American Journal of Analytical Chemistry
Vol.4 No.8(2013), Article ID:35435,7 pages DOI:10.4236/ajac.2013.48045
Complex Formation of 1,6-Anhydro-β-Maltose and Sodium Ions Using Single-Crystal X-Ray Crystallography and NMR Spectroscopy
1Department of Chemistry, School of Sciences and Engineering, Meisei University, Hino, Japan
2Solution & Marketing Division, JEOL RESONANCE Inc., Akishima, Japan
Email: tashiro@chem.meisei-u.ac.jp
Copyright © 2013 Takashi Fujimoto et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received May 15, 2013; revised June 17, 2013; accepted July 15, 2013
Keywords: 1,6-Anhydro-β-Maltose; Complex Formation; Single-Crystal X-Ray Crystallography; Solid-State NMR Spectroscopy
ABSTRACT
Complex formation of 1,6-anhydro-β-maltose and sodium ions was characterized using single-crystal X-ray crystallography and solutionand solid-state NMR spectroscopy. The 7-coordination structure, comprising two 1,6-anhydro- β-maltoses, a thiocyanate ion and a sodium ion, was identified in the crystal of the complex, where a sodium ion was positioned in the center of the pentagon. In the NMR study, the line broadening of 23Na signals and the decrease of the spin-lattice relaxation times (T1) of 23Na were observed in CD3OD in the presence of 1,6-anhydro-β-maltose, indicating complex formation.
1. Introduction
The abilities of carbohydrates to form complex with cations have been used in the separation of sugar mixtures and the isolation of single tautomeric forms in solution. One of its application is separation of sugars using cation-exchange chromatography [1,2]. For instance, Dglucose and D-fructose were separated using a Ca2+ column [3], and α and β anomers of D-allose (both as pyranose and furanose forms) were isolated owing to the difference of ability to coordinate with cations [4]. The structural feature of carbohydrates is hydroxyl groups, which is feasible to form complexes in solution with monovalent cations [5,6]. Electrophoretic studies demonstrated that alkali metal ions were able to form complexes with polyhydroxy compounds. The migration rate of 1,6-anhydro-β-D-glucopyranose in the presence of metal ion was extremely higher than that alone, indicating complex formation [6].
Our group has been studying the complex formation of alkali metal ions and 1,6-anhydro sugars [7,8], which were prepared from the corresponding free sugars through intramolecular dehydration. In past studies, the complexation of 1,6-anhydro-β-maltose with rubidium, which of 1,6-anhydro-β-D-glucopyranosewith rubidium [9] and which of 1,6-anhydro-β-maltotriose with potassium [10], was characterized using NMR spectroscopy. Besides these studies, interactions between sucrose and various metal ions were investigated by Rondeau et al. using mid-infrared and 13C NMR spectroscopies [11,12]. The crystal structure of the complex formed by two 1,6-anhydro-β-maltoses with a peroxide and two sodium ions, [Na2(1,6-anhydro-β-maltose)2(H2O)3]O2, was determined by single-crystal X-ray crystallography [13]. The O2 moiety was identified as a discrete peroxide dianion, whereas two sodium and two carbohydrate molecules constituted a binuclear complex counter cation. The crystal structure of 1,6-anhydro-β-maltose forming an inclusion complex with a potassium ion was also determined [14]. In the crystal structure, both 8- and 9-coordination forms were identified. The 8-coordination structure, which can be defined as the distorted capped pentagonal bipyramidal structure, comprised two 1,6-anhydro-β-maltoses, a thiocyanate ion, a methanol and a potassium ion. A potassium ion was positioned in the center of the bipyramidal structure, where two 1,6-anhydro-β-maltoses, possessing identical conformation, surrounded a potassium ion. The 9-coordination structure, which can be defined as the capped hexagonal bipyramidal structure, comprised three 1,6-anhydro-β-maltoses, a thiocyanate ion and a potassium ion. Although a potassium ion was positioned in the center of the bipyramidal structure as observed in 8-coordination structure, three 1,6-anhydro-β-maltoses, possessing identical conformation, surrounded a potassium ion. For the purpose of elucidating the mechanism of 1,6-anhydro sugar-forming complexes with alkali metal ions, we have investigated the complex formation of 1,6-anhydro-β-maltose (Figure 1) with sodium ions using single-crystal X-ray crystallography and NMR spectroscopy. The crystals of the free form of 1,6-anhydro- β-maltose and those of an inclusion complex with a sodium ion were also used in the analysis of the solid-state NMR spectroscopy.
2. Experimental
2.1. Single-Crystal X-Ray Crystallography
Equal molar amounts of 1,6-anhydro-β-maltose (324 mg) and NaSCN (78.6 mg) were dissolved in 3 ml methanol, and the solution was kept at room temperature for 48 hr. After filtration, a colorless prism, having approximate dimensions of 0.20 × 0.20 × 0.20 mm, was mounted in a loop. The 1,6-anhydro-β-maltose (324 mg) was dissolved in 5 ml methanol, and the solution was kept at room temperature for 48 hr. After filtration, a colorless prism, having approximate dimensions of 0.20 × 0.20 × 0.20 mm, was mounted in a loop. All measurements were carried out using a Rigaku RAXIS RAPID imaging plate area detector with graphite monochromated Mo-Kα radiation at 123 K. The crystal and experimental data of the 1,6-anhydro-β-maltose forming an inclusion complex with a sodium ion and its free form are given in Table 1.
2.2. Solid-State NMR Spectroscopy
Crystals of an inclusion complex with a sodium ion and

Figure 1. Structure of 1,6-anhydro-β-maltose.
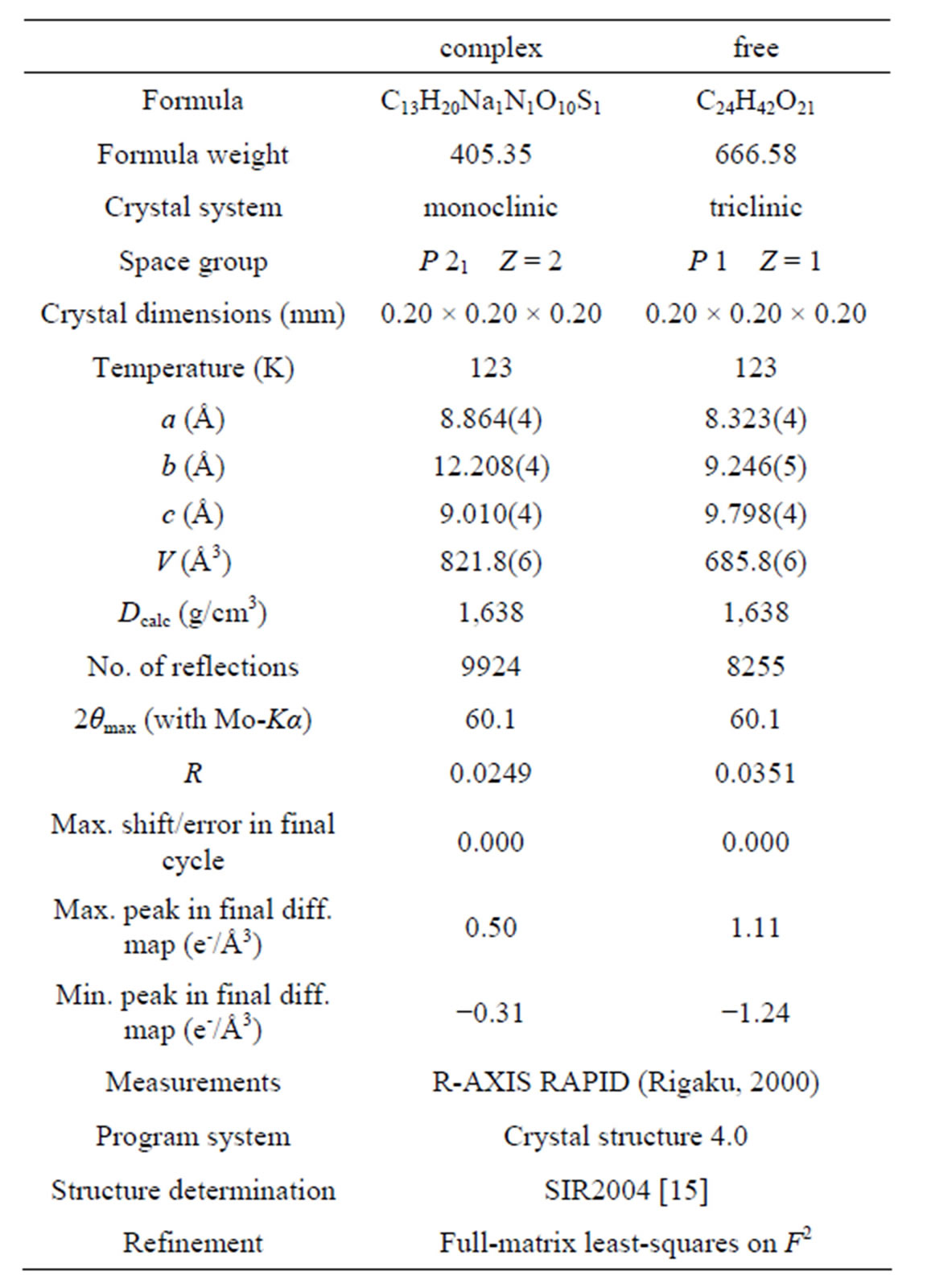
Table 1. Crystal and experimental data of 1,6-anhydro- β-maltose forming an inclusion complex with a sodium ion and the free form of 1,6-anhydro-β-maltose.
