Causes oxidative Stress in Mn2+-deficient cultures
To understand whether the oxidative stress in Mn2+-deficient cultures, needed for LIP expression [7] [8] , is caused by the deficiency of Mn2+ or by low MnSOD activity or a combination of them, MnSOD silenced mutants (MSC), prepared as previously reported in [18] , were used. The MSC mutants were grown in the presence of Mn2+ and molecular oxygen. Significantly higher level of oxidative stress was confirmed in MnSOD silenced mutant (MSC1) in comparison to MnSOD non-silenced mutant (Bar5), by measuring lower GSH/GSSG ratio and higher level of oxidized lipids, in spite of the presence of Mn2+ in the medium. The total GSH levels measured in BAR5 and MSC1 were 142 nM and 270 nM, respectively. The GSH/GSSG ratios measured in BAR5 and MSC1 were 1.85 and 1.6, respectively (Figure 1(a), Figure 1(c)). Lipid oxidation was determined by measuring MDA levels in MSC mutants. MDA level in MSC1 was 2.5 times higher than in control BAR5 (Figure 1(d)). Catalase activity was analyzed in protein extracts prepared from the MSC mutants’ biomasses. While in BAR5 catalase activity was high (3574 U/mg protein), in MSC1 catalase activity was 1930 U/mg protein (Figure 1(b)).
3.2. H2O2 Is involved in triggering of lip-H2 Expression
As was reported before, P. chrysosporium produce LIP under oxidative stress [6] [7] . Scavenging more molecules of H2O2 by increasing concentrations of pyruvate in Mn2+-deficient cultures of P. chrysosporium resulted in lower lip-H2 expression (Figure 2), indicating the essential dependence role of those molecules in the production of the enzyme by the fungus. At 2 mM pyruvate approximately 70% of H2O2 were scavenged (data not shown).

Figure 1. Measurement of the oxidative stress level in MSC mutants. Total GSH concen- trations (a) and GSH/GSSG ratio (c) in extracts, prepared from the MSC mutants’ biomasses, were determined by using the Glutathione assay kit (Sigma). The reaction was based on monitoring the production of 5-thio-2-nitrobenzoic acid (TNB2−) at 412 nm. The values are means ± standard deviations of 3 replicates. Lipid damage in MSC mutants (d) was dete- rmined in cell extracts of the mutants, grown in oxygenated cultures, by measuring MDA with the LPO-586 kit (OxisResearch). The means ± SD (error bars) of six replicates are shown. Catalase activity (b) was determined in MSC mutants protein extracts by following the degradation of H2O2 at 240 nm. The means ± SD (error bars) of six replicates are shown.

Figure 2. Influence of H2O2 scavenging on lip-H2 transcription in Mn2+ deficiency cultures of P. chrysosporium. Hydrogen peroxide was scavenged by increased concentrations (0 - 5 mM) of pyruvate. The expression of lip-H2 transcripts relative to 18S rRNA gene transcript was measured by real-time PCR. The means ± SD (error bars) of six replicates are shown.
3.3. H2O2 Production in Mn2+-Deficient cultures
Since one of the consequences of Mn2+ deficiency in P. chrysosporium cultures are low levels of MnSOD activity [7] [8] , H2O2 needed for LIP expression should be produced by alternative generators besides MnSOD.
In Mn2+-deficient cultures most of H2O2 molecules involved in lip-H2 expression are probably produced by glyoxal oxidase, which was detected as the predominant peroxide producer enzyme expressed in those cultures (Figure 3). CDH level was higher than MnSOD. All the enzymes were similarly expressed in aerated cultures (Figure 3).
3.4. Role of PKC activity in Mn2+-Deficient Cultures
High production of ROS in Mn2+-deficient cultures may affect lip-H2 expression by affecting signal transduction pathways enzymes, such as PKC. As presented in Figure 4, PKC activity was significantly lower in Mn2+- deficient cultures, indicating that for LIP production under high levels of ROS, PKC activity should be low. This was confirmed by inhibition of PKC by different inhibitors in Mn2+-deficient cultures, which provoked the enhancement of lip-H2 expression (Figure 5).
The PKC inactivation by staurosporin A, calphostin C or H7 caused a significant elevation in lip-H2 expression, up to 4-fold increase in Mn2+-deficient cultures. The effect of PKC inhibition on lip-H2 expression in aerated cultures was minimal (Figure 5).

Figure 3. Expression of H2O2 producer enzymes in Mn2+-deficient high LIP- producing cultures in comparison to aerated low LIP-producing cultures. The expression of MnSOD1, glox and cdh1 transcripts relative to 18S rRNA gene transcript was measured by real-time PCR. The means ± SD (error bars) of six replicates are shown.

