Computational Molecular Bioscience
Vol. 2 No. 2 (2012) , Article ID: 20463 , 11 pages DOI:10.4236/cmb.2012.22004
A DFT/ECP-Small Basis Set Modelling of Cisplatin: Molecular Structure and Vibrational Spectrum
Acad. R. Tsanev Institute of Molecular Biology, Bulgarian Academy of Sciences, Sofia, Bulgaria
Email: dodoff@obzor.bio21.bas.bg
Received March 25, 2012; revised April 25, 2012; accepted May 2, 2012
Keywords: Cisplatin; DFT Calculations; Basis Set; Effective Core Potential; Molecular Structure; Vibrational Analysis
ABSTRACT
A DFT conformational and vibrational analysis of a single molecule of cisplatin (cis-[Pt(NH3)2Cl2]) was performed by means of PW91 functional and LANL08 ECP basis set for the Pt atom. 3-21G and 3-21G* Basis sets were used for the remaining atoms. All the initially chosen conformations were found to converge to the global minimum conformation of C2v symmetry with H atoms lying in the coordination plane and pointing to the Cl atoms. The computational results were compared with the newest experimental structural data and with the vibrational spectroscopic data for cisplatin, obtained by other workers. The chosen level of theory was found to describe satisfactory the molecular structure (r. m. s. of the relative deviations ≤ 6%) and the harmonic vibrational frequencies (r. m. s. of the relative deviations ≤ 5%) of cisplatin.
1. Introduction
Cisplatin (cis-diamminedichloroplatinum(II)) is the first inorganic compound introduced in clinical use for the treatment of cancer [1-4]. It is a prototype of several thousands platinum [5-12] and other metal [6,8,9,13-16] coordination compounds synthesized and tested so far in the search for novel cytostatic agents with improved therapeutic characteristics with respect to the parent compound. Among them, only five more Pt(II) complexes— carboplatin, oxaliplatin, nedaplatin, lobaplatin and heptaplatin—have gained international or local marketing approval [8,17]. At present, cisplatin remains amongst the most widely used platinum chemotherapeutics, with particular effectiveness in the treatment of testicular, ovarian and bladder cancer [9,18,19].
Quantum-chemical studies of the molecular and electronic structure, and prediction of spectroscopic characteristics of pharmacologically active compounds, have always been challenging for computational chemists. Moreover, such data are quite useful for better understanding the reactivity of the drugs with physiological target molecules. Cisplatin molecule has been studied at different levels of theory using the Effective Core Potential [20] (ECP) approximation: Hartree-Fock (HF) [21- 24], Møler-Plesset (MP) [22,23] and Density Functional Theory (DFT) [22-25], as well as by the all-electron DFT approach [26]. The small molecule of cisplatin serves as a useful example for testing the computational accuracy and effectiveness of different computational schemes in predicting molecular geometry and vibrational frequencies [22-25].
Although the vibrational spectra of cisplatin have been studied by many researchers [22-25,27-29], the assignment of some low-frequency bands is ambiguous. Curiously, for more than forty years, the only available experimental structure of cisplatin was that of Milburn and Truter of 1966 [30]. Last year, Weller et al. [31] published a re-examination of the crystal structure of cisplatin, and showed that it exists in two polymorphic modifications. They found, inter alia, that the values of the Pt-N bond lengths were underestimated in the earlier study. Moreover, Weller et al. firstly located the positions of the hydrogen atoms by neutron powder diffraction, thus revealing the orientation of the NH3 groups with respect to the coordination plane.
The appearance of the more detailed and accurate structural data prompted us to perform a DFT analysis of cisplatin molecule and to compare our computational results with the new experimental data, as well as with the computational results published earlier. There is no uniform opinion in the literature concerning the optimal combination of basis sets and density functional for reproducing the structural parameters and vibrational frequencies of cisplatin. Thus, Wysokiński and Michalska [22] claim the combination of mPW1PW functional [32] and LANL2DZ basis [33-36] (all atoms) to be among the best performing theory level for predicting both geometry optimization and vibrations; Amado et al. [24] also recommend mPW1PW functional for structural and vibrational calculations. Regarding the basis sets, the combination of LANL2DZ [36] (Pt), LANL2MB [35,37] (N and Cl) and 3-21G [38,39] (H) has given the best vibrational results, whereas LANL2DZ (Pt)/CEP-4G (N and Cl)/3-21G (H) has produced the best structural parameters. Finally, Gao et al. [25] have found the combinations LSDA functional [40]/SDD basis set [33,41] and PBE- 1PBE [42]/SDD to give the best results for structural and vibrational calculations, respectively.
Here we present our computational results on the geometry optimization and vibrational analysis of a single cisplatin molecule obtained with the Perdew-Wang exchange-correlation functional PW91 [43,44] and small basis sets for the Pt and the remaining atoms, and compare them with the new experimental data and with the computational results of other researchers.
2. Methods
The DFT calculations were performed on a personal computer (2.29 GHz, 2.96 GB RAM), as well as on a HPC Cluster Platform Express 7000 (36 blades BL 280 c, dual Intel Xeon X5560 at 2.8 GHz and 24 GB RAM per blade), using the Firefly quantum chemistry package [45], which is partially based on the GAMESS (US) [46] source code. All structures were optimized using the algorithm of direct inversion in the iterative subspace and gradient convergence tolerance of 1 × 10–5 Hartree/Bohr. The final values of the maximum and r. m. s. gradients were below 8 × 10–6 and 3 × 10–6 Hartree/Bohr, respectively. The optimized structures were further subjected to vibrational analysis at the same theory level, using numerical calculation of Hessian matrix elements (displacement size 1 × 10–3 Bohr). No negative eigenvalues were obtained, thus assuring that actual maxima on the potential energy surface were located. The Hessian matrix and the total energy distribution matrix were expressed in internal coordinates (the complete set of bond lengths, bond angles, torsion angles and out-of-plain angles). The vibrational modes were visualized by means of the Molekel programme [47]. The calculated vibrational wavenumbers were compared with the FT Raman data of Amado et al. [24] for solid cisplatin.
The PW91 functional [43,43] was used in the DFT calculations. The relativistic ECP and associated noncontracted basis set LANL08 [48] were used for Pt atom. The H and N atoms were described by the all electron split-valence 3-21G basis set [38,39]. For the description of the Cl atoms, the 3-21G basis set and the supplemented with d-type functions 3-21G* basis set [49] were applied. The ECP and the basis sets were taken from the EMSL Basis Set Exchange Library [50-52].
Initial conformations and their minimization. Four conformations (a, b, b– and c) differing in the torsion angles H-N-Pt-Cl around one of the N-Pt bonds were considered (Figure 1). Their combination in couples gives seven non-eqivalent and non-enantiomorphic conformations as depicted in Figure 2. Each of these starting conformations were subjected to DFT minimization and all of them were found to converge to the a,a comformer (C2v simmetry). The calculated molecular geometry was compared with the single crystal X-ray and neutron powder diffraction data of Weller et al. [31] for the two polymorphic modifications of cisplatin.
All calculations concern a single molecule in gas phase. Input and output files are deposited as Supplementary material.