of the free form of 1,6-anhydro-β-maltose, and NaSCN alone were used for the measurements of the solid-state NMR spectroscopy. The 23Na spectra were acquired on a JNM-ECA600 spectrometer equipped with a 1 mm CP-MAS probe at a spinning speed of 70 kHz. The experimental parameters of 23Na for an inclusion complex were number of scans = 640, recycle time = 1.0 s, and those for NaSCN alone were number of scans = 256, recycle time = 3.0 s. The 23Na T1ρ were measured using a saturation recovery method on a JNM-ECA920 spectrometer equipped with a 4 mm MQ-MAS probe at a spinning speed of 15 kHz. The experimental parameters were number of scans = 8, recycle time = 1.0 s. The pulse intervals were 15 steps. The 13C CP-MAS spectra were acquired on a JNM-ECA500 equipped with a 4 mm CP-MAS probe at a spinning speed of 15 kHz. The experimental parameters of 13C CP-MAS were number of scans = 319, recycle time = 1.0 s.
2.3. Solution-State NMR Spectroscopy
Conventional 1H, 13C and 23Na NMR spectra were acquired on a Varian 600 spectrometer using 250 μL sample solutions of 30 mM 1,6-anhydro-β-maltose in the presence and absence of 30 mM NaSCN and 30 mM NaSCN alone in 99.9% CD3OD in Shigemi tubes at 30˚C. In preparation of the sample solution of 1,6-anhydro- β-maltose in the presence of NaSCN, crystals of an inclusion complex with a sodium ion were dissolved in CD3OD. The 1H and 13C assignments were carried out using the conventional 1D and 2D NMR techniques. The 13C and 23Na-T1 measurements were carried out using the inversion recovery method, and the measurements of 1H diffusion coefficients were carried out as described previously [10].
3. Results and Discussion
3.1. Single-Crystal X-Ray Crystallography
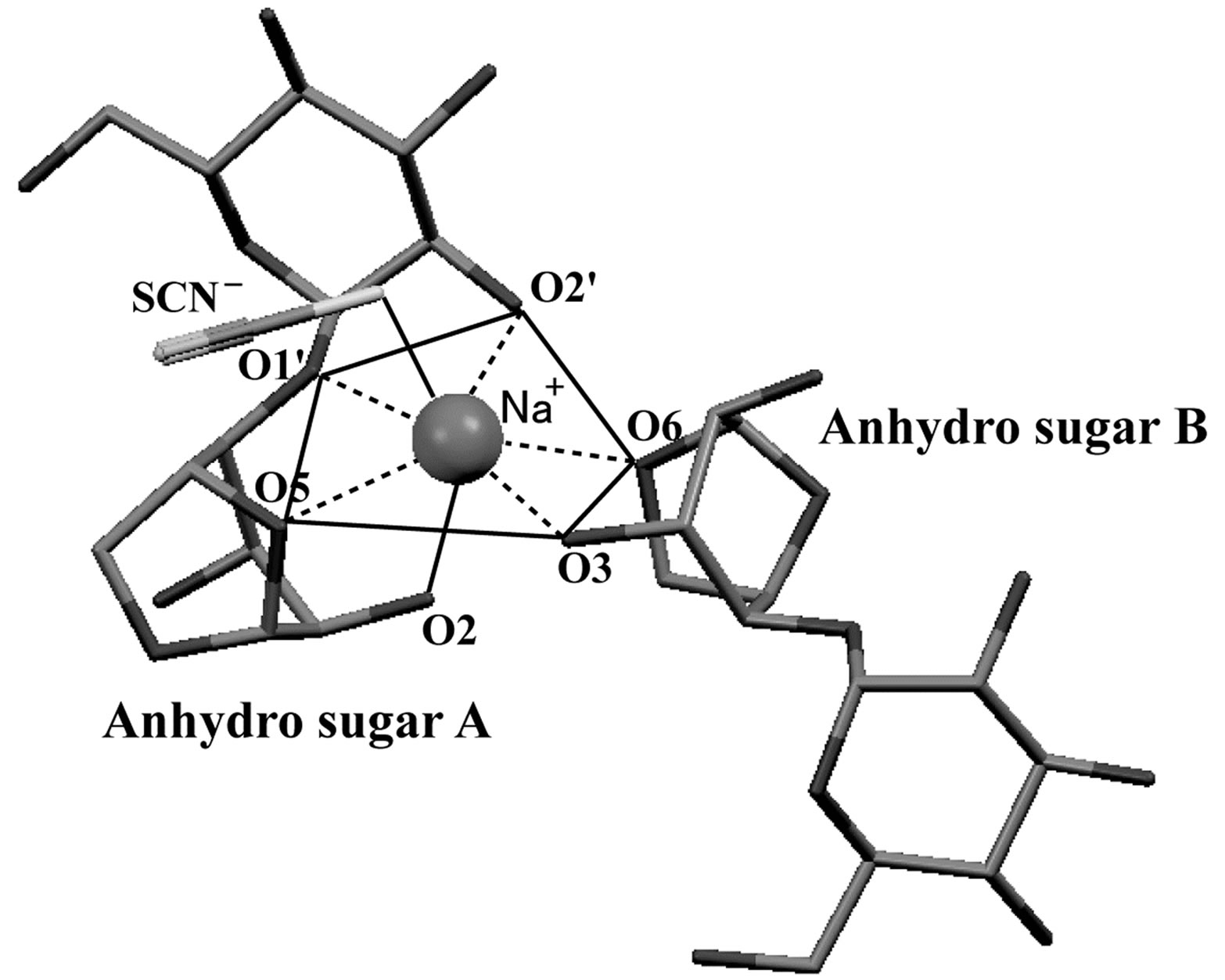
The crystal structures of 1,6-anhydro-β-maltose forming an inclusion complex with a sodium ion and its free form were determined usingsingle-crystal X-ray crystallography (Table 1). In the crystal structure of the complex, the 7-coordination structure, which can be defined as the distorted pentagonal bipyramidal structure, was identified as shown in Figure 2(a). It comprised two 1,6-anhydro-β-maltoses (designated as anhyro sugars A and B), a thiocyanate ion and a sodium ion. A sodium ion was positioned in the center of the pentagon, where two 1,6-anhydro-β-maltoses, possessing identical conformation, surrounded a sodium ion. The sodium ion made close contacts to all oxygen atoms involved in the 7-coordination structure. Its distances were in the range of 2.40 - 2.68 Å (Table 2). The pentagon comprised O5, O1’ and O2’ of anhyro sugar A and O3 and O6 of anhyro sugar B. The O2 of anhyro sugar A and a sulfur atom of a thiocyanate ion corresponded to the peak of each pyramid (Figures 2(a) and (b)). Four of these six oxygen atoms, O2, O3, O5 and O6 were involved in 1,6-anhydro moiety. The tendency forming bipyramidal structures was also identified in the crystal structure of 1,6-anhydro-β-maltose forming an inclusion complex with a potassiumion [14]. These results indicate the importance of 1,6-anhydro moiety in forming complex with alkali metal ions, and provide an insight into the accommodation pattern of 1,6-anhydro-β-maltose for an alkali metal ion.
