American Journal of Analytical Chemistry
Vol.05 No.05(2014), Article ID:44507,9 pages
10.4236/ajac.2014.55036
Determination of Hypoxanthine in the Presence of Copper by Adsorptive Stripping Voltammetry
Percio Augusto Mardini Farias1, Arnaldo Aguiar Castro2
1Department of Chemistry, Pontifícia Universidade Católica, Rua Marquês de São Vicente, Rio de Janeiro, Brazil
2Facultad Quimica, Quimica Analitica, Universidad de La Habana, Havana, Cuba
Email: pfarias@puc-rio.br
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 15 February 2014; revised 18 March 2014; accepted 26 March 2014
ABSTRACT
A stripping method for the determination of hypoxanthine in the presence of copper at the submi- cromolar concentration levels is described. The method is based on controlled adsorptive accu- mulation of hypoxanthine-copper at the thin-film mercury electrode followed by a fast linear scan voltammetric measurement of the surface species. Optimum experimental conditions were found to be the use of 1.0 × 10−3 mol·L−1 NaOH solution as electrolyte supporting, an accumulation poten- tial of −0.50 V and a linear scan rate of 200 mV·s−1. The response of hypoxanthine-copper is linear over the concentration ranges of 10 - 60 ppb. For an accumulation time of 30 minutes, the detection limit was found to be 250 ppt (1.8 × 10−9 mol·L−1). Adequate conditions for measuring the hy- poxanthine in the presence of metal ions, xanthine, uric acid and other nitrogenated bases were also investigated. The utility of the method is demonstrated by the presence of hypoxanthine asso- ciated in ATP or ssDNA.
Keywords:
Hypoxanthine Determination, Xanthine, Uric Acid, Copper Ion, ATP, ssDNA, Thin-Film Mercury Electrode, Fast Linear Scan Stripping Voltammetry

1. Introduction
Oxypurines, hypoxanthine, xanthine and uric acid are formed during purine metabolism and are found in tissues and body fluids such as blood and urine [1] . Determinations of extra cellular concentrations of purines are of considerable significance because changes in their concentrations can indicate several dysfunctions or diseases. For example, excessive accumulation of uric acid, produced as the end product from hypoxanthine and xanthine metabolism, may result in gout. Higher levels of purines in dialysates and heart perfusates can be an indicator of a heart attack [2] . Electroanalytical methods have been developed for the determination of xanthine and hypox- anthine in different matrices. Some have advantages over others regarding sensitivity and selectivity. But the demands for simpler, cheaper and with detection limits in the ppb level methods are still needed. The electroa- nalytical methods of controlled potential, with accumulation time and linear sweep with high scan rates have advantages over the speed and sensitivity [3] -[15] . Adsorptive stripping analysis has been shown to be an im- portant method in ultratrace organic analysis because of its broad scope of applications and relatively simple in- strumentation. The associated analytical costs are lower by orders of magnitude in comparison to spectrophoto- metry [16] or HPLC [17] -[19] . In addition, adsorptive stripping detection easily allows the development of in situ and continuous sensoring techniques. Using the great advances in voltammetry and using as supporting electrolyte alkaline solutions with low ionic strength, our group has developed new methodologies for the de- termination of trace and ultra-trace levels of adenine, thymine, guanine, ATP and DNA [20] -[23] . Then, using a dilute NaOH solution as electrolyte, the present work reports a new stripping voltammetric procedure for the measurement of hypoxanthine-copper based on its adsorption at the thin film mercury electrode. The advantages, instrumental parameters, and possible limitations of this procedure are also explained in this paper. Furthermore, the effects of a wide range of potentially interfering compounds such as xanthine, uric acid, other purine and py- rimidines compounds, ATP, ssDNA, and some metal ions were examined.
2. Experimental
2.1. Apparatus
Linear cyclic voltammograms were obtained with an EG & G PAR model 384-B Polarographic Analyser (Prin- ceton Applied Research, Princeton, NJ, USA), equipped with an external cell and a Houston Ametek-DMP-40 series digital plotter. The electrochemical cell was formed by a glassy carbon electrode (GCE) with thin-film mercury as a working electrode, an Ag/AgCl reference electrode with vicor tip and a platinum auxiliary elec- trode. A magnetic stirrer and a stirring bar (Nalgene Cat. No. 6600-0010, Rochester, NY, USA) provided con- vective transport during the accumulation step.