3. Results and Discussion
3.1. Conformational Considerations
The initial structures of cisplatin (Figure 2) were minimized with the two protocols: PW91/LANL08/3-21G (A) and PW91/LANL08/3-21G* (B) (vide supra). All of
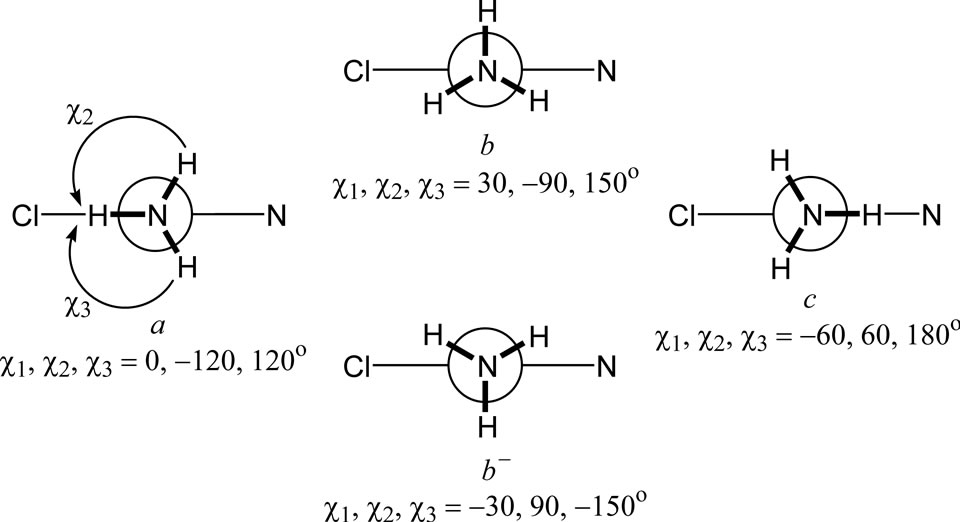
Figure 1. Newman projections along one of the N-Pt bonds for the conformations a, b, b- and c, of cisplatin differing in the torsion angles χ1, χ2 and χ3 (b and b- are enantiomorphic).
them converged to the a,a conformer (Figure 2), with energies (corrected with the vibrational zero-point energy) of –1147.5051 and –1147.9213 Hartree, respectively. Thus, within the given level of theory, we found a single minimum on the potential energy surface of the cisplatin molecule. Amado et al. [23,24] have pointed out that the number of local minima (1, 2 or 3) varies with the combination of density functional and basis sets. In all cases, however, including all electron basis set DFT calculations [26], the lowest energy conformation is of the a,a type.
Methods A and B give the same set of torsion angles around the N-Pt bonds (0, –117, 117). The calculated torsion angles are compared (Table 1) with the experimental data of Weller et al. [31] about the α- and β- polymorphs of cisplatin, the later containing two crystallographically non-equivalent molecules. As seen, in the solid state the torsion angles vary considerably, and no one conformation resembles enough the gas-phase minimumenergy conformation of a,a type. This is a consequence of the extended intermolecular H-bonding network of N-H×××Cl type [31]. Thus, it seems in the solid state the conformational preferences are governed by the H-bonding rather than by the torsion potential.
3.2. Bond Lengths and Bond Angles
The bond lengths and bond angles calculated by computational methods A and B are compared with the averaged structural data for the two crystal modifications of cisplatin [31]. The relative deviations of the individual structural parameters, as well as the arithmetic mean (s) and root mean square deviations (S) are summarized in Table 2. The accuracy obtained by the chosen theory levels is similar to or slightly better than that reported by Amado et al. [23,24]. Adding d-orbitals on the Cl atoms (method B) gives somewhat better overall description of the bond lengths and angles. The application of method
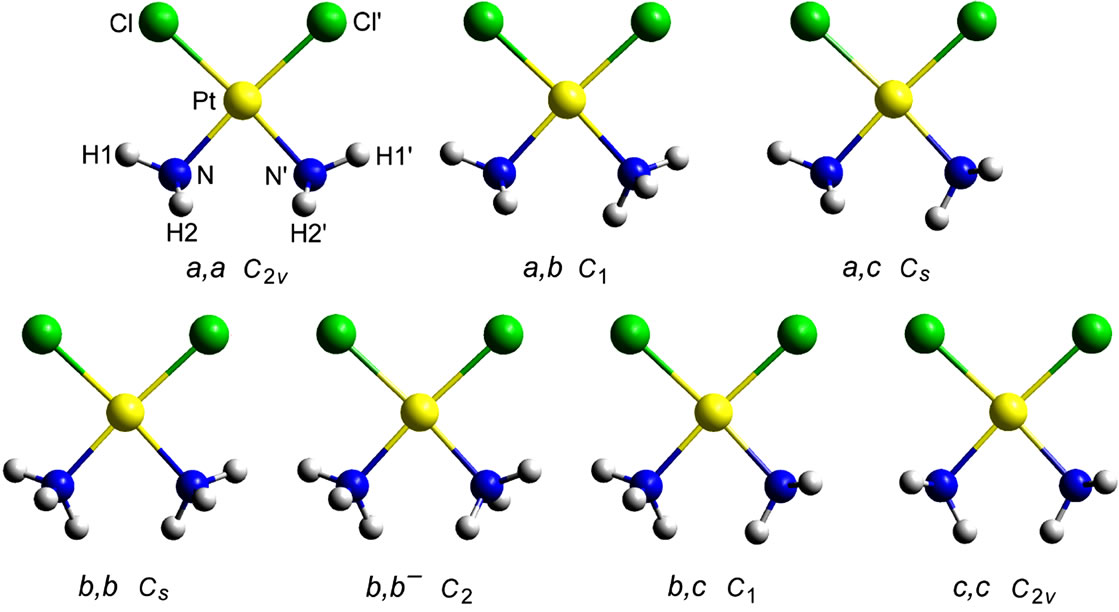
Figure 2. View of the seven non-equivalent and non-enantiomorphic conformations of cisplatin arising from the combination of the conformations a, b, b- and c in couples. Each of them was subjected to DFT minimization.
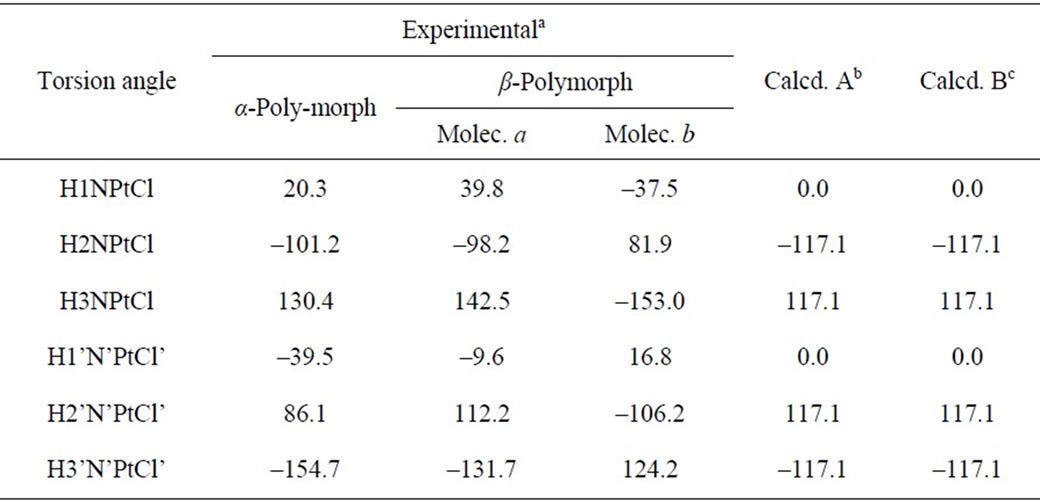
Table 1. Torsion angles (deg) for cisplatin.
aData from [31]; bValues calculated at PW91/LANL08/3-21G level; cValues calculated at PW91/LANL08/3- 21G* level.