In the crystal structure of the free form of 1,6-anhydro-β-maltose, asymmetric structures, designated as anhyro sugars A and B, were identified as shown in Figure 2(c). A water molecule was surrounded by three molecules of 1,6-anhydro-β-maltose, forming the 6-corrdination structure (Figures 2(c) and (d)). The close contacts between an oxygen atom of water and 1,6-anhydro-β- maltoses are summarized in Table 2. Only two oxygen atoms in 1,6-anhydro-moiety made close contacts to an oxygen atom of water. This structural feature was opposite to the 7-coordination structure identified in the
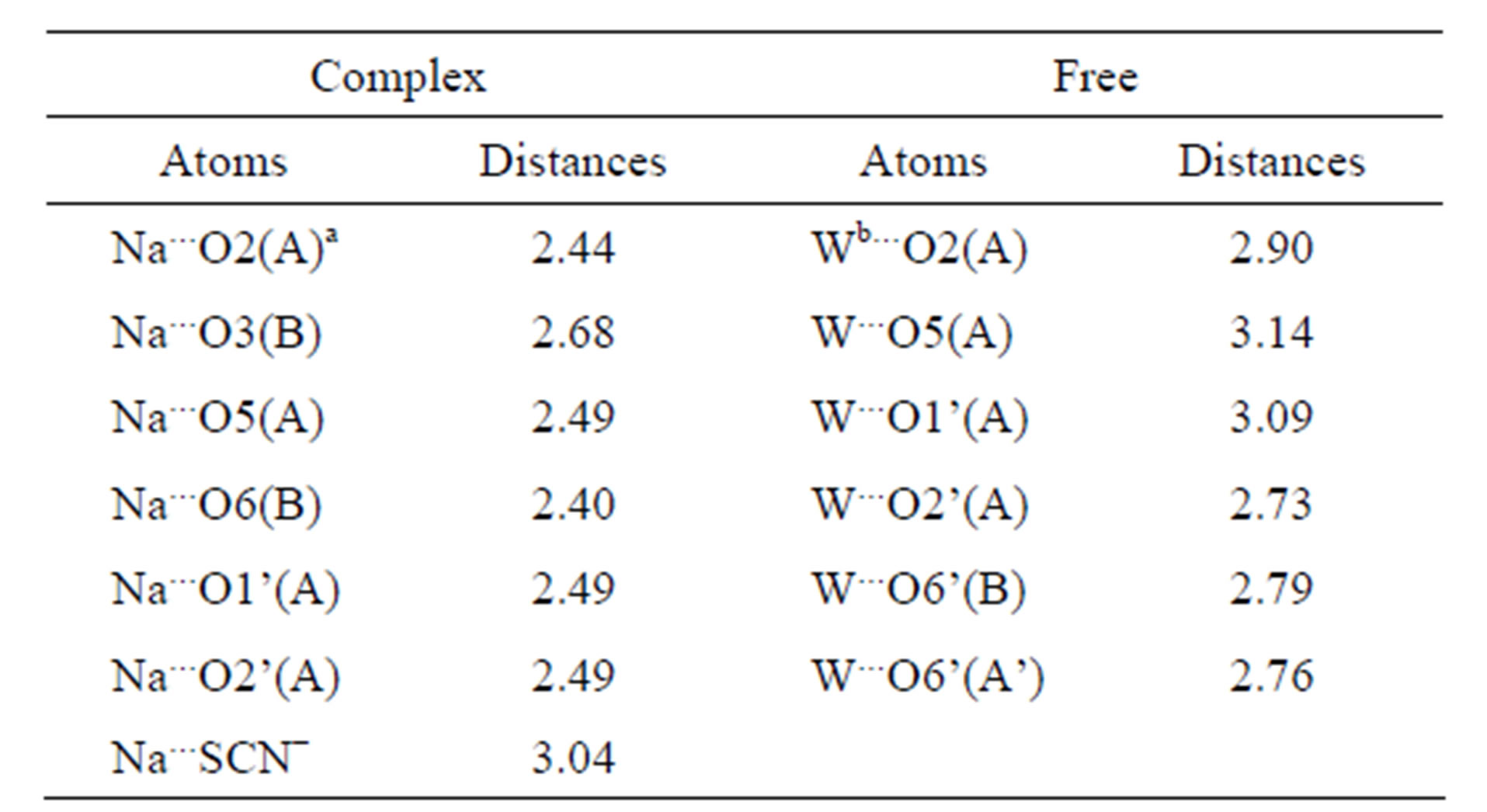
Table 2. Distances (Å) around a sodium ion in the crystal structure of the complex and those around an oxygen atom of the water in the crystal structure of the free form of 1,6-anhydro-β-maltose.
a“A” means anhydro sugar A defined in Figures 2(a) and (b). b“W” means an oxygen atom of the water defined in Figures 2(c) and (d).
crystal structure of the complex.
3.2. Solid-State NMR Spectroscopy
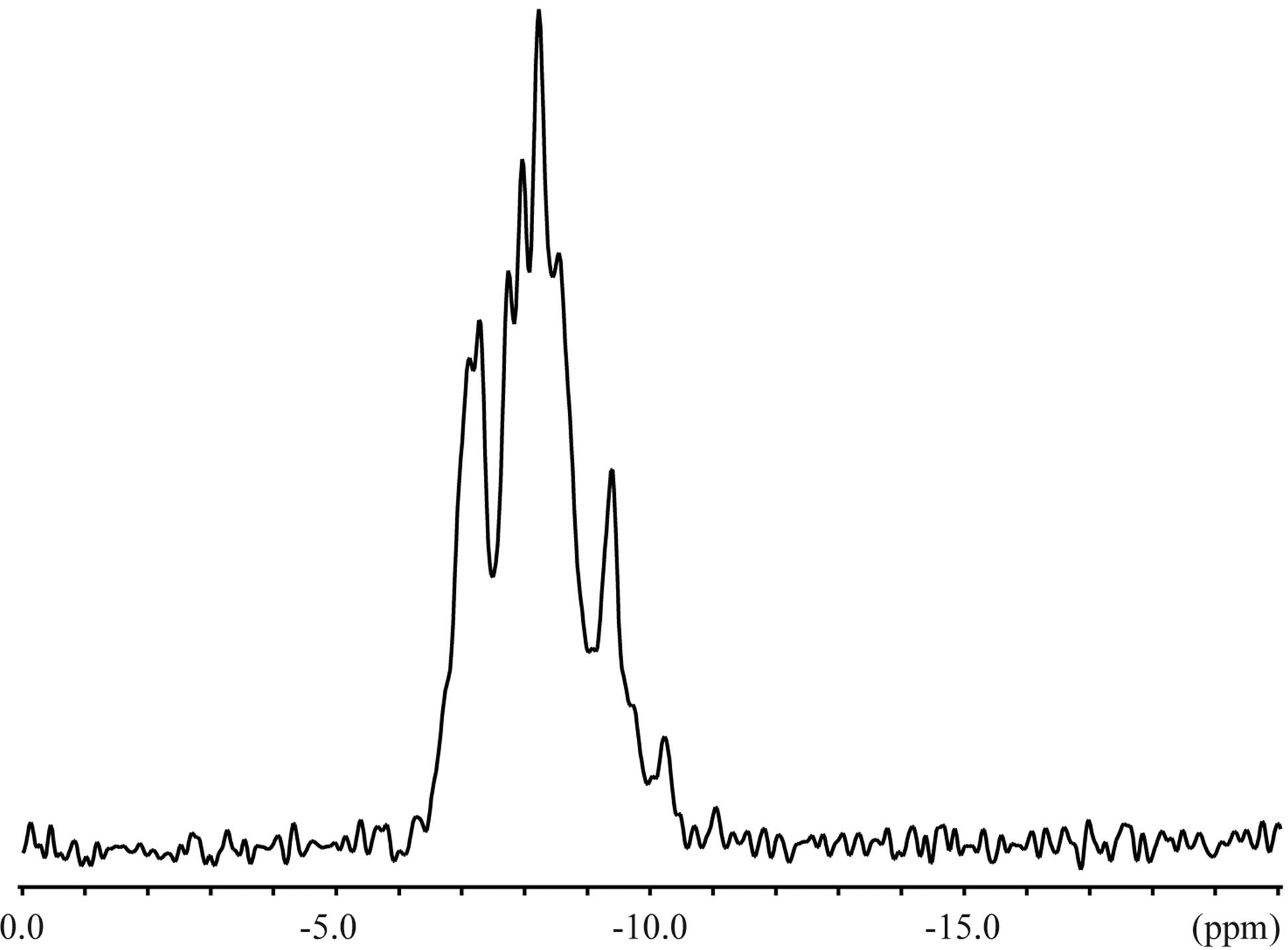
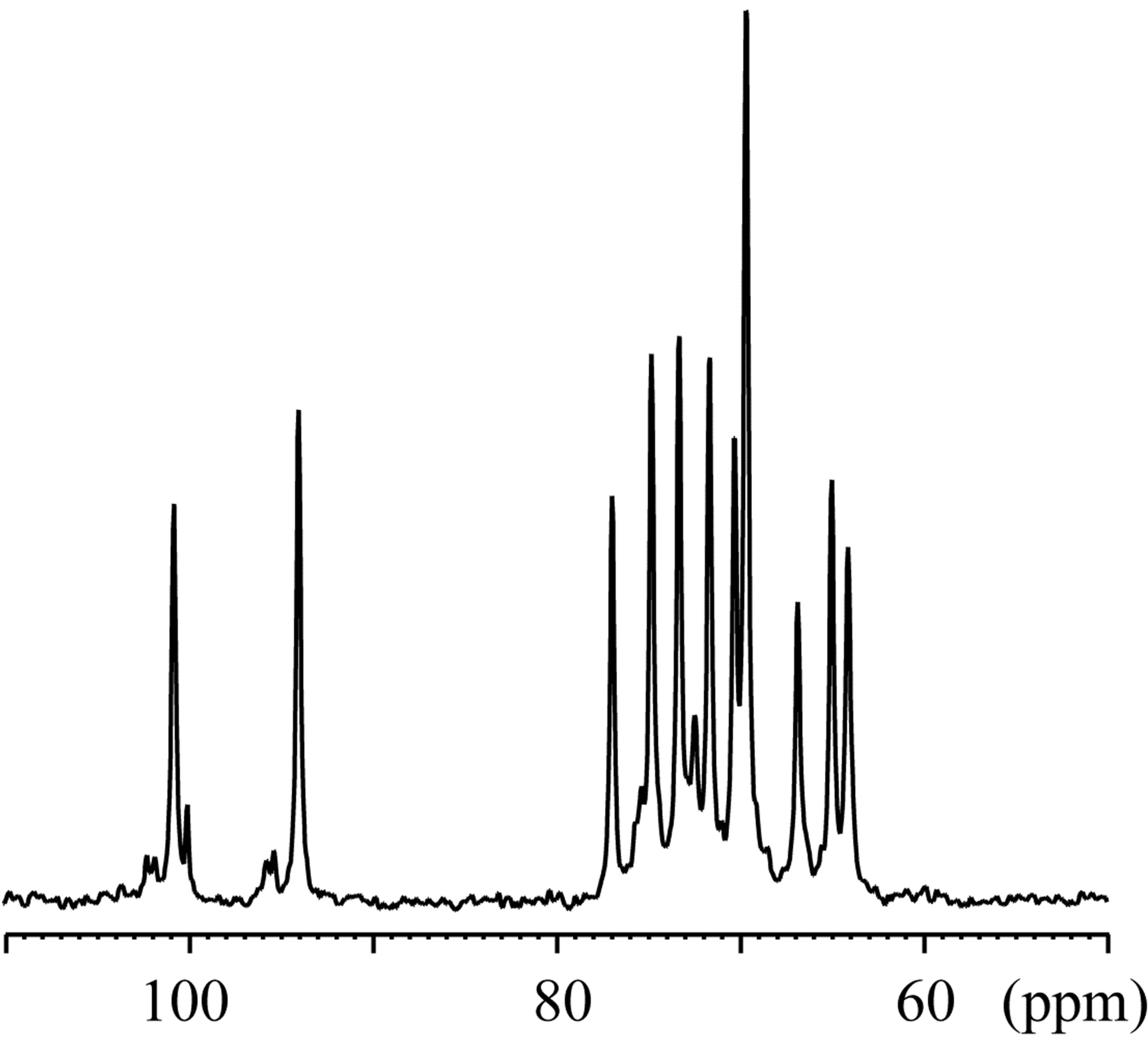