2.2. Forming the Thin-Film Mercury Electrode
Initially, a solution was prepared by dissolving 0.4 g of mercury (II) nitrate (II) in 100 ml of Milli-Q water aci- dified (5% HNO3). And, a GCE, ( 3.0 mm diameter, BAS-Bioanalytical Systems, Inc., West Lafayette, Indiana 47906, USA) was polished with alumina (BAS, PK-4). Then the GCE with the reference and the auxiliary elec- trode was mounted with the help of a Teflon holder in a voltammetric cell containing 1 mL of mercury (II) ni- trate solution, 1 mL of 10−1 mol·L−1 potassium nitrate solution and 8 mL of purified water. The solution in the cell was purged with nitrogen for 240 s to eliminate the oxygen present. The mercury plating on the surface of GCE was carried over for five minutes at −0.9 V. After a visual check that the mercury film was well done on the surface of GCE, it was rinsed with pure water along with the reference and the auxiliary electrode. Finally, the electrodes were properly engaged in the Teflon holder under a new and clean voltammetric cell containing only solutions with the NaOH electrolyte or already containing hypoxanthine.
2.3. Reagents
Water purified in a Milli-Q purification system (Millipore, Billerica, MA, USA) was used for all dilutions and sample preparations. All chemicals were of analytical reagent grade. Stock solutions of 1000 ppm were prepared from Sigma Chemicals (Sigma-Aldrich Brasil Ltda., São Paulo-SP, Brasil) by dissolving 25 mg of the target reagent hypoxanthine or xanthine plus solid NaOH with 25 mL of water (to achieve a final concentration of 0.1 mol·L−1 NaOH). Stock solutions of uric acid and other bases were similarly prepared. Solutions were stored in the dark at 4˚C. A 1000 ppm stock solution (atomic absorption standard solution, Sigma-Aldrich Brasil Ltda) was used for metal ions and diluted as required for standard additions. Stock solutions of 1000 ppm of adenosine 5’-triphosphate disodium salt hydrate (ATP) were prepared by dissolving 10 mg of the target reagent in 2 mL of diluted perchloric acid (10−1 mol·L−1). The subsequent solution was heated at 70˚C for 30 seconds. Thereafter, the sample was cooled down and diluted to 10 mL with water. A single-stranded calf thymus DNA (Cat. No. D-8899; Lot 43H67951) was used as received from Sigma. A 500 mg DNA/mL stock solution (around 5 mg/10 mL; Lyophilized powder containing 63% DNA) was prepared according to the procedure described for ATP. ATP and DNA were first treated with acid to obtain free purine and pyrimidine bases and other degradation products. The final solution was stored at 4˚C.
2.4. Procedure
A known volume (10 mL) of the supporting electrolyte solution (1.0 × 10−3 mol·L−1 sodium hydroxide) in pres- ence of 0.5 ppm of copper was added to the voltammetric cell and degassed with nitrogen for eight minutes (and for 60 seconds before each adsorptive stripping cycle). Initially the condition potential (–0.9 V) was applied to the electrode for a selected time (usually 60 s). After the initial potential (usually −0.50 V) was applied to the electrode over a selected time interval (usually 60 s), while the solution was slowed stirred. The stirring was then stopped, and after 30 s the voltammogram was recorded by applying a negative-going potential scan. The scan (at 200 mV·s−1; calculated drop time, 0.050 s; scan increment, 10 mV) was terminated at −1.10 V. The ad- sorptive stripping cycle was repeated using the same thin-film of mercury. After obtaining the background strip- ping voltammograms, aliquots of the hypoxanthine standards were introduced. A similar procedure was fol- lowed to test the interference of several metal ions, xanthine, uric acid, other purine and pyrimidine bases, and ATP or DNA degradation products. The entire procedure was automated and controlled by a 384-B Polaro- graphic Analyser. Throughout this operation nitrogen was passed over the solutions surface. All data was ob- tained at room temperature (25˚C).