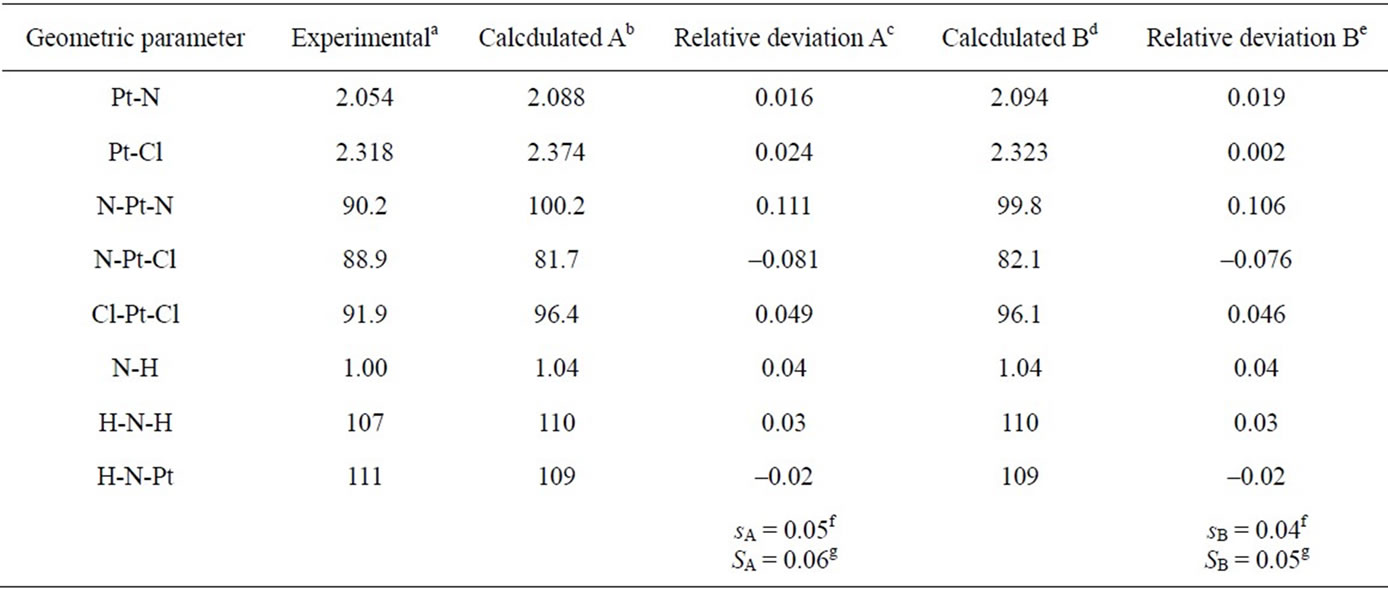
Table 2. Bond lengths (Å) and bond angles (deg) for cisplatin.
aAveraged data for α- and β-polymorphs of cisplatin [31]; bAveraged calculated values at PW91/LANL08/3-21G level (acldA); cRelative deviation (δaA) of the calculated (acldA) from the experimental (aexp) value: δaA = (acldA – aexp)/acldA; dAveraged calculated values at PW91/ LANL08/3-21G* level (acldB); eRelative deviation (δaB) of the calculated (acldB) from the experimental (aexp) value: δaB = (acldB – aexp)/acldB; fArithmetic mean of the relative deviations’ modules: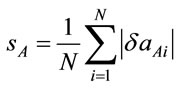 ,
, 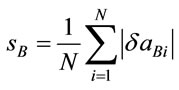 , N = number of geometric parametes; gRoot mean square of the relative deviations:
, N = number of geometric parametes; gRoot mean square of the relative deviations: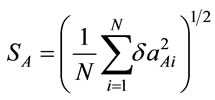 ,
,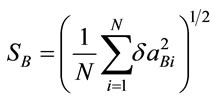 .
.
B results in Pt-Cl bond length very close to the experimental (2.323 vs. 2.318 Å, respectively), but worsens the results for the Pt-N bond as compared to method A. Both protocols overestimate the values of the angles N-Pt-N and N-Pt-Cl, and underestimating the N-Pt-Cl value with respect to the experimental data [31], the discrepancy being largest for N-Pt-N (ca. 10˚). The same trend has been noticed in the HF [23] and DFT [23-25] results with other ECP basis sets, and even when all-electron bases were applied [26]. These results can be explained by the existence of intramolecular interactions (H-bonds) Cl××× H-N which open the angles N-Pt-Nand Cl-Pt-Cl [23-26]. Indeed, the distances Cl×××H calculated by the A and B schemes are short: 2.354 and 2.349 Å, respectively. In the solid state it seems, however, that the intermolecular H-bonding network [31] leads to conformational changes which diminish the effect of the intramolecular Cl×××H-N contacts.
3.3. Vibrational Analysis
The number of the normal vibrations of a single cisplatin molecule is 27, distributed in C2v point group as follows: 9A1 + 5A2 + 8B1 + 5B2. All of them are Raman active vibrations. The assignments of the vibrational spectra of cisplatin available in the literature [22-25], are made under the approximation of a single molecule, and the effects of the solid state have not been considered. Because of comparability considerations, here we also hold to this approach. In Table a of the Supplementary material, however, a factor group analysis of the number and symmetry of the expected vibrational modes in the crystalline state is given.
In Tables 3 and 4, the harmonic vibrational modes of cisplatin calculated by methods A and B, respectively are compared with the experimental Raman sprecrtroscopic data of Amado et al. [24]. The coorespondence between the theoretical and experimental wave number is good even without scaling, the r. m. s. of the relative deviations (S) being 4.5%. Following the approach of other workers [22-25], the vibrational modes were divided in groups and the wavenumbers scaled. The individual scaling factors (ν̃exp/ν̃cld) within each group were averaged to obtain the group scaling factor σ (see the footnotes d-f to Table 3). For both protocols A and B, we have applied grouping into three sets with the corresponding scaling factors as follows: 1) δa(NH3) B1, ρ(NH3) A2 and δ(N-P-tN) – σ1 = 0.92; 2) νa(NH3), δa(NH3) A1, δa(NH3) A2, δs(NH3), ρ(NH3) A1, ρ(NH3) B2, ρ(NH3) B1, ν(Pt-Cl) and πs(Pt) – σ2 = 0.97; 3) νs(NH3), δs(NH3), ν(Pt-Ν) and δ(N-Pt-Cl) – σ3 = 1.03. The scaled wave numbers fit closely to the experimental data as indicated from the arithmetic mean (s) and the r. m. s. (S) of the relative deviations: 1.1% and 1.4%, respectively (Tables 3 and 4). The former was included for better comparison with the data of Amado et al. [24] who use this parameter. Our s-value (1.1%) is the same as the value obtained by them from HF calculations and is slightly better as compared with the analogous values (1.3% and 1.5%) from their DFT calculations [24]. Our calculations show interchange of the order of the
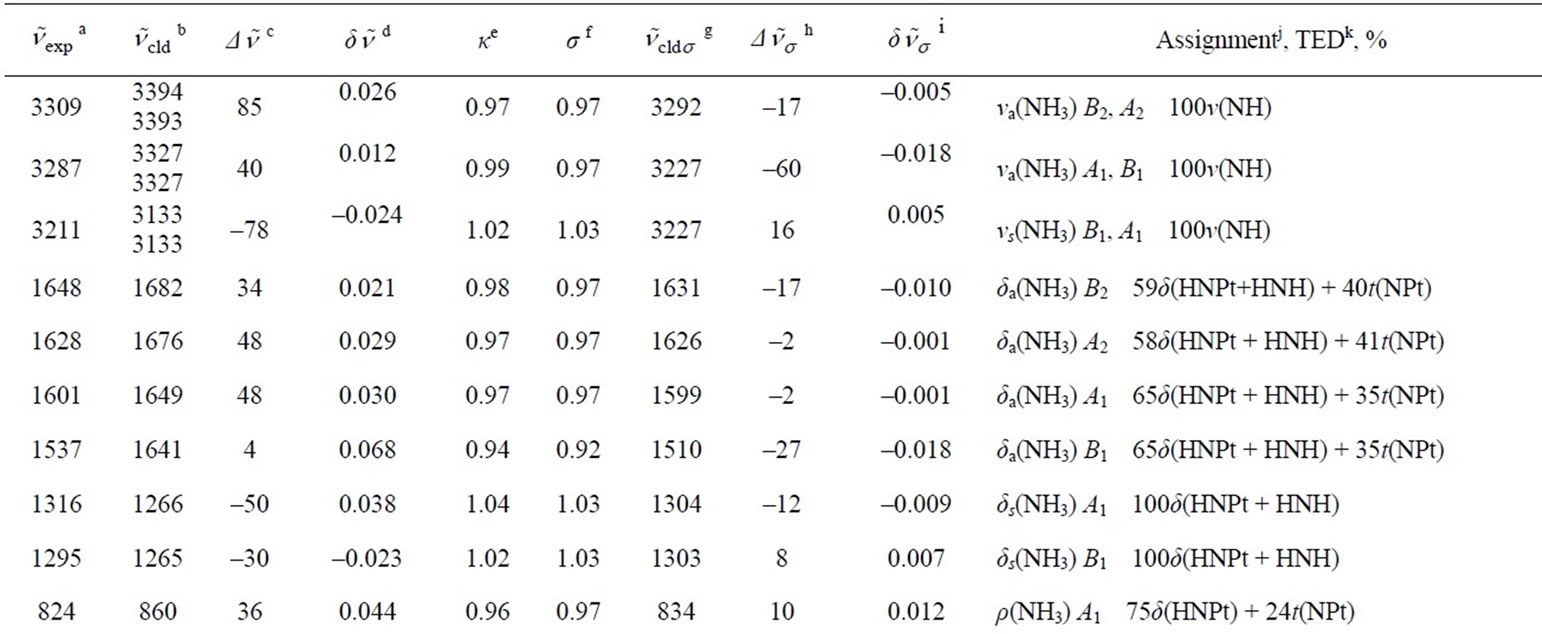

Table 3. Calculated (PW91/LANL08/3-21G) normal vibrational modes (wavenumbers , cm–1) of cisplatin molecule, compared with the experimental data.