The 23Na solid-state spectra were acquired for the crystals of an inclusion complex with a sodium ion and NaSCN alone as shown in Figure 3. The signal resonating at −0.06 ppm in the free form of NaSCN could be water (Figure 3(b)), considering the hygroscopic property of NaSCN. The 23Na T1ρ were 0.649 s and 6.26 s for the complex and NaSCN alone, respectively. Comparisons of 23Na spectra and T1ρ indicated the various close contacts between 23Na and other nuclei in the inclusion complex. The 13C CP-MAS spectra were acquired for crystals of an inclusion complex of 1,6-anhydro-β-maltose with a sodium ion and those of its free from (Figure 4). As indicated in the single-crystal X-ray crystallography, the free form adopted asymmetric structures. This structural feature was reflected in the 13C CP-MAS spectrum (Figure 4(b)). The signals resonating at 102.2 and 95.6 ppm were assigned as C1 and C1’, respectively, referring the assignments in CD3OD (Table 4). Both signals split as doublet, and its chemical shift difference was 0.39 ppm. However, this doublet pattern could not clearly be identified in the other 13C signals resonating in the upfield region of 64 - 75 ppm. For instance, the 13C signals resonating in the region of 64 - 66 ppm, corresponding to C6 and C6’, did not show the same splitting pattern. In the 13C CP-MAS spectrum of the complex, the singlet pattern was observed for C1 and C1’ (Figure 4(a)), which was expected in consideration of the identical conformation of 1,6-anhydro-β-maltose.