3. Results and Discussion
3.1. Parameters Affecting the Adsorptive Stripping Behavior
In the presence of hypoxanthine, the copper (II) can be initially reduced to copper (I). The copper (I) is com- plexed with hypoxanthine and the complex is adsorbed on the film mercury electrode. Thus, the copper (I) in the complex adsorbed may be reduced to copper (0) using cathodic stripping voltammetry or oxidized to copper (II) using anodic stripping voltammetry. A possible mechanism of reduction is shown below [24] -[27] .
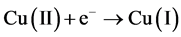
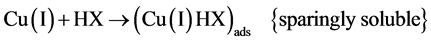
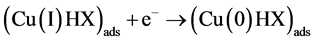
Figure 1 shows a linear cyclic voltammogram obtained after the adsorptive stripping of hypoxanthine (0.60 ppm) in a 1 × 10−3 mol·L−1 NaOH solution in the presence of 0.25 ppm of copper. Two reduction peaks ap- peared during the forward cathodic scan at −0.170 (a-due to the reduction of copper II to copper I) and at −0.640 V (b-suggests the presence of Cu (I) hypoxanthine complex). A sharp peak also appeared on the reverse sweep at –0.300 V (c-oxidation of copper zero to II). The hypoxanthine peak did not appear in the absence of copper. Occasionally, a cathodic peak with very poor stability appeared at −0.390 V which is credited to the reduction of copper II to zero. The formation of Cu(OH)2 from Cu(II) in the NaOH solution should also be considered.
The effect of the copper concentration in the hypoxanthine-copper stripping peak at two different accumula- tion potentials also was studied. In the first experiment the copper concentration was fixed at 0.25 ppm and the hypoxanthine varied from 0.10 to 0.60 ppm. The hypoxanthine-copper peaks were obtained using an accumula- tion time of 120 s at 0.0 V. (Other conditions as in Figure 1) The current peak was increased up to 0.40 ppm (copper), the potential peak was shifted from −560 to −640 V; and the half-width, b1/2 was broadened from 70 to 80 mV. In the second experiment, the hypoxanthine was fixed at 0.50 ppm and the copper varied from 0.05 to 0.35 ppm. The hypoxanthine-copper peaks were obtained using an accumulation time of 60 s at −0.5 V. (Other conditions as in Figure 1) Higher hypoxanthine-copper stripping peaks were observed in this second experiment. The current peak was increased up to 0.25 ppm (copper); the potential peak was shifted from −800 to −870 V; and the half-width, b1/2, was broadened from 65 to 80 mV.
One of the important parameters to the adsorptive stripping voltammetry first studied in this work was the

Figure 1. Linear CV adsorptive stripping vol- tammogram of 0.60 ppm hypoxanthine in pre- sence of 0.25 ppm copper in a solution 1.0 × 10−3 mol·L−1 NaOH. Condition time, 60 s at −0.9 V. Accumulation time, 120 s at −0.0 V. Final potential, −1.1 V. Equilibrium time, 30 s. Scan rate, 50 mV·s−1. Thin-film mercury elec- trode (5 min at −0.9 V). “a” is due to the reduc- tion of Cu (II) to (I); “b” suggests the presence of Cu (I) hypoxanthine complex; and “c” the oxidation of Cu(0) to (II) as an anodic peak.
comparison between the different scanning processes. Using accumulation potential at −0.5 V, a comparison between the linear scan and differential pulse mode was tested for hypoxanthine-copper (II) determination. The linear scan mode offers better signal-to-background characteristics and greater speed, and is recommended for the determination of hypoxanthine-copper. Also was observed which at a higher linear scan rate the hypoxan- thine-copper peak current was significantly increased while peak potentials were strongly shifted.
The study of the potential for accumulation in these techniques with stripping voltammetry is very important to eliminate possible interferences and improve the sensitivity and selectivity of the analytical method. The ef- fect of the accumulation potential on the hypoxanthine-copper stripping peak was examined over the 0.0 to −0.5 V range. A gradual increase in the hypoxanthine-copper current is observed as the preconcentration potential is changed from 0.0 and −0.3 V. At higher accumulation potentials (greater than −0.3 V) two processes were ob- served; a peak associated with the reduction and oxidation of copper disappears, and the hypoxanthine-copper stripping peak is maintained and with signal constant. Thus, was verified which stripping measurements using more negative accumulation potentials yielded a hypoxanthine-copper peak of higher analytical interest (with low detection limits).