, cm–1) of cisplatin molecule, compared with the experimental data.
aExperimental Raman wavenumbers [24]; bTheoretical values; cDeviation of the theoretical values from the experimental ones: ; dRelative deviation of the theoretical values from the experimental ones: δ
; dRelative deviation of the theoretical values from the experimental ones: δ ; eIndividual scaling factor:
; eIndividual scaling factor: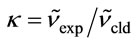 ; fGroup scaling factor:
; fGroup scaling factor: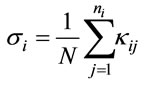 , where ni is the number of the individual scaling factors in the i-th group. Τhus σ1 = 0.92 for δa(NH3) B1, ρ(NH3) A2 and δ(NPtN); σ2 = 0.97 for νa(NH3), δa(NH3) A1, δa(NH3) A2, δs(NH3), ρ(NH3) A1, ρ(NH3) B2, ρ(NH3) B1, ν(PtCl) and πs(NPtCl); σ3 = 1.03 for νs(NH3), δs(NH3), ν(PtΝ) and δ(NPtCl); gTheoretical scaled values; hDeviation of the theoretical scaled values from the experimental ones:
, where ni is the number of the individual scaling factors in the i-th group. Τhus σ1 = 0.92 for δa(NH3) B1, ρ(NH3) A2 and δ(NPtN); σ2 = 0.97 for νa(NH3), δa(NH3) A1, δa(NH3) A2, δs(NH3), ρ(NH3) A1, ρ(NH3) B2, ρ(NH3) B1, ν(PtCl) and πs(NPtCl); σ3 = 1.03 for νs(NH3), δs(NH3), ν(PtΝ) and δ(NPtCl); gTheoretical scaled values; hDeviation of the theoretical scaled values from the experimental ones: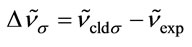 ; iRelative deviation of the theoretical scaled values from the experimental ones:
; iRelative deviation of the theoretical scaled values from the experimental ones: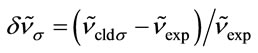 ; jNotations for the modes: ν—stretching, δ—bending, ρ—rocking, π—out-of-plane bending, t— torsion; the indices s and as stand for symmetric and asymmetric respectively, and the index op denotes out-of-plane; kTotal energy distribution over internal coordinates; lArithmetic mean of the relative deviations’ modules:
; jNotations for the modes: ν—stretching, δ—bending, ρ—rocking, π—out-of-plane bending, t— torsion; the indices s and as stand for symmetric and asymmetric respectively, and the index op denotes out-of-plane; kTotal energy distribution over internal coordinates; lArithmetic mean of the relative deviations’ modules: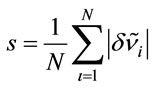 ;
;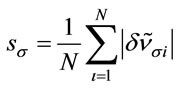 ; N = 27 is the number of vibrational modes; mRoot mean square of the relative deviations:
; N = 27 is the number of vibrational modes; mRoot mean square of the relative deviations: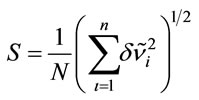 ,
,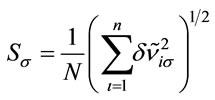 .
.
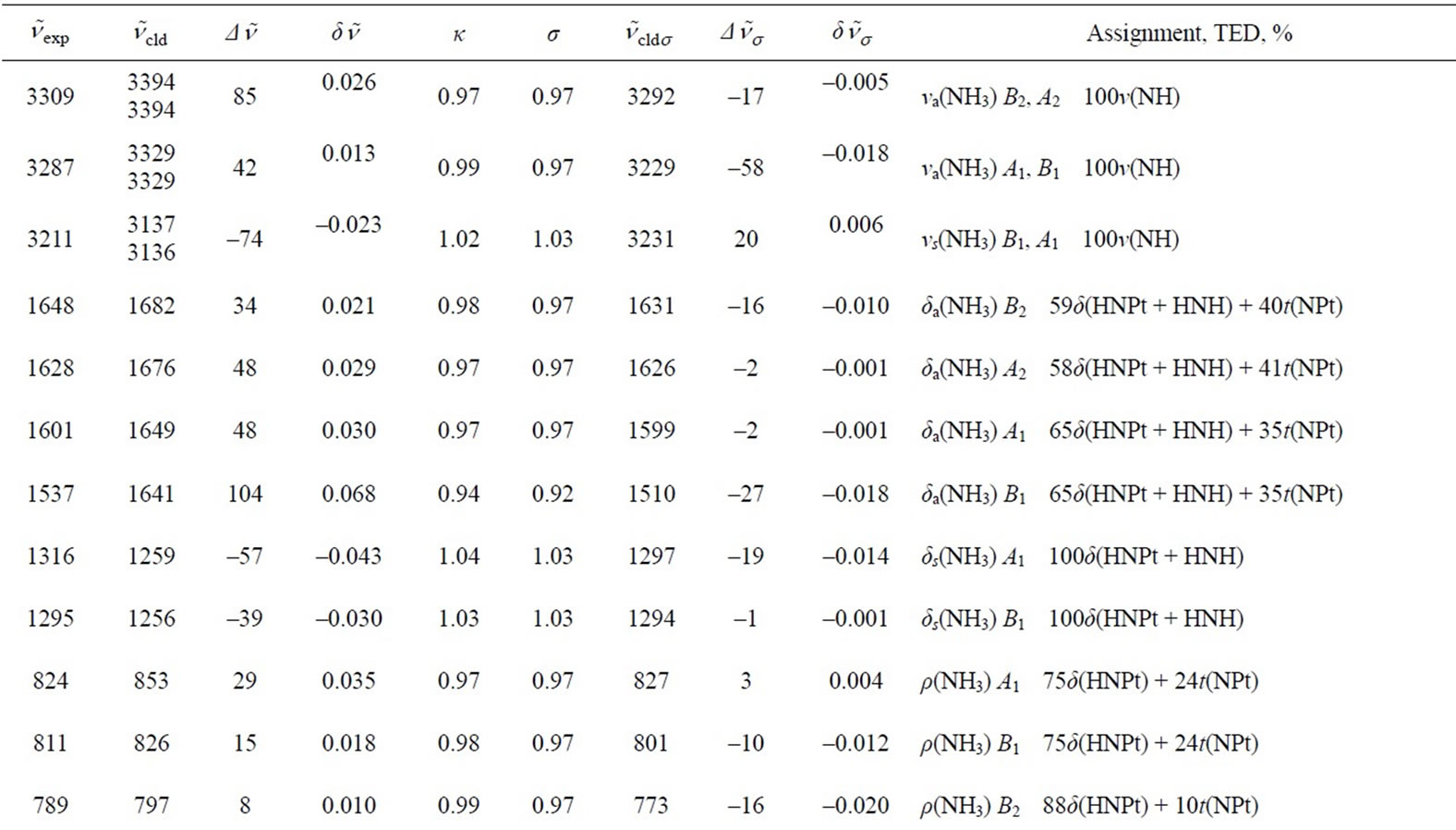
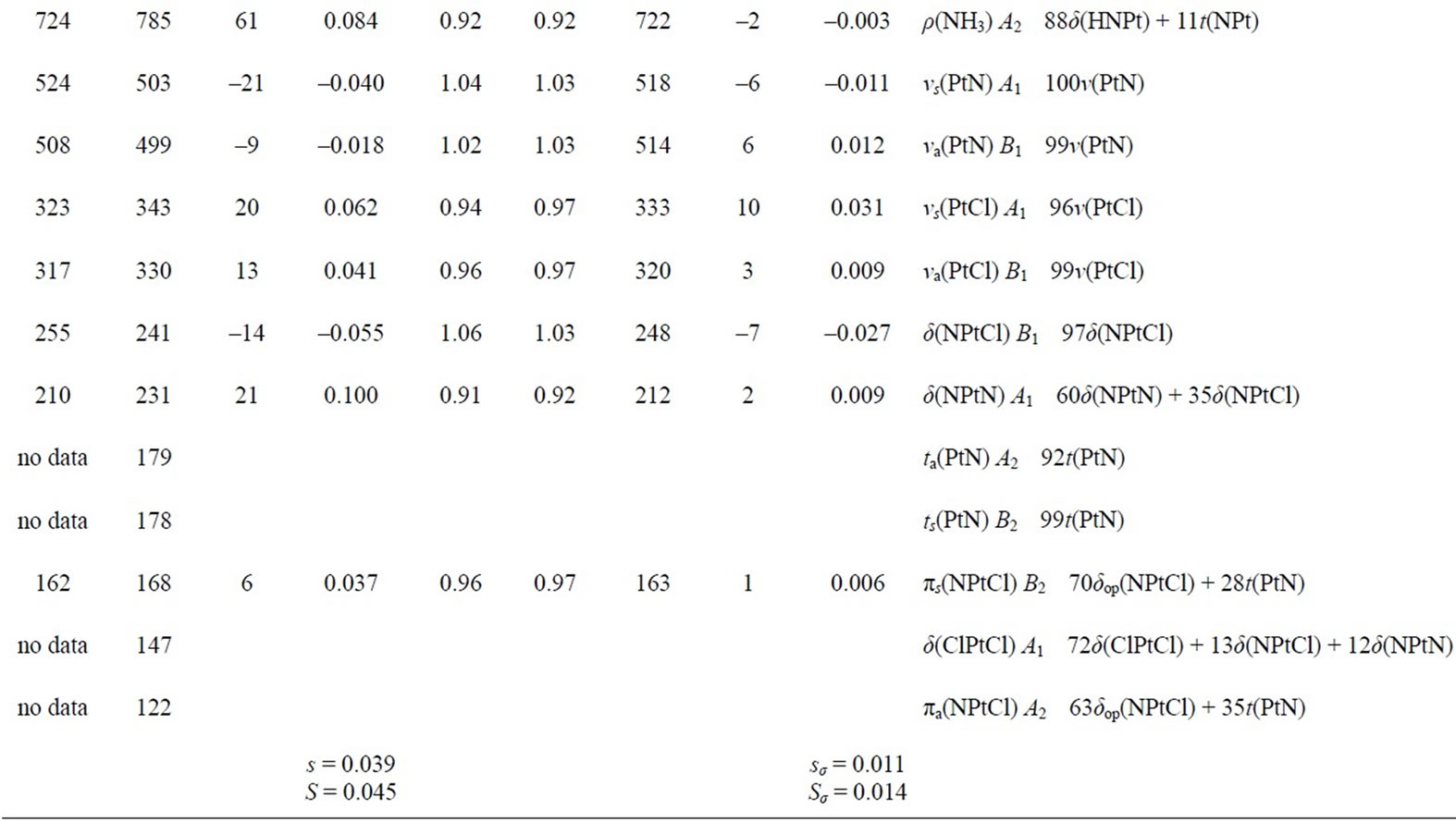
Table 4. Calculated (PW91/LANL08/3-21G*) normal vibrational modes (wavenumbers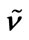 , cm–1) of cisplatin molecule, compared with the experimental dataa.
, cm–1) of cisplatin molecule, compared with the experimental dataa.
aThe notations are the same as in Table 3.
δ(N-Pt-Cl) and δ(N-Pt-N) modes as compared to the data of Amado et al. [23,24], and conform with the earlier assignments of Pavankumar et al. [21]. The other discrepany concerns the order of πs(N-Pt-Cl) and δ(Cl-Pt-Cl) modes: our results correlate the experimental band at 162 cm–1 [23,24] with πs(N-Pt-Cl), whereas δ(Cl-Pt-Cl) vibration should be expected at ca. 140 cm–1 (Tables 3 and 4).
As concerns the two protocols, adding d-orbitals to Cl atoms (protocol B), slightly worsens the fit to the experimental wavenumbers, especially with respect to the ν(Pt-N) and ν(Pt-Cl) vibrations. The shape of the skeletal vibrations of cisplatin molecule is visualized in Figure 3. Selected harmonic force constants for cisplatim molecule are presented in Table 5. In agreement with the finding that adding d-orbitals to the Cl atoms shortens the bond Pt-Cl and lengthens the bond Pt-N (cf. Table 2), it is seen that it also strengthens the former and weakens the later, judging by the force constants given in Table 5.
Typically the tasks on geometry optimization and vibrational analysis take totally 15 min on a PC.