3.3. Solution-State NMR Spectroscopy
The 23Na NMR spectra were acquired in CD3OD in the presence and absence of 1,6-anhydro-β-maltose (Figure 5). The chemical shifts and half height widths of 23Na
 (a)
(a) (b)
(b)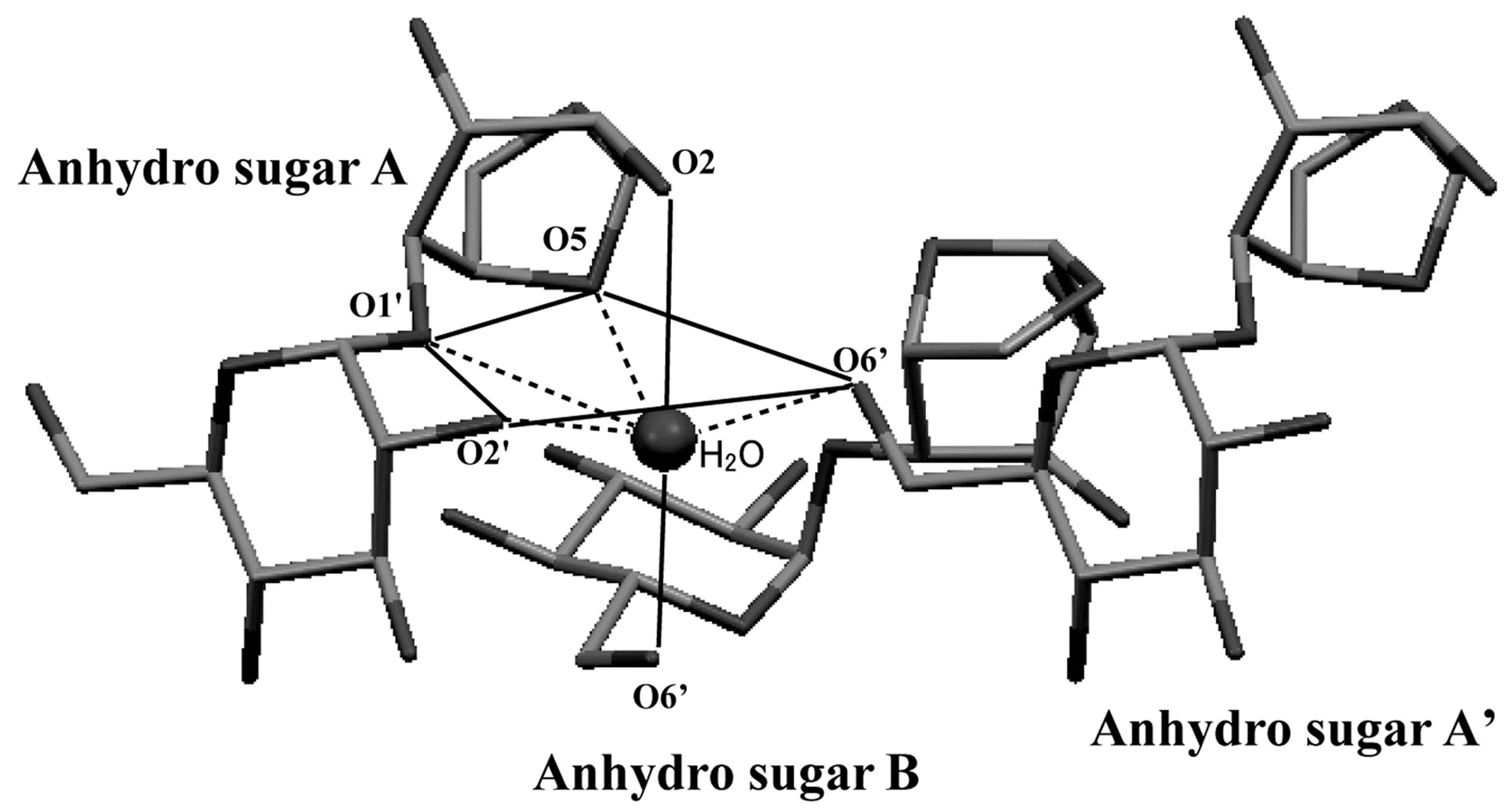 (c)
(c)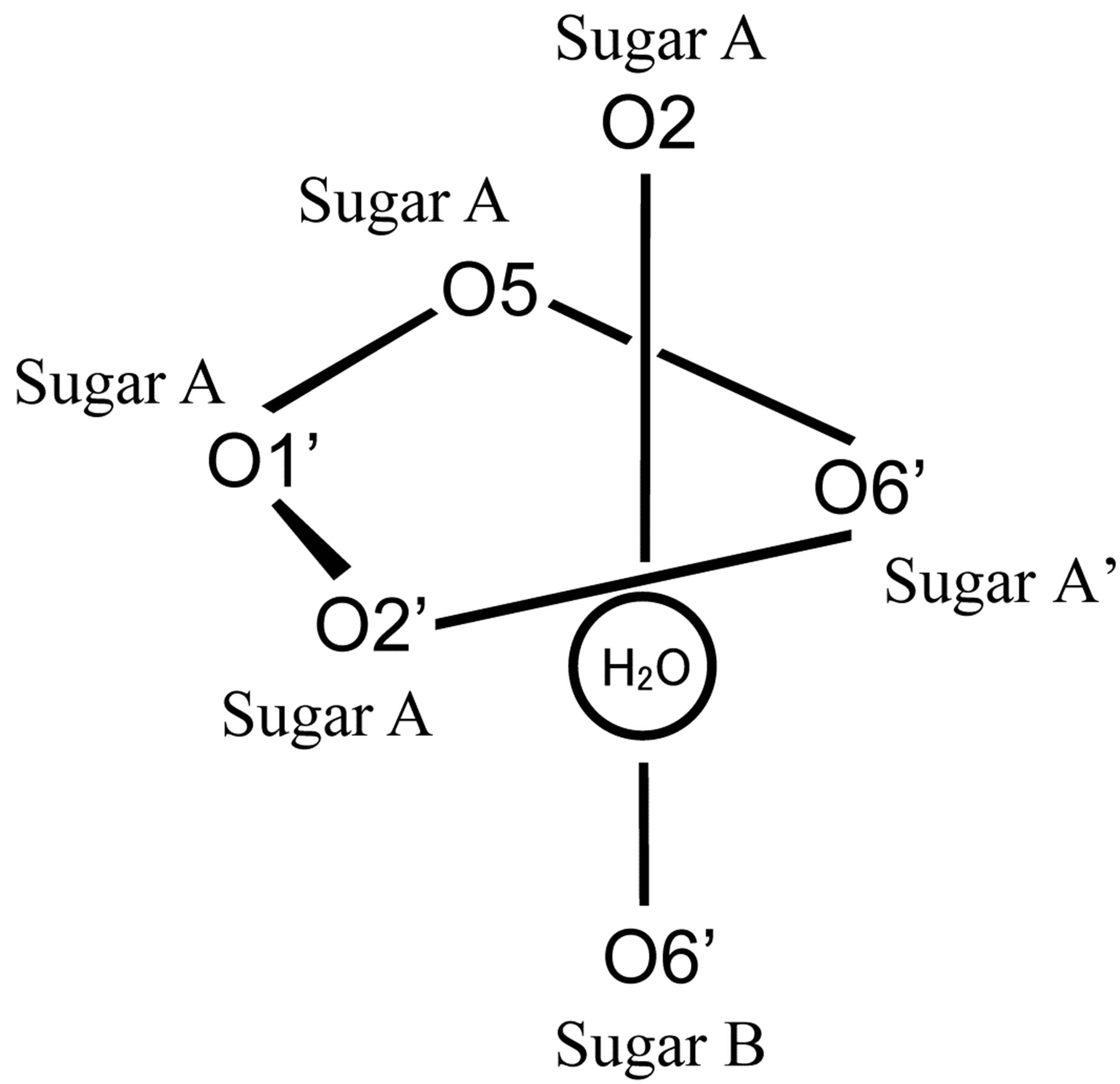 (d)
(d)
Figure 2. (a) The crystal structure of 1,6-anhydro-β-maltose forming an inclusion complex with a sodium ion and (b) its schematic drawing. (c) The crystal structure of the free form of 1,6-anhydro-β-maltose and (d) its schematic drawing. In (a), close contacts between 1,6-anhydro-β-maltoses and a sodium ion are shown by dashed lines, and an outline of 7-coordination structure, corresponding to the schematic drawing of (b), is shown by solid lines. In (c), close contacts between 1,6-anhydro-β-maltoses and an oxygen atom of water are shown by dashed lines, and an outline of 6-coordination structure, corresponding to the schematic drawing of (d), is shown by solid lines.
 (a)
(a)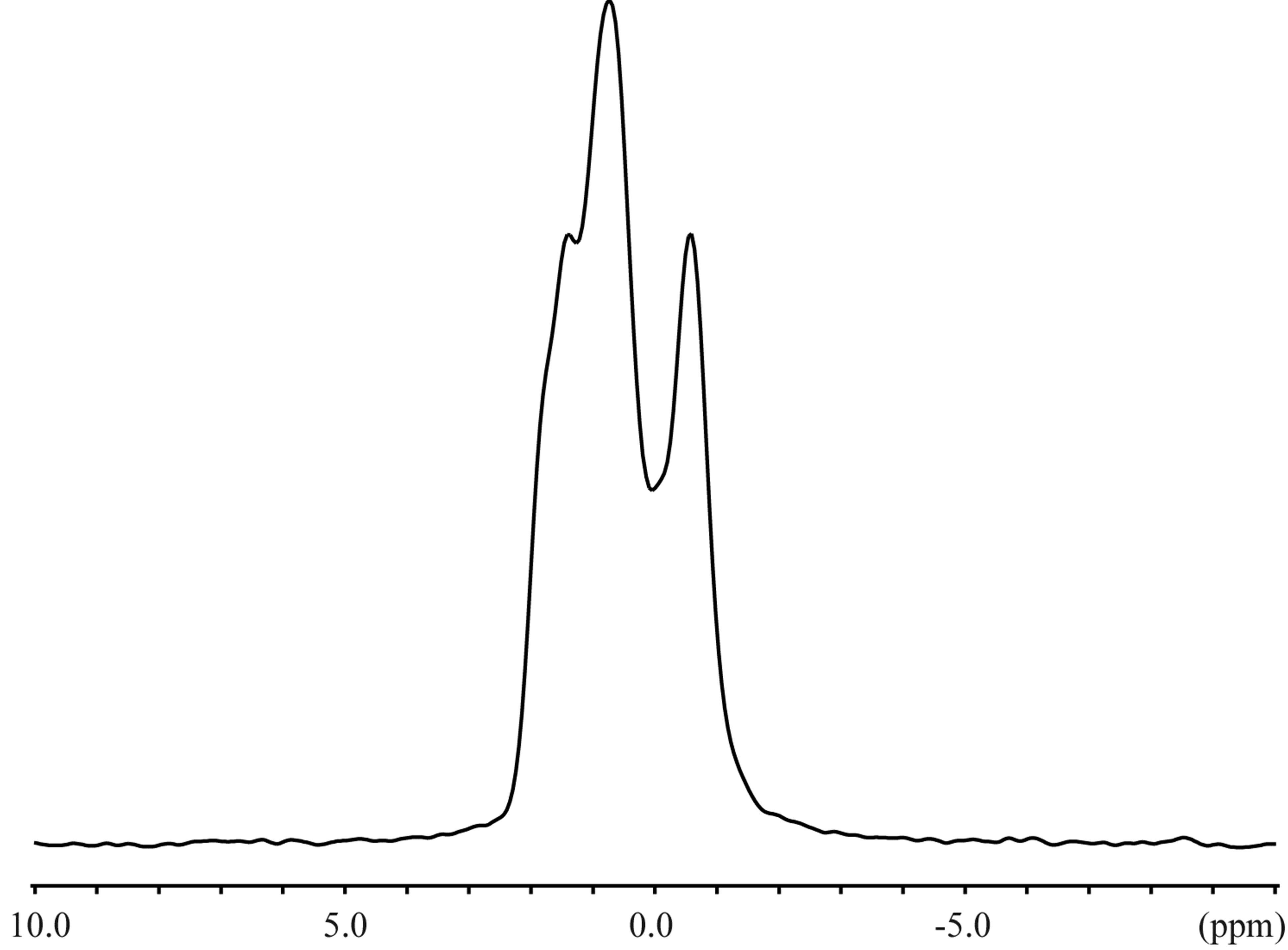 (b)
(b)
Figure 3. The 23Na solid-state NMR spectra of the (a) inclusion complex of 1,6-anhydro-β-maltose with a sodium ion and (b) NaSCN.