The scan rate (υ) is a parameter of great importance to understand the electrochemical behavior of hypoxan- thine-copper complex on the surface of the mercury-film electrode. Then, the effect of the scan rate (υ) in the hypoxanthine-copper voltammograms was studied (experimental conditions: hypoxanthine (0.50 ppm) in the presence of copper (0.50 ppm) in 1.0 × 10−3 mol·L−1 NaOH; accumulation time, 60 s at −0.5 V; scan rate, 20− 200 mV·s−1; other conditions as in Figure 1). As expected, there was a strong dependence of hypoxanthine- copper peak currents (Ip) on the scan rate. The peak current for the surface-adsorbed hypoxanthine-copper is di- rectly proportional to the scan rate. The plot of log Ip vs log υ is linear (correlation coefficient, 1.000), with a slope of 0.9 which is close to that (1.0) expected for an ideal redox couple immobilized on an electrode surface. The potential shifts from −0.750 to −0.810 V when the scan rate increases from 20 to 200 mV·s−1. Over this scan rate interval, there is also an increase in b1/2 (from 27 to 67 mV). At 500 mV·s−1 uncommon fluctuations of the baseline were observed in the standard measurements. Hence, the rate of 200 mV·s−1, which yielded opti- mum results with respect to both sensitivity and resolution, was used in all subsequent scans.
To find the highest peak current at a given concentration of hypoxanthine-copper complex, the current study was conducted with the increasing accumulation time. At one point the saturation of certain amount of hypo- xanthine-copper complex under the surface of the mercury-film electrode should occur, following the concepts of the Langmuir isotherm. Figure 2 shows the dependence of the linear CV hypoxanthine-copper peak current on the pre-concentration time. Initially (up to 60 s) the current increases, and then level off at longer accu- mulation times. Such time-dependent profiles represent the corresponding adsorption isotherms since the peak current depends on the amount adsorbed. As in all types of stripping measurements, the choice of accumulation time requires a trade-off between sensitivity and speed.
3.2. Quantitative Utility
Like any other method of analytical chemistry, some parameters must be studied and interpreted, as the limit of detection and quantification, the reproducibility of the method and the linear variation of the current with the quantitative increase of the concentration of hypoxanthine-copper complex. Figure 3 shows the linear CV ad- sorptive stripping voltammogram for hypoxanthine-copper at the subnanomolar concentration. Using a 900 s pre-concentration time set at −0.5 V were developed well-defined peaks for 10 ppb of hypoxanthine concentra- tion in presence of 0.50 ppm of copper. Experiments using lower hypoxanthine concentration were also per- formed. The effective pre-concentration, derived from the adsorption process, resulted in the significant lower- ing of the detection limit to 0.25 ppb (1.8 × 10−9 mol·L−1). This was estimated from the quantitation of 1 ppb af- ter a 15-min accumulation (S/N = 2). Thus, 25 pg can be detected in 10 mL of solution.
The highly reproducible linear CV adsorptive stripping response of 0.50 ppm hypoxanthine in presence of 0.50 ppm of copper is adequate for analytical determinations. Eight successive measurements (using 30 s accu- mulation times at –0.50 V; final potential at −1.1 V; 60 s condition times at −0.9 V; scan rate, 200 mV·s−1 and 1.0 × 10−3 mol·L−1 of NaOH as supporting electrolyte ) yielded a mean peak current of 8453 ± 97 nA and a range of 8312 - 8625 nA. Such precision compares favorably with that reported for other compounds measured by adsorptive stripping analysis [28] [29] .
Figure 4 shows linear CV voltammograms for solutions of increasing hypoxanthine concentration (steps of 10 ppb), in the presence of 0.50 ppm copper (II), after 120 s of accumulation time at −0.5 V. Well-defined strip- ping peaks were observed over this concentration range. In contrast, the corresponding solution-phase

Figure 2. Effect of accumulation time on the linear CV adsorptive stripping voltammo- grams of hypoxanthine (0.50 ppm) in presence of copper (0.50 ppm) in a solution of 1.0 × 10−3 mol·L−1 NaOH. Accumulation time, a (0 and without equilibrium time), b(0), c(30), d(60),e(90), f(120), g(150) s at −0.5 V. Other conditions as in Figure 1. Also shown is the resulting accumulation time plot.