4. Conclusion and Perspective
The DFT computational protocol involving the PW91 exchange-correlation functional, the relativistic ECP and associated non-contracted basis set LANL08 for description of the Pt atom and 3-21G or 3-21G* basis sets for the remaining atoms, gives satisfactory results for the
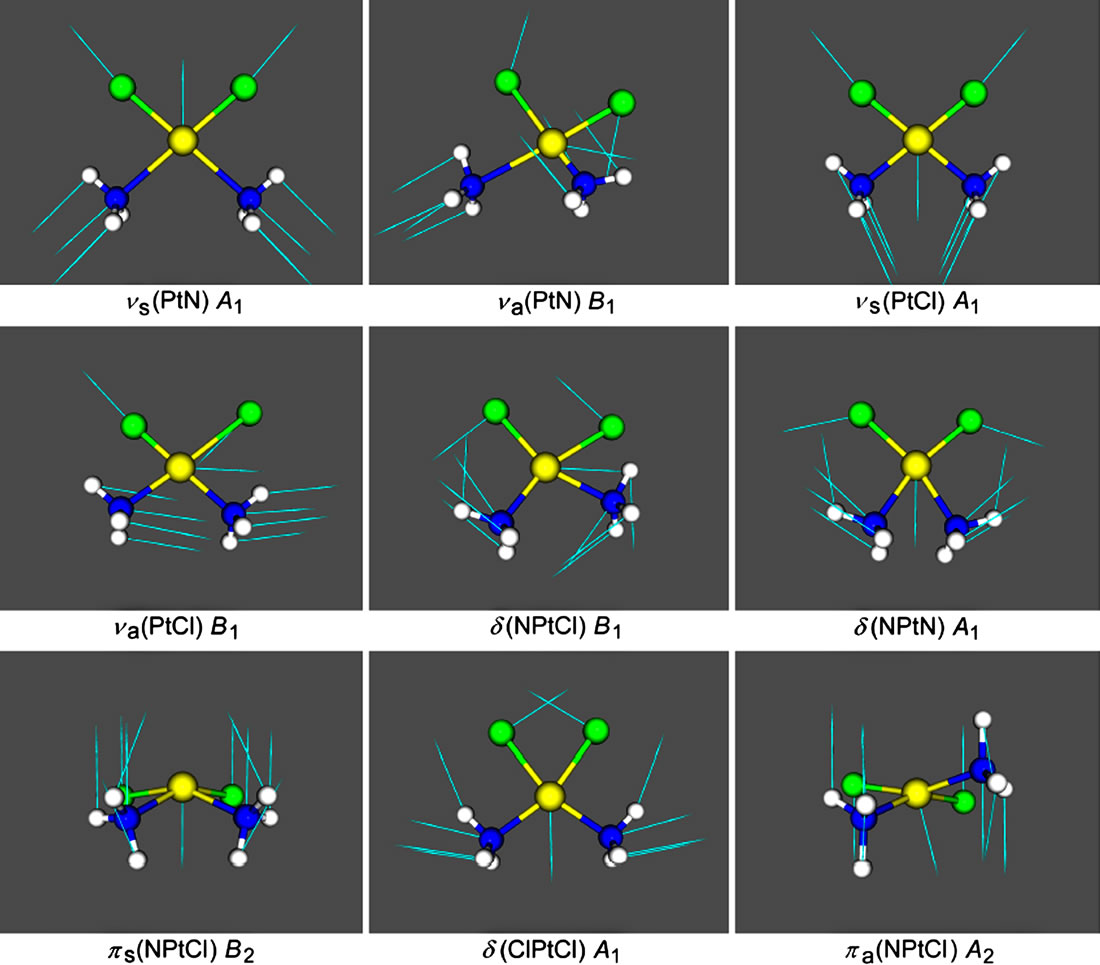
Figure 3. View of the nine skeletal vibrations of cisplatin molecule with their symmetry. The arrows represent the displacement vectors. The vibrational modes were visualized with the aid of Molekel programme using the Firefly output.
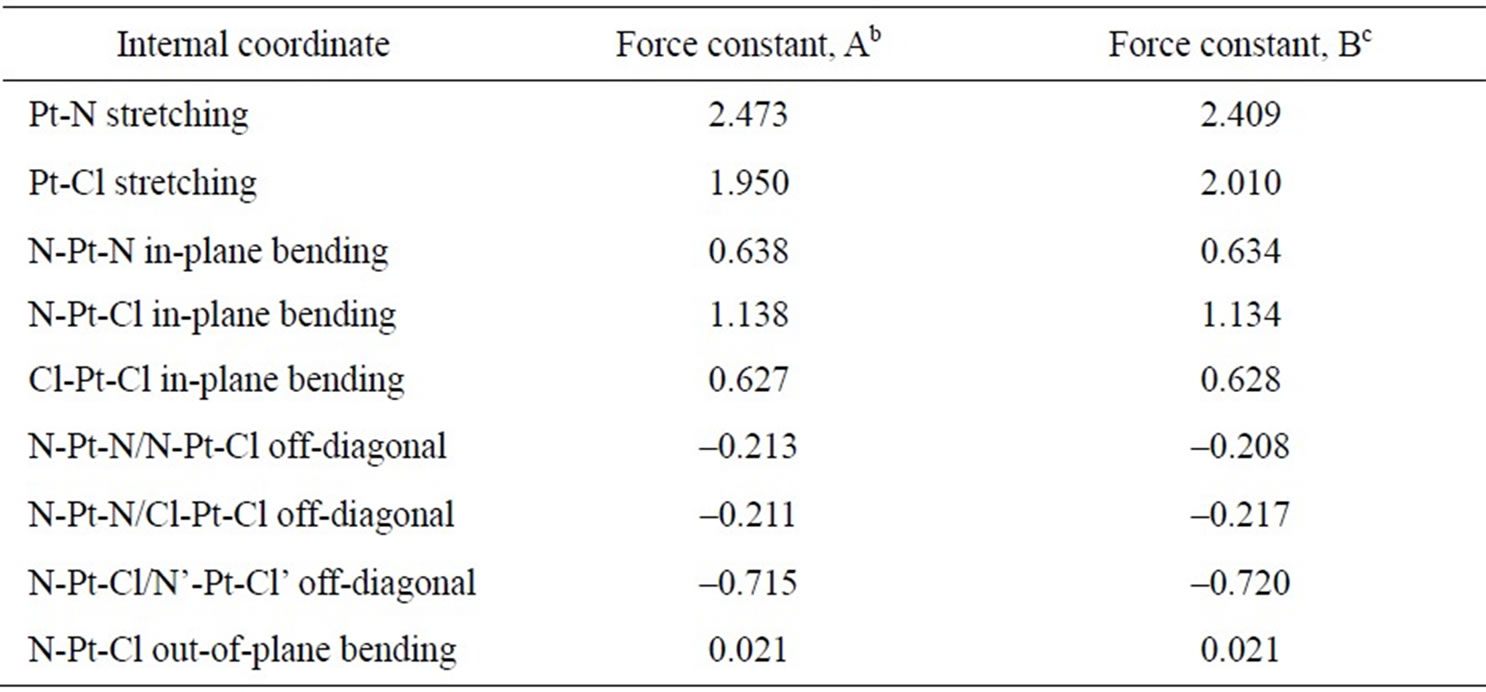
Table 5. Selected harmonic force constantsa for cisplatin.
aUnits: mdyn·Å–1 for the stretching and mdyn·Å·rad–2 for the bending and off-diagonal bending-bending force constants; bPW91/LANL08/3-21G level; cPW91/LANL08/3-21G* level.
equilibrium geometry of cisplatin molecule. The calculated wave numbers of the normal vibrations of the molecule are in very good agreement with the experimental data. The procedure used seems to be a good compromise between accuracy and computational resources. It will be used for DFT modelling of other platinum complexes that are of interest as potential cytostatic agents, and in particular towards some of the newly synthesized in our laboratory Pt(II) [53] and Pt(IV) complexes with sulfonamide ligands.
5. Acknowledgements
Thanks are due to Professor Alex Granovsky for kindly providing us with the Firefly programme. The work is part of the FP7 HP-SEE Project (Contract No RI-261499) and of the bilateral cooperation between the Bulgarian Academy of Sciences and the Aristotle University of Thessaloniki (2012-14).
REFERENCES
- B. Lippert “Chemistry and Biochemistry of a Leading Anticancer Drug,” Verlag Helvetica Chimica Acta, Zürich, 1999.
- S. M. Cohen and S. J. Lippard, “Cisplatin: From DNA Damage to Cancer Chemotherapy,” Progress in Nucleic Acids Research and Molecular Biology, Vol. 67, 2001, pp. 93-130. doi:10.1016/S0079-6603(01)67026-0
- R. A. Alderden, M. D. Hall and T. W. Hambley, “The Discovery and Development of Cisplatin,” Journal of Chemical Education, Vol. 83, No. 5, 2006, pp. 728-734. doi:10.1021/ed083p728
- A.-M. Florea and D. Büsselberg, “Cisplatin as an AntiTumor Drug: Cellular Mechanisms of Activity, Drug Resistance and Induced Side Effects,” Cancers, Vol. 3, 2011, pp. 1351-1371. doi:10.3390/cancers3011351
- T. Boulikas and M. Vougiouka, “Cisplatin and Platinum Drugs at the Molecular Level,” Oncology Reports, Vol. 10, No. 6, 2003, pp. 1663-1682.