Figure 4. PKC activity in aerated and Mn2+-deficient cultures. Protein sam- ples (5 µg) were assayed for PKC activity using the PepTag non-radioactive kit (Promega). The reaction was performed for 30 min at 30˚C. The non- phosphorylated peptide substrate was then separated from the phosphorylated substrate by electrophoresis on 0.8% agarose gel. PKC activity was estimated by the fluorescence intensity of the band corresponding to the non-phos- phorylated substrate. Intense band represent low activity of PKC.

Figure 5. Influence of PKC inhibition on lip-H2 transcription in cell extracts of Mn2+-deficient cultures. The PKC inhibitors (40 ng/ml staurosporin A, 122 ng/ml calphostin C or 5 µg/ml H7) were added to 120-h-old aerated and Mn2+- deficient high LIP-producing cultures for 2 h. The expression of lip-H2 tran- scripts relative to 18S rRNA gene transcript was measured by real-time PCR. The means ± SD (error bars) of six replicates are shown.
4. Discussion
It was previously shown that scavenging of
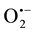 and H2O2 by tiron and pyruvate, respectively, caused a significant reduction in lip-H2 expression in oxygenated cultures, indicating that the presence of
and H2O2 by tiron and pyruvate, respectively, caused a significant reduction in lip-H2 expression in oxygenated cultures, indicating that the presence of
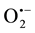 and H2O2 is essential for the induction of lip-H2 expression, probably as precursors for the formation of the OH• needed to trigger lip-H2 expression [6] [7] [16] . In this work we show that the presence of H2O2 is essential for lip-H2 expression, in Mn2+-deficient cultures, in a dose dependent manner. Despite the low level of MnSOD, the major enzyme responsible for H2O2 production, in Mn2+-deficient cultures in comparison to oxygenated and aerated cultures [6] [7] , there is a sufficient amount of H2O2, needed for lip-H2 expression. The H2O2 present in Mn2+- deficient cultures might be produced by different mechanisms generating less quantity of H2O2 than MnSOD mechanism. P. chrysosporium produce a variety of oxidases that are capable of generating H2O2, among them are glyoxal oxidase, glucose oxidase, veratryl alcohol oxidase and methanol oxidase [2] [17] . These enzymes may compensate the absence of MnSOD activity in Mn2+-deficient cultures. In this work we show a significantly higher glox expression in high-LIP-producing cultures than low-LIP-producing cultures, indicating a compensation of the deficiency in MnSOD.
and H2O2 is essential for the induction of lip-H2 expression, probably as precursors for the formation of the OH• needed to trigger lip-H2 expression [6] [7] [16] . In this work we show that the presence of H2O2 is essential for lip-H2 expression, in Mn2+-deficient cultures, in a dose dependent manner. Despite the low level of MnSOD, the major enzyme responsible for H2O2 production, in Mn2+-deficient cultures in comparison to oxygenated and aerated cultures [6] [7] , there is a sufficient amount of H2O2, needed for lip-H2 expression. The H2O2 present in Mn2+- deficient cultures might be produced by different mechanisms generating less quantity of H2O2 than MnSOD mechanism. P. chrysosporium produce a variety of oxidases that are capable of generating H2O2, among them are glyoxal oxidase, glucose oxidase, veratryl alcohol oxidase and methanol oxidase [2] [17] . These enzymes may compensate the absence of MnSOD activity in Mn2+-deficient cultures. In this work we show a significantly higher glox expression in high-LIP-producing cultures than low-LIP-producing cultures, indicating a compensation of the deficiency in MnSOD.
Recently, an inverse correlation between ROS/LIP and PKC activities was reported [16] . Significantly low levels of PKC were obtained in oxygenated cultures (containing high ROS levels) relative to aerated cultures (containing low ROS levels). Inhibition or activation of PKC activity in oxygenated cultures was accompanied by a further increase or decrease in lip-H2 expression, respectively. Inhibition of PKC activity in oxygenated cultures also caused an increase in MnSOD1 expression [16] . Here we show that the same correlation between ROS/LIP and PKC activities exist in Mn2+-deficient cultures. Significantly lower PKC activity was measured in Mn2+-deficient cultures vs. aerated cultures. Inactivation of PKC activity by staurosporin A, calphostin C or H7 caused significant elevation in lip-H2, once again indicating that enhancement of LIP production by high levels of ROS is not directly connected with the signal transduction pathway enzyme, PKC, as the direct relationship usually reported for PKC and ROS for other biologic systems [22] [23] .
To further understanding the role of Mn2+ deficiency in oxidative stress produced in Mn2+-deficient cultures, MSC mutants previously prepared by using RNAi [18] were used. The MSC mutants have slower growth rate, different MnSOD activity level influenced the fungus growth. Addition of tiron or H2O2 to the medium improved its growth rate. The MSC mutants exhibit an unusual hyphal growth (data not shown). MSC1 hyphae are bulbous and thicker at the tip of the hyphae, MSC10 exhibit a highly branched phenotype. Low temperature abolishe this unusual phenotype (data not shown). The involvement of MnSOD in hyphal elongation and branching is known [24] .