Figure 3. Linear CV adsorp- tive stripping voltammograms of the hypoxanthine (0-A and 10-B ppb) in presence of cop- per (0.50 ppm) in a solution of 1.0 × 10−3 mol L−1 NaOH. Accumulation time, 900 s at −0.5 V. Other conditions as in Figure 2.
response (accumulation time, 0 s) was not useful for quantitative work at this level. The stripping peak current increases linearly with the concentration. A least-squares analysis of the standard addition data yields a slope of 42 nA/ppb and a correlation coefficient of 0.997.
The main sources of interference are the several possibilities of some metal ions compete with the copper ion in the complexing with hypoxanthine as well as the presence of organic surfactants. These species can produce new peak reduction or overlap with the peaks of hypoxanthine-copper complex, thus obscuring the measurement. The effects of ions (Ni (II), Co(II), Cd(II), Zn (II) and Fe (III)), some nitrogenated bases (guanine, adenine, thymine cytosine, uracil and xanthine) and uric acid (which could interfere with the analytical process) were studied for hypoxanthine-copper determination. The determination of 0.10 ppm of hypoxanthine was not af- fected by the addition of up to 0.5 ppm of Cd (II), up to 0.4 ppm of Zn (II) or Fe (III), up to 0.3 ppm of Ni (II) and up to 0.2 ppm of Co (II). The determination of 0.10 ppm of hypoxanthine was not affected by the addition of up to 0.10 ppm of thymine, xanthine or uric acid, up to 0.06 ppm of methyl cytosine, up to 0.04 ppm of uracil and up to 0.02 ppm of guanine, cytosine or adenine. The great advantage of this method for the determination of hypoxanthine-copper is little interference of xanthine and uric acid, which are always present during the forma- tion of purine metabolism. In a similar experiment using 60 s as the accumulation time at −0.5 V (other condi- tion same as Figure 4), the presence of ssDNA (0.12 ppm) did not change the current and potential of hypoxan- thine (0.50 ppm)-copper (2.0 ppm) peak.
Several compounds of biological importance and its metabolites have similar molecular structures, as well as hypoxanthine and xanthine, so it is interesting to use the artifice of their simultaneous determinations. Figure 5 shows well-defined xanthine and hypoxanthine-copper peaks at −0.150 and −0.800 V, respectively: (A) the xanthine peak in the presence of hypoxanthine; and (B) the hypoxanthine-copper peak in the presence of xan- thine. The distinct peak potentials suggest the possibility of simultaneous xanthine and hypoxanthine-copper de- terminations. In preliminary studies of our research group, using similar chemical and instrumental parameters,

Figure 4. Linear CV adsorptive stripping voltammo- grams obtained after increasing the hypoxanthine con- centration in 10 ppb steps (b-g) in presence of copper (0.50 ppm) in a solution of 1.0 × 10−3 mol·L−1 NaOH. (a) blank; without hypoxanthine. Accumulation time, 120 s at −0.5 V. Other conditions as in Figure 3. Also shown is the resulting calibration plot.
it was found that the compound of the xanthine is not complexed by the presence of copper ion [5] .
To verify the applicability of this new method electroanalytical of the determination of hypoxanthine, syn- thetic matrices were developed. Figure 6 illustrates the method for the determination of hypoxanthine-copper in a synthetic sample containing xanthine, uric acid and other nitrogenated bases (guanine, thymine, cytosine, me- thyl cytosine, adenine and uracil), ATP and DNA (all at 1 ppm concentration). Four successive standard addi- tions to the sample resulted in well-shaped adsorptive stripping peaks. Thus, the hypoxanthine-copper peak in the original sample (curve b) was quantified based on the resulting standard addition plot (also show in Figure 6). Because of the inherent sensitivity of the method, short (30 s) accumulation times can be used. Five consecu- tive analyses of the sample yielded an average value of 1.1 ppm with a standard deviation of the 0.1 ppm. This results value agrees with the expected hypoxanthine concentration (1.0 ppm).