- J. Reedijk, “New Cues for Patinum Atitumor Chemistry: Kinetically Controlled Metal Binding to DNA,” Proceedings of the National Academy of Sciences of the USA, Vol.100, No. 7, 2003, pp. 3611-3616. doi:10.1073/pnas.0737293100
- I. Kostova, “Platinum Complexes as Anticancer Agents,” Recent Patents on Anti-Cancer Drug Discovery, Vol. 1, No. 1, 2006, pp. 1-22.
- M. J. Hannon, “Metal-based Anticancer Drugs: From a Past Anchored in Platinum Chemistry to a Post-Genomic Future of Diverse Chemistry and Biology,” Pure and Applied Chemistry, Vol. 79, No. 12, 2007, pp. 2243-2261. doi:10.1351/pac200779122243
- T. Boulikas, A. Pantos, E. Bellis and P. Christofis, “Designing Platinum Compounds in Cancer: Structures and Mechanisms,” Cancer Therapy, Vol. 5, 2007, pp. 537- 583.
- X. Wang and Z. Guo, “Towards the Rational Design of Platinum(II) and Gold(III) Complexes as Antitumour Agents,” Dalton Transactions, No. 12, 2008, pp. 1521-32. doi:10.1039/B715903J
- J. Reedijk, “Platinum Anticancer Coordination Compounds: Study of DNA Binding Inspires New Drug Design,” European Journal of Inorganic Chemistry, Vol. 2009, No, 10, 2009, pp. 1303-1312. doi:10.1002/ejic.200900054
- J. Reedijk, “Increased Understanding of Platinum Anticancer Chemistry,” Pure and Applied Chemistry, Vol. 83, No. 9, 2011, pp. 1709-1719. doi:10.1351/PAC-CON-10-11-03
- I. Kostova, “Gold Coordination Complexes as Anticancer Agents,” Anti-cancer Agents in Medicinal Chemistry, Vol. 6, No. 1, 2006, pp. 19-32.
- I. Ott and R. Gust, “Non Platinum Metal Complexes as Anti-cancer Drugs,” Archiv der Pharmazie, Vol. 340, No. 3, 2007, pp. 117-26. doi:10.1002/ardp.200600151
- E. R. T. Tiekink, “Anti-cancer Potential of Gold Complexes,” Inflammopharmacology, Vol. 16, No. 3, 2008, pp. 138-142. doi:10.1007/s10787-007-0018-5
- L. Cattaruzza, D. Fregona, M. Mongiat, L. Ronconi, A. Fassina, A. Colombatti and D. Aldinucci, “Antitumor Activity of Gold(III)-Dithiocarbamato Derivatives on Prostate Cancer Cells and Xenografts,” International Journal of Cancer, Vol. 128, No. 1, 2011, pp. 206-215. doi:10.1002/ijc.25311
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, “The Status of Platinum Anticancer Drugs in the Clinic and Clinical Trials,” Dalton Transactions, Vol. 39, No. 35, 2010, pp. 8113-8127. doi:10.1039/C0DT00292E
- C. W. Helm and J. C. States “Enhancing the Efficacy of Cisplatin in Ovarian Cancer Treatment—Could Arsenic Have a Role,” Journal of Ovarian Research, Vol. 2, No. 2, 2009, pp. 1-7. doi:10.1186/1757-2215-2-2
- S. Usanova, A. Piée-Staffa, U. Sied, J. Thomale, A. Schneider, B. Kaina and B. Köberle, “Cisplatin Sensitivity of Testis Tumour Cells Is Due to Deficiency in Interstrand-crosslink Repair and Low ERCC1-XPF Expression,” Molecular Cancer, Vol. 9, No. 248, 2010, pp. 1-11. doi:10.1186/1476-4598-9-248
- M. Dolg, “Effective Core Potentials,” In: J. Grotendorst, Ed., Modern Methods and Algorithms of Quantum Chemistry, John von Neumann Institute for Computing, Jülich, 2000, pp. 507-540.
- P. N. V. Pavankumar, P. Seetharamulu, S. Yao, J. D. Saxe, D. G. Reddy and F. H. Hausheer, “Comprehensive ab initio Quantum Mechanical and Molecular Orbital (MO) Analysis of Cisplatin: Structure, Bonding, Charge Density, and Vibrational Frequencies,” Journal of Computational Chemistry, Vol. 20, No. 3, 1999, pp. 365-382. doi:10.1002/(SICI)1096-987X(199902)20:3<365::AID-JCC8>3.0.CO;2-1
- R. Wysokiński and D. Michalska, “The Performance of Different Density Functional Methods in the Calculation of Molecular Structures and Vibrational Spectra of Platinum(II) Antitumor Drugs: Cisplatin and Carboplatin,” Journal of Computational Chemistry, Vol. 22, No. 9, 2001, pp. 901-912. doi:10.1002/jcc.1053
- A. M. Amado, S. M. Fiuza M. P. M. Marques and L. A. E. Batista de Carvalho, “Conformational and Vibrational Study of Platinum(II) Anticancer Drugs cis-Diamminedichloroplatinum(II) as a Case Study,” The Journal of Chemical Physics, Vol. 127, No. 18, 2007, Article ID: 185104. doi:10.1063/1.2787528
- S. M. Fiuza, A. M. Amado, M. P. M. Marques and L. A. E. Batista de Carvalho, “Use of Effective Core Potential Calculations for the Conformational and Vibrational Study of Platinum(II) Anticancer Drugs. cis-Diamminedichloroplatinum(II) as a Case Study,” The Journal of Physical Chemistry, Vol. 112, No. 14, 2008, pp. 3253- 3259. doi:10.1021/jp710868p
- H. Gao, F. Y. Xia, C. J. Huang and K. Linc, “Density Functional Theory Calculations on the Molecular Structures and Vibration Spectra of Platinum(II) Antitumor Drugs,” Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, Vol. 78, No. 4, 2011, pp. 1234-1239. doi:10.1016/j.saa.2010.12.003
- R. C. de Berrêdo and F. E. Jorge, “All-electron Double Zeta Basis Sets for Platinum: Estimating Scalar Relativistic Effects on Platinum(II) Anticancer Drugs,” Journal of Molecular Structure: THEOCHEM, Vol., 961, No. 1-3, 2010, pp. 107-112. doi:10.1016/j.theochem.2010.09.007
- K. Nakamoto, P. J. McCarthy, J. Fujita, R. A. Condrate and G. T. Behnke, “Infrared Studies of Ligand-Ligan Interaction in Dihalogenodiammineplatinum(II) Complexes,” Inorganic Chemistry, Vol. 4, No. 1, 1965, pp. 36-43. doi:10.1021/ic50023a008
- G. Raudaschl, B. Lippert, J. D. Hoeschele, H. E. Howard-Lock, C. J. L. Lock and P. Pilon, “Adduct Formation of cis-(NH3)2PtX2 (X = Cl–, I–) with Formamides and the Crystal Structures of cis-(NH3)2PtCl2×(CH3)2NCHO. Application for the Purification of the Antitumor Agent Cisplatin,” Inorganica Chimica Acta, Vol. 106, No. 3, 1985, pp. 141-149. doi:10.1016/S0020-1693(00)87550-7
- I. A. Degen and A. J. Rowlands, “The Fourier Transform Raman Spectra of a Series of Platinum(II), Palladium(II) and Gold(III) Square-Planar Complexes,” Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, Vol. 47, No. 9-10, 1991, pp. 1263-1268. doi:10.1016/0584-8539(91)80213-3
- G. H. W. Milburn and M. R. Truter, “The Crystal Structures of cisand trans-Dichlorodiammineplatinum(II),” Journal of the Chemical Society A, 1966, pp. 1609-1616. doi:10.1039/J19660001609
- V. P. Ting, M. Schmidman, C. C. Wilson and M. T. Weller, “Cisplatin: Polymorphism and Structural Insight into an Important Chemotherapetic Drug,” Angewandte Chemie, International Edition, Vol. 49, No. 49, 2011, pp. 9408-9411. doi: 10.1002/anie.201003185
- C. Adamo and V. Barone, “Exchange Functionals with Improved Long-range Behavior and Adiabatic Connection Methods without Adjustable Parameters: the mPW and mPW1PW Models,” The Journal of Chemical Physics, Vol. 108, No. 2, 1998, pp. 664-675. doi:10.1063/1.475428
- T. H. Dunning and P. J. Hay, “Gaussian Basis Sets for Molecular Calculations,” In: H. F. Schaefer, Ed., Methods in Electronic Structure Theory (Modern Theoretical Chemistry), Plenum Press, New York, 1977, pp. 1-28.