Mutants having high and low activity of MnSOD [18] were grown in oxygenated conditions in the presence of Mn2+. In those conditions mutants having low activity of MnSOD were subjected to a more severe oxidative stress in comparison to mutants having high activity of MnSOD, which was shown by higher total GSH level, lower GSH/GSSG ratio, higher lipid oxidation and lower level of catalase activity.
Results obtained with BAR5 (non MnSOD silenced mutants) are comparable to that reported in the past for the highest LIP-producing oxygenated cultures [6] . The MnSOD silenced mutants (MSC) might simulate the fungus in cultures grown under Mn2+ deficiency, both having low levels of MnSOD activity. MnSOD silenced mutant which was grown under oxygenated cultures conditions used for wild type, was subjected to severe oxidative stress, despite of the presence of Mn2+. The oxidative stress was even more severe in comparison to wild type oxygenated and Mn2+-deficient cultures [6] [7] due to the direct oxygen flushing to the cultures not containing MnSOD. In aerated cultures, in which MnSOD activity was higher than in Mn2+-deficient cultures due to the presence of Mn2+, a moderate oxidative stress was detected and less LIP was produced [7] . Thus, the lowering of MnSOD by excluding Mn2+, produce an effect similar to that obtained in oxygenated cultures.
5. Conclusions
In conclusion, the exclusion of Mn2+ from the culture medium of the fungus probably causes the lowering of MnSOD activity, which in consequence provokes oxidative stress similar to the obtained by oxygen flushing to the medium, needed for the induction of lip-H2 expression and LIP enzyme production.
The oxidative stress mechanism in Mn2+-deficient cultures influences LIP gene in the same way as was showed for oxygenated cultures. Probably, the high levels of ROS, preferentially OH•, generated by reduction of H2O2, mainly catalyzed by GLOX, inhibit PKC activity as a regulatory strategy in the cells, inducing LIP.
All the results obtained in this work can contribute to the understanding of LIP regulation in P. chysosporium.
Cite this paper
AviMatityahu,11,AyaSitruk,YitzhakHadar,Paula A.Belinky, (2015) Factors Affecting the Induction of Lignin Peroxidase in Manganese-Deficient Cultures of the White Rot Fungus Phanerochaete chrysosporium. Advances in Microbiology,05,83-92. doi: 10.4236/aim.2015.52009
References
- 1. de Jong, E. (1993) Physiological Roles and Metabolism of Fungal Aryl Alcohols. Wageningen Agricultural University, Wageningen.
- 2. Gold, M.H. and Alic, M. (1993) Molecular Biology of the Lignin-Degrading Basidiomycete Phanerochaete chrysosporium. Microbiological Reviews, 57, 605-622.
- 3. Cullen, D. (2002) Molecular Genetics of Lignin-Degrading Fungi and Their Applications in Organopollutant Degradation. In: The Mycota: A Comprehensive Treatise on Fungi as Experimental Systems for Basic and Applied Research, Vol. 11, Agricultural Applications, Kempken, Springer, Berlin, 71-90.
- 4. Kirk, T.K. and Farrell, R.L. (1987) Enzymatic “Combustion”: The Microbial Degradation of Lignin. Annual Review of Microbiology, 41, 465-505.
http://dx.doi.org/10.1146/annurev.mi.41.100187.002341 - 5. Tien, M. and Kirk, T.K. (1984) Lignin-Degrading Enzyme from Phanerochaete chrysosporium: Purification, Characterization, and Catalytic Properties of a Unique H2O2-Requiring Oxygenase. Proceedings of the National Academy of Sciences of the United States of America, 81, 2280-2284.
http://dx.doi.org/10.1073/pnas.81.8.2280 - 6. Belinky, P.A., et al. (2003) Reactive Oxygen Species and Induction of Lignin Peroxidase in Phanerochaete chrysosporium. Applied and Environmental Microbiology, 69, 6500-6506.
http://dx.doi.org/10.1128/AEM.69.11.6500-6506.2003 - 7. Belinky, P.A., Flikshtein, N. and Dosoretz, C.G. (2006) Induction of Lignin Peroxidase via Reactive Oxygen Species in Manganese-Deficient Cultures of Phanerochaete chrysosporium. Enzyme and Microbial Technology, 39, 222-228.
http://dx.doi.org/10.1016/j.enzmictec.2005.10.023 - 8. Rothschild, N., et al. (1999) Manganese Deficiency Can Replace High Oxygen Levels Needed for Lignin Peroxidase Formation by Phanerochaete chrysosporium. Applied and Environmental Microbiology, 65, 483-488.