4. Conclusion
An effective means for the determination of trace levels of hypoxanthine in presence of copper has been de- scribed. The use of the simple and diluted alkaline electrolyte provided a sensitive and selective adsorptive strip- ping voltammetric method. The NaOH medium (Ep hypoxanthine-copper = −0.800 V) seems to be advantageous compared with the borate buffer medium [30] (Ep hypoxanthine = +0.04 V) for studies of hypoxanthine behavior at the electrode because the peak potential shifts further away from the mercury wave. The peak po-

Figure 5. Linear CV adsorptive stripping voltammo- grams of xanthine (0.70 ppm) in presence of hypoxan- thine (0.70 ppm) (a), and of hypoxanthine (1.1 ppm) in presence of xanthine (0.70 ppm) and copper (0.60 ppm) (B) in a solution of 5.0 × 10−3 mol·L−1 NaOH. Accu- mulation time, 120 s at −0.0 V (A) and −0.5 V (b). Other conditions as in Figure 4.

Figure 6. Illustration of hypoxanthine determination in a synthetic sample containing several nitrogenated bases, ATP and ssDNA (all with 1.0 ppm of concentration) by linear cyclic adsorp- tive stripping voltammetry. a-Electrolyte support, 10 mL of 1.0 × 10−3 mol·L−1 NaOH with 2.0 ppm of copper (II). b-addition of 4.0 × 10−1 mL of synthetic sample; c-g steps addition of 0.01 ppm of standard hypoxanthine. Accumulation time, 30 s at −0.5 V. Potential final, −1.0 V. Oth- er conditions as in Figure 4. Also shown is the resulting calibration curve for the sample.
tentials of xanthine and hypoxanthine-copper are separated by 0.65 V. This difference allows the simultaneous determination of these bases in the same array. The use of mercury film in the presence of copper (II) offers an attractive alternative to electroanalytical methods using carbon electrodes [3] [4] . The methods use these elec- trodes, providing similar detection limits, but the use of mercury film reduces interference of various metal ions and other purines and pyrimidines. The uric acid presence, which is main metabolite of purine metabolism, also doesn’t interfere. The detection limit of 1.8 × 10−9 mol·L−1 for hypoxanthine-copper is comparable to those found for HPLC methods [17] -[19] . Further studies using alkaline solutions as supporting electrolytes in the presence copper can be conducted for the detection of HIV drugs, DNA-intercalating dyes, amino-acids, peptides and proteins determinations.
Acknowledgements
The authors gratefully acknowledge the CNPq and CAPES of the Government of Brazil and PUC-Rio for their support of this work.
References
- Harkness, R.A. (1988) Hypoxanthine, Xanthine and Uridine in Body-Fluids, Indicators of ATP Depletion. Journal of Chromatography-Biomedical Applications, 429, 255-278. http://dx.doi.org/10.1016/S0378-4347(00)83873-6
- Crushing, D. and Mustafa, S. (1991) Adenosine and Adenosine Nucleotides as Regulators of Cellular Functions. CRC Press, Boca Raton.
- Goyal, R.N., Mittal, A. and Sharma, S. (1994) Simultaneous Voltammetric Determination of Hypoxanthine, Xanthine, and Uric-Acid. Electroanalysis, 6, 609-611. http://dx.doi.org/10.1002/elan.1140060715
- Wang, Z.H. and Zhang, H. (2005) An Inlaid Super-Thin Carbon Paste Film Modified Electrode for Simultaneous Determination of Xanthine and Hypoxanthine. Chinese Journal of Analytical Chemistry, 33, 671-674.