signals in the presence and absence of 1,6-anhydro- β-maltose were 0.748 ppm and 32.3 Hz, and 0.479 ppm and 16.8 Hz, respectively. The significant line broadening was observed in the presence of 1,6-anhydro-β-
 (a)
(a)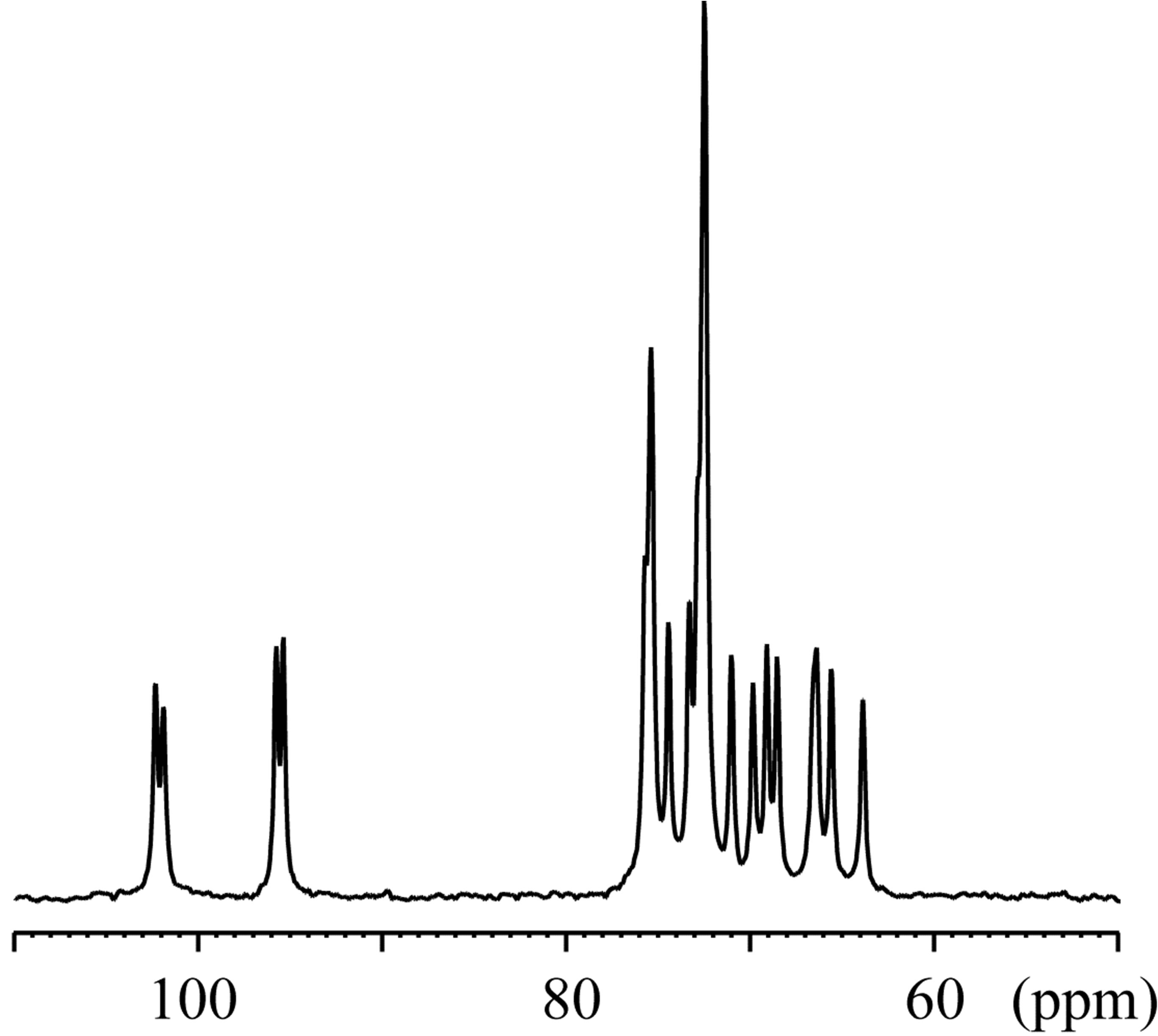 (b)
(b)
Figure 4. The 13C CP-MAS NMR spectra of the crystals of the (a) inclusion complex of 1,6-anhydro-β-maltose with a sodium ion and (b) free form of 1,6-anhydro-β-maltose.
 (a)
(a)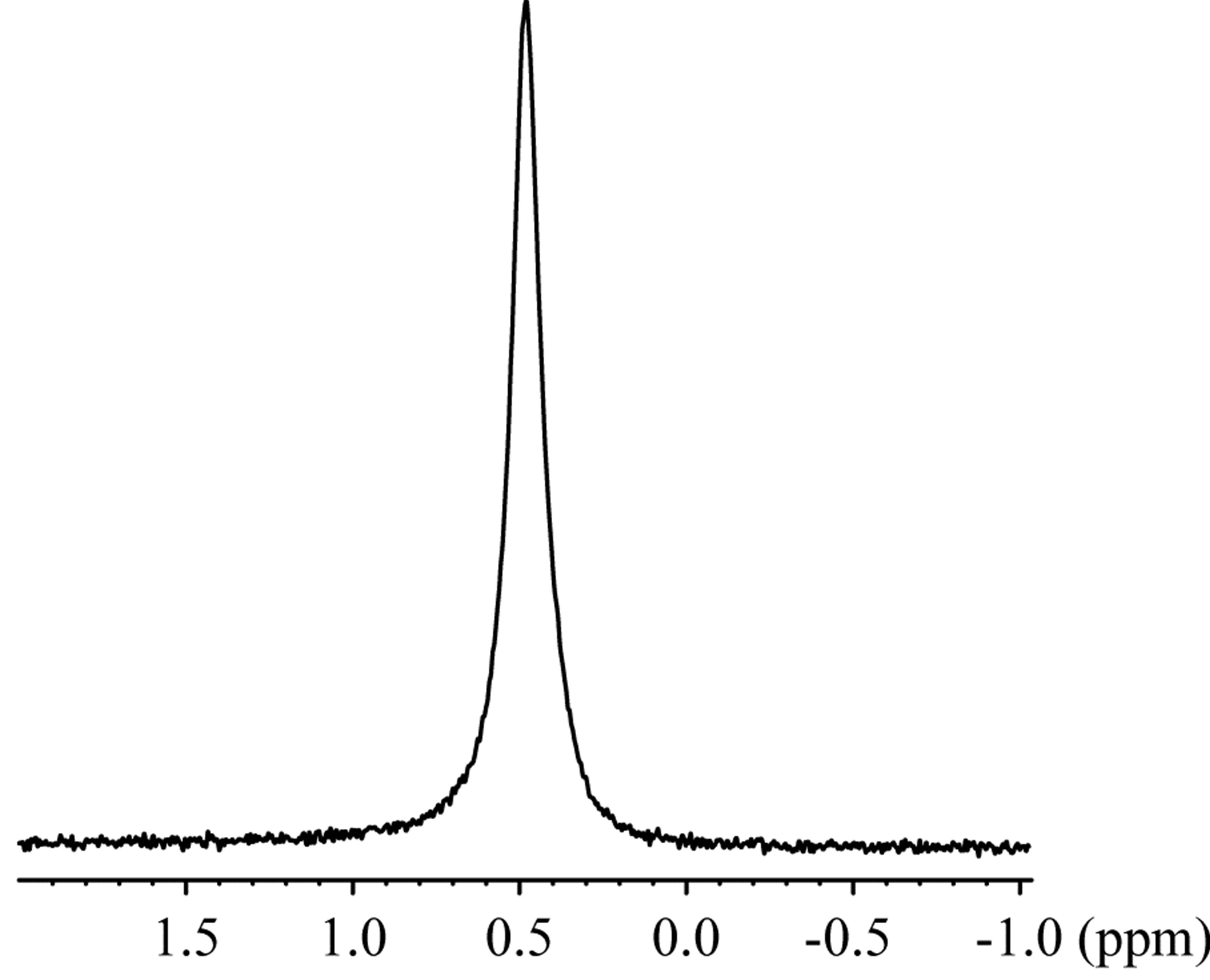 (b)
(b)
Figure 5. The 23Na NMR spectra of the (a) inclusion complex of 1,6-anhydro-β-maltose with a sodium ion and (b) NaSCN in CD3OD.