- 9. Boominatan, K. and Reddy, C.A. (1992) cAMP-Mediated Differential Regulation of Lignin Peroxidase and Manganese-Dependent Peroxidase Production in the White-Rot Basidiomycete Phanerochaete chrysosporium. Proceedings of the National Academy of Sciences of the United States of America, 89, 5586-5590.
http://dx.doi.org/10.1073/pnas.89.12.5586 - 10. Bar-Lev, S.S. and Kirk, T.K. (1981) Effects of Molecular Oxygen on Lignin Degradation by Phanerochaete chrysosporium. Biochemical and Biophysical Research Communications, 99, 373-378.
http://dx.doi.org/10.1016/0006-291X(81)91755-1 - 11. Dosoretz, C.G., Chen, A.H.C. and Grethlein, H.E. (1990) Effect of Oxygenation Conditions on Submerged Cultures of Phanerochaete chrysosporium. Applied Microbiology and Biotechnology, 34, 131-137.
http://dx.doi.org/10.1007/BF00170937 - 12. Dosoretz, C.G., Rothschild, N. and Hadar, Y. (1993) Overproduction of Lignin Peroxidase by Phanerochaete chrysosporium (BKM-F-1767) under Nonlimiting Nutrient Conditions. Applied and Environmental Microbiology, 59, 1919-1926.
- 13. Dosoretz, C.G. and Grethlein, H.E. (1991) Physiological Aspects of the Regulation of Extracellular Enzymes of Phanerochaete chrysosporium. Applied Biochemistry and Biotechnology, 28-29, 253-265.
http://dx.doi.org/10.1007/BF02922605 - 14. Forney, L.J., Reddy, C.A., Tien, M. and Aust, S.D. (1982) The Involvement of Hydroxyl Radical Derived from Hydrogen Peroxide in Lignin Degradation by the White Rot Fungus Phanerochaete chrysosporium. Journal of Biological Chemistry, 257, 11455-11462.
- 15. Tien, M. and Tu, C.P. (1987) Cloning and Sequencing of a cDNA for a Ligninase from Phanerochaete chrysosporium. Nature, 326, 520-523.
http://dx.doi.org/10.1038/326520a0 - 16. Matityahu, A., Hadar, Y. and Belinky, P.A. (2010) Involvement of Protein Kinase C in Lignin Peroxidase Expression in Oxygenated Cultures of the White Rot Fungus Phanerochaete chrysosporium. Enzyme and Microbial Technology, 47, 59-63.
http://dx.doi.org/10.1016/j.enzmictec.2010.05.002 - 17. Cullen, D. and Kersten, P.J. (2004) Enzymology and Molecular Biology of Lignin Degradation. In: The Mycota III: Biochemistry and Molecular Biology, Springer-Verlag, Berlin, 249-273.
- 18. Matityahu, A., Hadar, Y., Dosoretz, C.G. and Belinky, P.A. (2008) Gene Silencing by RNA Interference in the White Rot Fungus Phanerochaete chrysosporium. Applied and Environmental Microbiology, 74, 5359-5365.
http://dx.doi.org/10.1128/AEM.02433-07 - 19. Tien, M. and Kirk, T.K. (1988) Lignin Peroxidase of Phanerochaete chrysosporium. Methods in Enzymology, 161, 238-249.
http://dx.doi.org/10.1016/0076-6879(88)61025-1 - 20. Rothschild, N., Hadar, Y. and Dosoretz, C.G. (1995) Ligninolytic System Formation by Phanerochaete chrysosporium in Air. Applied and Environmental Microbiology, 61, 1833-1838.
- 21. Lee, J.S., Hah, Y.C. and Roe, J.H. (1993) The Induction of Oxidative Enzymes in Streptomyces coelicolor upon Hydrogen Peroxide Treatment. Journal of General Microbiology, 139, 1013-1018.
http://dx.doi.org/10.1099/00221287-139-5-1013 - 22. Schmitz, H.P. and Heinisch, J.J. (2003) Evolution, Biochemistry and Genetics of Protein Kinase C in Fungi. Current Genetics, 43, 245-254.
http://dx.doi.org/10.1007/s00294-003-0403-6 - 23. Maher, P. (2001) How Protein Kinase C Activation Protects Nerve Cells from Oxidative Stress-Induced Cell Death. Journal of Neuroscience, 21, 2929-2938.
- 24. Georgiou, C.D., Patsoukis, N., Papapostolou, I. and Zervoudakis, G. (2006) Sclerotial Metamorphosis in Filamentous Fungi Is Induced by Oxidative Stress. Integrative and Comparative Biology, 46, 691-712.
http://dx.doi.org/10.1093/icb/icj034
NOTES

*Corresponding author.