- Farias, P.A.M., Castro, A.A. and Wagener, A.D.R. (2009) Ultratrace Xanthine Determination in Diluted Alkaline Electrolyte by Adsorptive Stripping Voltammetry at the Mercury Film Electrode. Analytical Letters, 42, 2011-2025. http://dx.doi.org/10.1080/00032710903082853
- Wang, Y. and Tong, L.L. (2010) Electrochemical Sensor for Simultaneous Determination of Uric Acid, Xanthine and Hypoxanthine Based on Poly (bromocresol purple) Modified Glassy Carbon Electrode. Sensors and Actuators B- Chemical, 150, 43-49. http://dx.doi.org/10.1016/j.snb.2010.07.044
- Wang, Y. (2011) Simultaneous Determination of Uric Acid, Xanthine and Hypoxanthine at Poly(pyrocatechol violet)/Functionalized Multi-Walled Carbon Nanotubes Composite Film Modified Electrode. Colloids and Surfaces B- Biointerfaces, 88, 614-621. http://dx.doi.org/10.1016/j.colsurfb.2011.07.051
- Wang, Y., Wang, L., Tian, T., Yao, G.J., Hu, X.Y., Yang, C. and Xu, Q. (2012) A Highly Sensitive and Automated Method for the Determination of Hypoxanthine Based on Lab-on-Valve Approach Using Fe3O4/MWCNTs/beta-CD Modified Electrode. Talanta, 99, 840-845. http://dx.doi.org/10.1016/j.talanta.2012.07.040
- Zhang, F.Y., Wang, Z.H., Zhang, Y.Z., Zheng, Z.X., Wang, C.M., Du, Y.L. and Ye, W.C. (2012) Simultaneous Electrochemical Determination of Uric Acid, Xanthine and Hypoxanthine Based on Poly(L-arginine)/Graphene Composite Film Modified Electrode. Talanta, 93, 320-325. http://dx.doi.org/10.1016/j.talanta.2012.02.041
- Lawal, A.T. and Adeloju, S.B. (2012) Mediated Xanthine Oxidase Potentiometric Biosensors for Hypoxanthine Based on Ferrocene Carboxylic Acid Modified Electrode. Food Chemistry, 135, 2982-2987. http://dx.doi.org/10.1016/j.foodchem.2012.07.052
- Gong, W., Dou, Z.Y., Cui, L.L., Liu, D.J. and He, X.Q. (2012) Electrocatalytic Oxidation and Simultaneous Determination of Uric Acid, Xanthine, Hypoxanthine and Dopamine Based on Beta-Cyclodextrin Modified Glassy Carbon Electrode. Chemical Research in Chinese, 28, 1047-1053.
- Ojani, R., Alinezhad, A. and Abedi, Z. (2013) A Highly Sensitive Electrochemical Sensor for Simultaneous Detection of Uric Acid, Xanthine And Hypoxanthine Based on Poly(L-methionine) Modified Glassy Carbon Electrode. Sensors and Actuators B-Chemical, 188, 621-630. http://dx.doi.org/10.1016/j.snb.2013.07.015
- Raj, M.A. and John, S.A. (2013) Simultaneous Determination of Uric Acid, Xanthine, Hypoxanthine and Caffeine in Human Blood Serum and Urine Samples Using Electrochemically Reduced Graphene Oxide Modified Electrode. Analytica Chimica Acta, 771, 14-20. http://dx.doi.org/10.1016/j.aca.2013.02.017
- Torres, A.C., Ghica, M.E. and Brett, C.M.A. (2013) Design of a New Hypoxanthine Biosensor: Xanthine Oxidase Mo- dified Carbon Film and Multi-Walled Carbon Nanotube/Carbon Film Electrodes. Analytical and Bioanalytical Chemistry, 405, 3813-3822. http://dx.doi.org/10.1007/s00216-012-6631-1
- Chen, C.H., Lin, Y.T. and Lin, M.S. (2013) Low-Potential Amperometric Determination of Purine Derivatives through Surface Oxide Regeneration Method. Analytica Chimica Acta, 796, 42-47. http://dx.doi.org/10.1016/j.aca.2013.08.011
- Khajehsharifi, H. and Pourbasheer, E. (2011) Simultaneous Spectrophotometric Determination of Xanthine, Hypoxanthine and Uric Acid in Real Matrix by Orthogonal Signal Correction-Partial Least Squares. Journal of the Iranian Che- mical Society, 8, 1113-1119.