maltose. The 23Na-T1 values were 9.49 and 21.8 ms in the aforesaid order. The changes of the half height widths and 23Na-T1 unambiguously indicated complex formation of 1,6-anhydro-β-maltose with sodium ions in CD3OD.
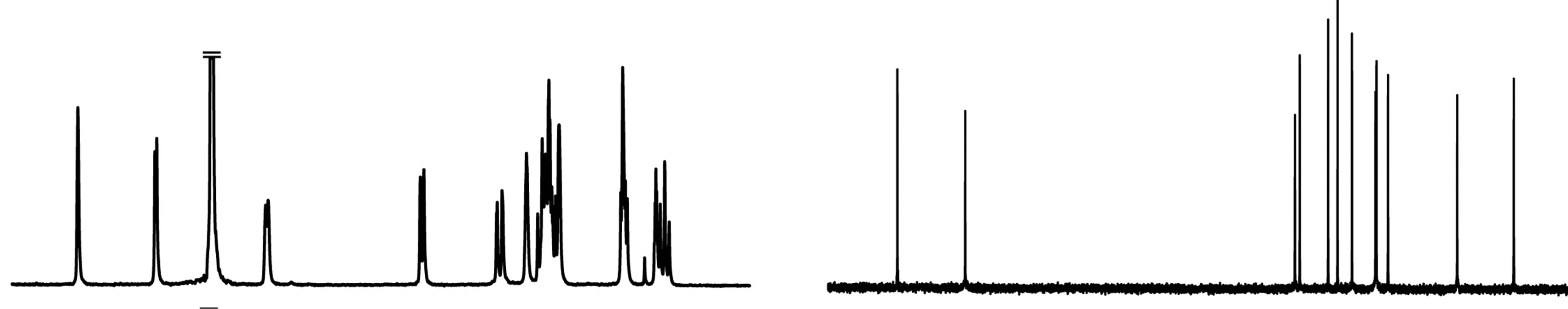
The 1H NMR spectra of 1,6-anhydro-β-maltose in the presence and absence of NaSCN are shown in Figures 6(a) and (c). The distinct changes of the spectral pattern were observed in the chemical shift range of 3.24 - 3.75 ppm. The maximum difference of 1H chemical shift was 0.04 ppm in H4, and the overall 1H chemical shift changes were relatively small (Table 3). The 1H diffusion coefficients were calculated only for the isolated signals, and ca. 14% decrease was identified in the presence of NaSCN (Table 3). The diffusion coefficient of a molecule is inversely proportional to the molecular radius, and the lowered diffusion is attributable to the complex formation. The 13C NMR spectra of 1,6-anhydro-β-maltose in the presence and absence of NaSCN are shown in Figures 6(b) and (d), and comparisons of 13C chemical shifts and 13C-T1 are listed in Table 4. The 13C chemical shifts of C3 and C1’ were both upfield shifted for 0.7 ppm in the presence of NaSCN. In comparison of 13C-T1, a ca. 14% decrease was observed for C1, C2 and C3 in the presence of NaSCN (Table 4). The C1, C2 and C3 are positioned adjacent to the oxygen atoms forming the 7-coordination structure (Figure 2(b)), which could be the reason of affecting its 13C-T1. Because the distinct decrease of 13C-T1 was identified in a portion of 1,6-anhydro moiety, the results of 13C-T1 studies indicated the importance of the 1,6-anhydro moiety in the complex formation, which was also supported in the analysis of the X-ray crystal structure.
4. Conclusion
Complex formation of 1,6-anhydro-β-maltose and sodium ions has been elucidated using single-crystal X-ray crystallography and1H, 13C and 23Na NMR spectroscopy.
 (a)
(a)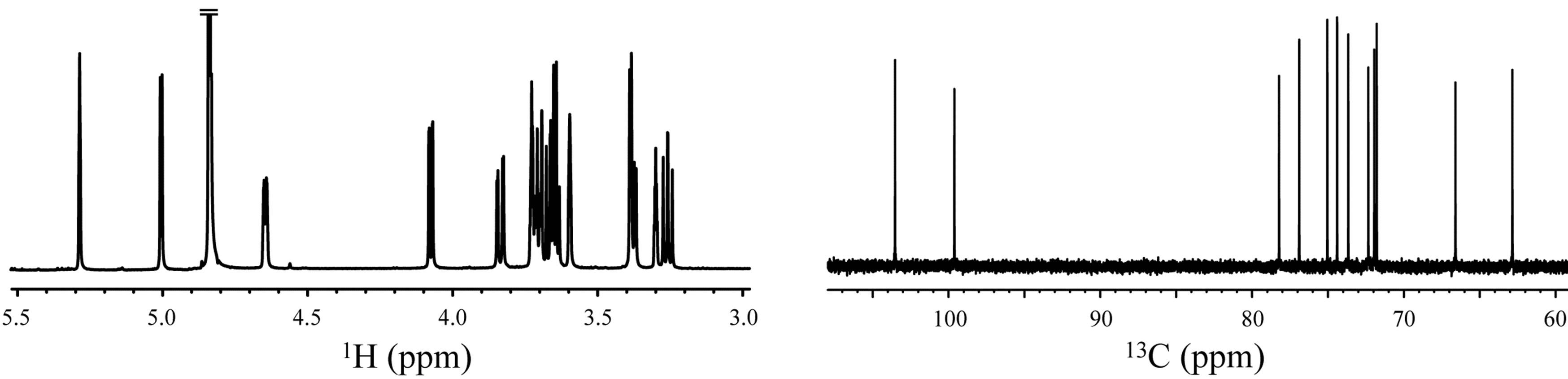 (b)
(b)
Figure 6. The 1H and 13C NMR spectra of 1,6-anhydro-β-maltose in the (a,b) presence and (c,d) absence of NaSCN in CD3OD.