- P. J. Hay and W. R. Wadt, “Ab initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg,” The Journal of Chemical Physics, Vol. 82, No. 1, 1985, pp. 270-283. doi:10.1063/1.448799
- W. R. Wadt and P. J. Hay, “Ab initio Effective Core Potentials for Molecular Calculations. Potentials for Main Group Elements Na to Bi,” The Journal of Chemical Physics, Vol. 82, No. 1, 1985, pp. 284-298. doi:10.1063/1.448800
- P. J. Hay and W. R. Wadt, “Ab initio Effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitals,” The Journal of Chemical Physics, Vol. 82, No. 1, 1985, pp. 299-310. doi:10.1063/1.448975
- W. J. Hehre, R. F. Stewart and J. A. Pople, “Self-Consistent Molecular Orbital Methods. I. Use of Gaussian Expansions of Slater—Type Atomic Orbitals,” The Journal of Chemical Physics, Vol. 51 No. 6, 1969, pp. 2657- 2664. doi:10.1063/1.1672392
- J. S. Binkley, J. A. Pople and W. J. Hehre, “Self-Consistent Molecular Orbital Methods. 21. Small Split-Valence Basis Sets for First-Row Elements,” Journal of the American Chemical Society, Vol. 102, No. 3, 1980, pp. 939- 947. doi:10.1021/ja00523a008
- M. S. Gordon, J. S. Binkley, J. A. Pople, W. J. Pietro and W. J. Hehre, “Self-Consistent Molecular-Orbital Methods. 22. Small Split-Valence Basis Sets for Second-Row Elements,” Journal of the American Chemical Society, Vol. 104, No. 10, 1982, pp. 2797-2803. doi:10.1021/ja00374a017
- S. H. Vosko, L. Wilk and M. Nusair, “Accurate SpinDependent Electron Liquid Correlation Energies for Local Spin Density Calculations: a Critical Analysis,” Canadian Journal of Physics, Vol. 58, No. 8, 1980, pp. 1200-1211. doi:10.1139/p80-159
- D. Andrae, U. Häussermann, M. Dolg, H. Stoll and H. Preuss, “Energy-Adjusted ab initio Pseudopotentials for the 2nd and 3rd Row Transition-Elements,” Theoretical Chemistry Accounts, Vol. 77, No. 2, 1990, pp. 123-141. doi:10.1007/BF01114537
- J. P. Perdew, K. Burke and M. Ernzerhof, “Generalized Gradient Approximation Made Simple,” Physical Reviews Letters, Vol. 77, No. 18, 1996, pp. 3865-3868. doi:10.1103/PhysRevLett.77.3865
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, “Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation,” Physical Review B, Vol. 46, No. 11, 1992, pp. 6671-6687. doi:10.1103/PhysRevB.46.6671
- J. P. Perdew, K. Burke and Y. Wang, “Generalized Gradient Approximation for the Exchange-Correlation Hole of a Many-electron System,” Physical Review B, Vol. 54, No. 23, 1996, pp. 16533-16539. doi:10.1103/PhysRevB.54.16533
- A. A. Granovsky, “Firefly version 7.1.G,” 2009. http://classic.chem.msu.su/gran/firefly/index.html
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, “General Atomic and Molecular Electronic Structure System,” Journal of Computational Chemistry, Vol. 14, No. 11, 1993, pp. 1347-1363. doi:10.1002/jcc.540141112
- Molekel, 2009. http://molekel.cscs.ch/wiki/pmwiki.php
- L. E. Roy, P. J. Hay and R. L. Martin, “Revised Basis Sets for the LANL Effective Core Potentials,” Journal of Chemical Theory and Computations, Vol. 4, No. 7, 2008, pp. 1029-1031. doi:10.1021/ct8000409
- W. J. Pietro, M. M. Francl, W. J. Hehre, D. J. DeFrees, J. A. Pople and J. S. Binkley, “Self-Consistent Molecular Orbital Methods. 24. Supplemented Small Split-Valence Basis Sets for Second-Row Elements,” Journal of the American Chemical Society, Vol. 104, No. 19, 1982, pp. 5039-5048. doi:10.1021/ja00383a007
- EMSL Basis Set Exchange Library. https://bse.pnl.gov/bse/portal
- D. Feller, “The Role of Databases in Support of Computational Chemistry Calculations,” Journal of Computational Chemistry, Vol. 17, No. 13, 1996, pp. 1571-1586. doi:10.1002/(SICI)1096-987X(199610)17:13<1571::AID-JCC9>3.0.CO;2-P
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, “Basis Set Exchange: A Community Database for Computational Sciences,” Journal of Chemical Information and Modeling, Vol. 47, No. 3, 2007, pp. 1045-1052. doi:10.1021/ci600510j
- N. I. Dodoff, M. Lalia-Kantouri, M. Gdaniec, A. Czapik, N. G. Vassilev, L. S. Markova and M. D. Apostolova, “trans-Dichloro(η2-ethylene)(N-3-pyridinylmethanesulfonamide)platinum(II). Crystal Structure, Spectroscopic, and Thermoanalytical Characterization, and Cytotoxicity Assays,” Journal of Coordination Chemistry, Vol. 65, No. 4, 2012, pp. 688-704. doi:10.1080/00958972.2012.659729
- R. Beattie, “Vibrational Spectra of Solids,” In: A. J. Barnes and W. J. Orville-Thomas, Eds., Vibrational Spectroscopy. Modern trends, (Russian Edition), Mir, Moscow, 1981, pp. 341-358.
Appendix
Factor Group Analysis of the Vibrations of Crystalline Cisplatin
Weller et al. [31] found that cisplatin exists in two polymorphs: low-temperature α- and high-temperature β- modifications. The crystals of both forms are triclinic, space group P1̃ with two and four asymmetric molecules for the α- and β-form, respectively. The relation between the point, site and factor group symmetry for both crystal forms is: C1 ® C1 ® Ci, respectively. A factor group analysis of the number and symmetry of the vibrational modes [54] of the two polymorphs is given in Table A.
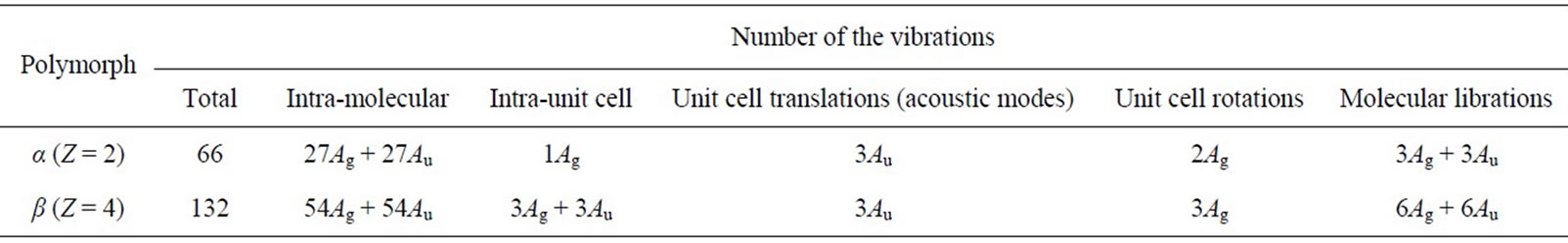
Table A. Factor group analysis for the crystals of α- and β-polymorphs of cisplatin.