- Czauderna, M. and Kowalczyk, J. (1997) Simultaneous Measurement of Allantoin, Uric Acid, Xanthine and Hypoxanthine in Blood by High-Performance Liquid Chromatography. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences, 704, 89-98. http://dx.doi.org/10.1016/S0378-4347(97)00459-3
- Cooper, N., Khosravan, R., Erdmann, C., Fiene, J. and Lee, J.W. (2006) Quantification of Uric Acid, Xanthine and Hy- poxanthine in Human Serum by HPLC for Pharmacodynamic Studies. Journal of Chromatography B-Analytical Te- chnologies in the Biomedical and Life Sciences, 837, 1-10. http://dx.doi.org/10.1016/j.jchromb.2006.02.060
- Lin, L., Song, C.Y., Xie, L.Y., Yu, L., Wu, L.M., Zhang, M.L., Yang, S.L., Gao, H.C. and Li, X.K. (2010) Electroche- mical Determination of Xanthine and Hypoxanthine in Rat Striatum with an Acetylene Black-Dihexadecyl Hydrogen Phosphate Composite Film Modified Electrode by HPLC coupled with in Vivo Microdialysis. Microchimica Acta, 170, 47-52. http://dx.doi.org/10.1007/s00604-010-0374-5
- Farias, P.A.M., Wagener, A.D.R. and Castro, A.A. (2001) Adsorptive Voltammetric Behavior of Adenine in Presence of Guanine and Some Trace Elements at the Static Mercury Drop Electrode. Analytical Letters, 34, 2125-2140. http://dx.doi.org/10.1081/AL-100106844
- Farias, P.A.M., Wagener, A.D.R. and Castro, A.A. (2001) Adsorptive Voltammetric Behavior of Thymine in Presence of Guanine at the Static Mercury Drop Electrode. Analytical Letters, 34, 1295-1310. http://dx.doi.org/10.1081/AL-100104154
- Farias, P.A.M., Wagener, A.D.R., Junqueira, A.A. and Castro, A.A. (2007) Adsorptive Stripping Voltammetric Beha- vior of Adenosine Triphosphate (ATP) in Presence of Copper at the Mercury Film Electrode. Analytical Letters, 40, 1779-1790. http://dx.doi.org/10.1080/00032710701384717
- Farias, P.A.M., Castro, A.A., Wagener, A.D.R. and Junqueira, A.A. (2007) DNA Determination in the Presence of Copper in Diluted Alkaline Electrolyte by Adsorptive Stripping Voltammetry at the Mercury Film Electrode. Electro- analysis, 19, 1207-1212. http://dx.doi.org/10.1002/elan.200703845
- Glodowski, S., Bilewicz, R. and Kublik, Z. (1986) Determination of Traces of Purine by Cathodic Stripping Voltam- metry at the Hanging Copper Amalgam Drop Electrode. Analytica Chimica Acta, 186, 39-47. http://dx.doi.org/10.1016/S0003-2670(00)81772-9
- Househam, B.C., Van den Berg, C.M.G. and Riley, J.P. (1987) The Determination of Purines in Fresh and Sea-Water by Cathodic Stripping Voltammetry after Complexation with Copper (I). Analytica Chimica Acta, 200, 291-303. http://dx.doi.org/10.1016/S0003-2670(00)83777-0
- Bilewicz, R., Glodowski, S. and Kublik, Z. (1989) The Influence of Adenine on the Electrochemical-Behavior of the Cu(II)/Cu(0) System. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, 274, 201-212. http://dx.doi.org/10.1016/0022-0728(89)87041-X
- Correia dos Santos, M.M., Freire Lopes, C.M.L. and Simões Gonçalves, M.L. (1996) Voltammetric Studies of Purine Bases and Purine Nucleosides with Copper. Bioelectrochemistry and Bioenergetics, 39, 55-60. http://dx.doi.org/10.1016/0302-4598(95)01857-3
- Castro, A.A., de Souza, M.V.N., Rey, N.A. and Farias, P.A.M. (2011) Determination of Efavirenz in Diluted Alkaline Electrolyte by Cathodic Adsorptive Stripping Voltammetry at the Mercury Film Electrode. Journal of the Brazilian Society, 22, 1662-1668.
- Castro, A.A., Aucelio, R.Q., Rey, N.A., Miguel, E.M. and Farias, P.A.M. (2011) Determination of the Antirretroviral Drug Nevirapine in Diluted Alcaline Electrolyte by Adsorptive Stripping Voltammetry at the Mercury Film Electrode. Combinatorial Chemistry & High Throughput Screening, 14, 22-27. http://dx.doi.org/10.2174/1386207311107010022
- Palecek, E., Jelen, F., Hung, M.A. and Lasovsky, J. (1981) Reaction of the Purine and Pyrimidine-Derivatives with the Electrode Mercury. Bioelectrochemistry and Bioenergetics, 8, 621-631. http://dx.doi.org/10.1016/0302-4598(81)87005-5