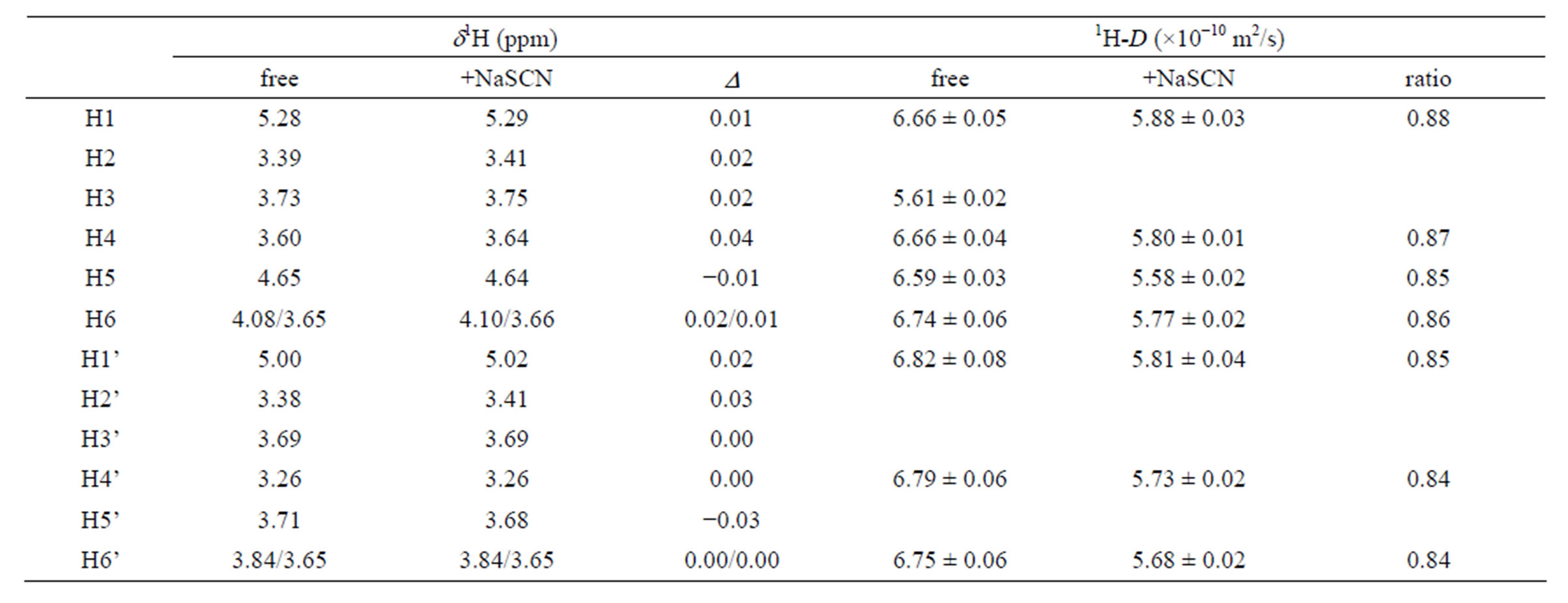
Table 3. Comparison of the 1H chemical shifts and 1H diffusion coefficients (D) of 1,6-anhydro-β-maltose in the presence and absence of NaSCN.

Table 4. Comparison of the 13C chemical shifts and 13C-T1 of 1,6-anhydro-β-maltose in the presence and absence of NaSCN.
In solution, the comparisons of 1H diffusion coefficients and 23Na-T1 unambiguously indicated complex formation, and 13C NMR studies indicated importance of 1,6-anhydro moiety in the complex formation. A contribution of 1,6-anhydro moiety was also recognized in the 7-corrdination structure identified in the single-crystal X-ray structure.
5. Supplementary Data
Crystallographic data, excluding structure factors, have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication with CCDC No. 927906 and 922475 for the free form of 1,6-anhydro- β-maltose and its complex with NaSCN, respectively. Copies of the data can be obtained free of charge on application with the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax +44-1223-336-033; e-mail: deposit@ccdc.cam.ac.uk).
6. Acknowledgements
The authors wish to thank Dr. M. Nakano for measuring 23Na-T1ρ. This work was partly supported by Nanotechnology Platform Program (Molecule and Material Synthesis) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), the Joint Studies Program of the Institute for Molecular Science (IMS), Japan. This work was also supported by an Ito Science Foundation and a Grant-in-Aid for Scientific Research (No. 21550092 and 24550108 for M.T.) from the Ministry of Education, Culture, Sports, Science, and Technology.
REFERENCES
- S. J. Angyal, “Complexes of metal cations with carbohydrates in solution,” Advances in Carbohydrate Chemistry & Biochemistry, Vol. 47, 1989, pp. 1-43. doi:10.1016/S0065-2318(08)60411-4
- B. Gyurcsik and L. Nagy, “Carbohydrates as Ligands: Coordination Equilibria and Structure of the Metal Complexes,” Coordination Chemistry, Vol. 203, No. 1, 2000, pp. 81-149. doi:10.1016/S0010-8545(99)00183-6
- P. E. Barker and S. Thawait, “Separation of Fructose from Carbohydrate Mixtures by Semi-Continuous Chromatography,” Chemistry and Industry (London), Vol. 21, 1983, pp. 817-821.
- S. J. Angyal, G. S. Bethell and R. J. Beveridge, “The Separation of Sugars and of Polyols on Cation-Exchange Resins in the Calcium Form,” Carbohydrate Research, Vol. 73, No. 1, 1979, pp. 9-18. doi:10.1016/S0008-6215(00)85471-3
- M. D. Diaz and S. Berger, “Studies of the Complexation of Sugars by Diffusion-Ordered NMR Spectroscopy,” Carbohydrate Research, Vol. 329, No. 1, 2000, pp. 1-5. doi:10.1016/S0008-6215(00)00239-1
- K. F. Morris, P. Stilbs and C. S. Johnson, Jr. “Analysis of Mixtures Based on Molecular Size and Hydrophobicity by Means of Diffusion-Ordered 2D NMR,” Analytical Chemistry, Vol. 66, No. 2, 1994, pp. 211-215. doi:10.1021/ac00074a006
- C. Schuerch, “Synthesis and Polymerization of Anhydro Sugars,” Advances in Carbohydrate Chemistry & Biochemistry, Vol. 39, 1981, pp. 157-212. doi:10.1016/S0065-2318(08)60206-1
- T. Tanaka, W. C. Huang, M. Noguchi, A. Kobayashi and S. Shoda, “Direct Synthesis of 1,6-Anhydro Sugars from Unprotected Glycopyranoses by Using 2-Chloro-1,3-Dimethylimidazolinium Chloride,” Tetrahedron Letters, Vol. 50, No. 18, 2009, pp. 2154-2157. doi:10.1016/j.tetlet.2009.02.171
- T. Fujimoto, S. Sakuraki, A. Tsutsui, K. Furihata, T. Machinami and M. Tashiro, “Observation of 1,6-anhydro- β-D-Maltose and 1,6-Anhydro-β-D-Glucopyranosecomplexed with Rubidium by NMR Spectroscopy and Electrospray Ionization Mass Spectrometry,” Analytical Sciences, Vol. 21, No. 10, 2005, pp. 1245-1247. doi:10.2116/analsci.21.1245
- T. Kato, K.Tsubono, O.Kamo, T. Kato, K. Furihata, T. Fujimoto, T. Machinami and M. Tashiro, “Characterization of the Complex Formation of 1,6-Anhydro-β-Maltotriose with Potassium using 1H and 39K NMR Spectroscopy,” Magnetic Resonance in Chemistry, Vol. 47, No. 11, 2009, pp. 948-952. doi:10.1002/mrc.2495
- P. Rondeau, S. Sers, D. Jhurry and F. Cadet, “Sugar Interaction with Metals in Aqueous Solution: Indirect Determination from Infrared and Direct Determination from Nuclear Magnetic Resonance Spectroscopy,” Applied Spectroscopy, Vol. 57, No. 4, 2003, pp. 466-472. doi:10.1366/00037020360626023
- P. Rondeau, D. Jhurry, S. Sers and F. Cadet, “Study of the Interactions between Sucrose and Metal Ions (Mg2+ and K+) and Their Simultaneous Quantification in Ternary Mixture by Mid-Infrared and 13C Nuclear Magnetic Resonance Spectroscopies,” Applied Spectroscopy, Vol. 58, No. 7, 2004, pp. 816-822. doi:10.1366/0003702041389238
- T. Kato, T. Fujimoto, A. Tsutsui, M. Tashiro, Y. Mitsutsuka and T. Machinami, “Identification of a Descrete Peroxide Dianion, O22-, in a Two Sodium-(1,6-Anhydro- β-Maltose)2-Peroxide Complex,” Chemistry Letters, Vol. 39, No. 2, 2010, pp. 136-137. doi:10.1246/cl.2010.136
- T. Fujimoto, T. Kato, Y. Usui, O. Kamo, K. Furihata, K. Tsubono, T. Kato, T. Machinami and M. Tashiro, “Characterization of the Complex Formation of 1,6-Anhydro- β-Maltose and Potassium Ions Using NMR Spectroscopy and Single-Crystal X-Ray Crystallography,” Carbohydrate Research, Vol. 346, No. 13, 2011, pp. 1991-1996. doi:10.1016/j.carres.2011.06.017
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori and R. Spagna, “SIR2004: An Improved Tool for Crystal Structure Determination and Refinement,” Journal of Applied Crystallography, Vol. 38, No. 2, 2005, pp. 381-388. doi:10.1107/S002188980403225X